Truncated mutants of the putative Wnt receptor LRP6/Arrow can stabilize β-catenin independently of Frizzled proteins (original) (raw)
. Author manuscript; available in PMC: 2008 Aug 4.
Published in final edited form as: Oncogene. 2004 Jun 17;23(28):4873–4884. doi: 10.1038/sj.onc.1207642
Abstract
Secreted signaling proteins of the Wnt family are known to regulate a diverse range of developmental processes and their signaling pathway through β-catenin is frequently activated in cancer. The identification of both Frizzled and LRP5/6 proteins as components of cell surface receptors for Wnt proteins has raised questions about their individual functions. We have investigated this issue through a structure-function analysis of Frizzled and LRP proteins that have been implicated in Wnt1 signaling. Consistent with other reports, we find that LRP6/Arrow proteins deleted for their extracellular domain are able to activate the Wnt/β-catenin signaling pathway. Importantly, our results demonstrate that this signaling from LRP6/Arrow derivatives can occur in a Frizzled- and ligand-independent manner. Furthermore we show that the PPSP motifs within the intracellular domain of LRP6 are required for signaling. In contrast to results with LRP6, overexpression of Frizzled proteins did not activate the pathway. Based on evidence of ligand binding to both Frizzled and LRP6, current models suggest that both proteins are components of a Wnt receptor complex that signals to β-catenin. In light of these models, our data imply that LRP5/6/Arrow proteins constitute the distal signal initiating component of these receptors. The results also support the notion that LRP5/6 are candidate oncogenes.
Keywords: Wnt signaling, β-catenin, Receptors, Frizzled, LRP6, Arrow
INTRODUCTION
The Wnt family of secreted proteins is widely recognized as one of the major families of extracellular signaling factors in animal development (Cadigan & Nusse, 1997; Nusse & Varmus, 1992). Conserved among metazoans as diverse as humans and hydra, Wnt proteins have been shown to regulate numerous key aspects of development. In mammals there are 19 different Wnt proteins and their functions include crucial roles in gastrulation, body axis specification, brain morphogenesis, kidney formation, and limb patterning (reviewed in (Huelsken & Birchmeier, 2001; Uusitalo et al., 1999)). In addition, excessive Wnt signaling is associated with cancer, both in the formation of precancerous lesions at risk of progression (reviewed in (Polakis, 2000)), and in the progression of malignant tumors to a more invasive phenotype (Weeraratna et al., 2002). At the cellular level, Wnt-mediated signals have been shown to affect proliferation, transformation, aggregation, rescue from apoptosis and, most commonly, cell fate determination (Bradley & Brown, 1995; Cadigan & Nusse, 1997; Longo et al., 2002; Shimizu et al., 1997; Toyofuku et al., 2000; You et al., 2002).
Much progress has been made in elucidating the intracellular signaling mechanisms activated by Wnt proteins (reviewed in (Huelsken & Behrens, 2002)). By far the best characterized of these is the canonical Wnt/β-catenin pathway, which is activated by many different Wnt proteins (Cadigan & Nusse, 1997; Shimizu et al., 1997). A key component of this pathway is β-catenin, whose stability in the cytosol is controlled by the multiprotein Axin-APC complex. In response to an appropriate Wnt ligand at the cell surface, the activity of this complex is inhibited and β-catenin accumulates in the cytosol. β-catenin then enters the nucleus, where it interacts with members of the TCF protein family and regulates the transcription of specific target genes (Eastman & Grosschedl, 1999; Hecht & Kemler, 2000).
While many details of this intracellular signaling pathway are known, and activation of the pathway by mutation has been documented in a wide variety of human cancers (Polakis, 2000), relatively little is known about mechanisms of Wnt signal transduction across the plasma membrane. To date, three classes of protein have been implicated as components of cell surface receptors that transduce Wnt protein signals to the canonical β-catenin pathway: members of the Frizzled family of seven-transmembrane domain proteins (Bhanot et al., 1999; Deardorff et al., 2001; Gazit et al., 1999; Hsieh et al., 1999), the LDL-receptor related proteins LRP5 and LRP6 (Pinson et al., 2000; Tamai et al., 2000; Wehrli et al., 2000), and certain heparan sulfate proteoglycans (Lin & Perrimon, 1999; Tsuda et al., 1999). Although absence of specific proteoglycans can prevent Wnt signaling under certain circumstances, evidence from Drosophila indicates that the requirement for these molecules can be overcome by excess ligand (Binari et al., 1997). This suggests that proteoglycans facilitate Wnt signaling but are not essential receptor components. In contrast, there is genetic evidence that a Fzd family member and an LRP are absolutely required for signal transduction (Bhanot et al., 1999; Chen & Struhl, 1999; Wehrli et al., 2000).
Frizzled (Fzd/fz) family members were initially implicated as Wnt receptors because Schneider2 (S2) cells transfected with Dfz2 were shown to bind the Wnt1 homolog Wingless and undergo activation of downstream signaling (Bhanot et al., 1996). There is functional redundancy between Dfz2 and fz (Dfz1) in the Drosophila embryo, but concurrent mutation of both genes results in a wingless-like phenotype (Bhanot et al., 1999; Chen & Struhl, 1999). Further evidence consistent with Frizzled proteins acting as Wnt receptors has also been described in vertebrate systems (Deardorff et al., 1998; Deardorff et al., 2001; Gazit et al., 1999; He et al., 1997; Hsieh et al., 1999; Yang-Snyder et al., 1996), although the existence of 9 different Frizzled (Fzd) genes in mice adds potential complexity to the analysis of their individual functions by gene knock-out experiments.
In addition to the compelling evidence for Frizzleds as Wnt receptors, it was subsequently reported that the Drosophila segment polarity gene arrow behaves genetically as would be expected for a Wingless receptor gene (Wehrli et al., 2000). The product of arrow is a single pass transmembrane protein that is homologous to the LDL receptor-related proteins LRP5 and LRP6 in mammals. Remarkably, targeted gene disruption of LRP6 results in mice with multiple developmental defects that resemble a combination of several known Wnt gene knockout phenotypes (Pinson et al., 2000). Collectively these results suggest that both Frizzleds and LRP5/6 are necessary components of Wnt receptors. The extracellular domains of both types of protein have been shown to bind Wnts (Hsieh et al., 1999; Tamai et al., 2000) and it has been reported that Frizzled and LRP6 extracellular domains become physically associated in the presence of Wnt1, implying formation of a ternary complex (Tamai et al., 2000).
The above data implicating both Frizzled proteins and LRP5/6 as components of receptors for the canonical Wnt pathway raise important questions about the functional contribution of these different components. What, for example, are their respective roles in Wnt signal transduction, and are one or both components needed for signal transmission to the Axin-APC complex and β-catenin? To address these questions, we have examined the signaling potential of representative Frizzled and LRP6 derivatives in cultured cells. While we were unable to elicit signaling to β-catenin by expression of Frizzled proteins, our results show that mouse LRP6 or Drosophila Arrow derivatives that lack their extracellular domains are able to activate the canonical Wnt signaling pathway in both a ligand-independent and Frizzled-independent manner. These data imply that LRP5/6/Arrow proteins are the principal distal signal initiating components of Wnt receptors for the canonical Wnt/β-catenin pathway.
RESULTS
Inhibitory forms of Frizzled and LRP
We began our analysis of Wnt receptor components by evaluating the ability of Frizzled and LRP5/6 derivatives to antagonize ligand-induced canonical Wnt/β-catenin signaling. Fzd8 and LRP6 were chosen as representatives of each family and we generated constructs that expressed the N-terminal extracellular domain of each protein in a soluble form (Fzd8 Ex and LRP6 Ex, Figure 1). These were transfected into 293T cells, together with a Wnt1 expression vector, and cytosolic β-catenin levels were analyzed by Western blotting. Expression of Wnt1 alone in this system induced robust stabilization of β-catenin, but co-expression of Fzd8 Ex substantially antagonized this effect (Figure 2A). Similar results were obtained when cells were treated with conditioned media containing soluble forms of Wnt1 and Fzd8 Ex (data not shown). These results are consistent with previous reports that the ligand-binding domain of Fzd molecules can act as a competitive inhibitor of canonical Wnt signaling (Hsieh et al., 1999; Leyns et al., 1997; Uren et al., 2000; Wang et al., 1997). Expression of the extracellular domain of LRP6 also attenuated Wnt-mediated stabilization of β-catenin in co-transfection assays although, consistent with other reports, expression of a membrane-anchored form of this domain (LRP6-Δ173) was even more potent and resulted in complete antagonism of the Wnt1 signal (Figure 2B; (Mao et al., 2001a; Mao et al., 2001b; Tamai et al., 2000)). These results demonstrate that the extracellular domains of either a Frizzled or an LRP protein can inhibit Wnt-mediated signaling to β-catenin, and it is most likely that they do so by competing with essential components of the endogenous Wnt receptors in these cells.
Figure 1. Diagram of LRP6 and Frizzled constructs used in this study.
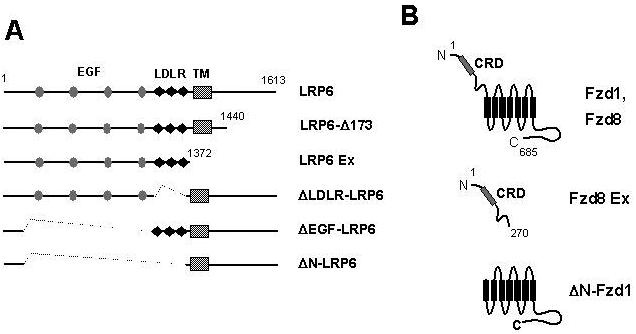
(A) Full-length LRP6 and deletion derivatives, showing EGF-like repeats (EGF, gray ovals), LDLR repeats (LDLR, black diamonds) and transmembrane domain (TM, hatched box). (B) Full-length Frizzled 1 and Frizzled 8 (Fzd1, Fzd8), showing the extracellular cysteine-rich domain (CRD) and seven transmembrane domains (black rectangles); the soluble extracellular domain construct Fzd8 Ex; and ΔN-Fzd1, from which the extracellular domain is deleted.
Figure 2. Dominant negative forms of both LRP6 and Frizzled inhibit Wnt/β-catenin signaling.
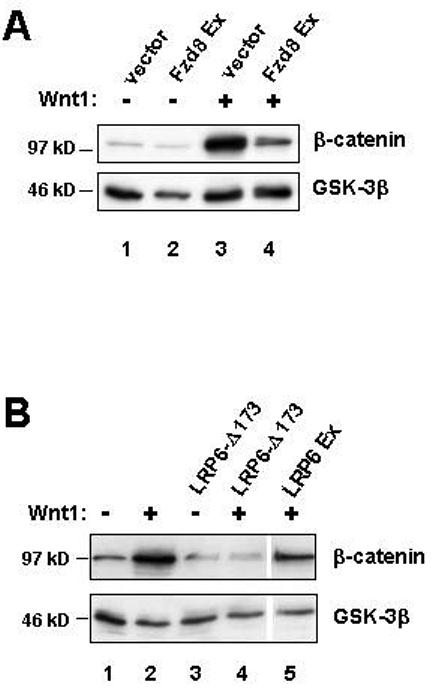
(A) Dominant negative Fzd8 inhibits cytosolic β-catenin stabilization in response to Wnt1 signaling. Western blot analysis of cytosolic β-catenin levels in 293T cells transiently transfected with either empty vector (lanes 1 and 2), or vector encoding Wnt1 (lanes 3 and 4), in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of a Fzd8 Ex expression construct. Analysis of GSK-3β provides a loading control (lower panel). (B) Dominant negative LRP6 inhibits cytosolic β-catenin stabilization. 293T cells were transiently transfected with empty vector (lanes 1 and 3) or a Wnt1 expression vector (lanes 2, 4, and 5) in the absence (lanes 1 and 2) or presence (lanes 3, 4 and 5) of the indicated LRP6 dominant negative constructs. Cytosolic β-catenin levels and GSK-3β were analyzed as in (A).
Effects of Fzd overexpression
Next we wished to test the hypothesis that overexpression of individual receptor components, or mutants thereof, might be sufficient to activate signaling in the absence of exogenous ligand. This approach was based on numerous precedents in which high level expression of growth factor receptors, either in full-length or truncated form, can result in constitutive signaling (Mumm & Kopan, 2000; Schlessinger, 2000). Several expression constructs were generated, each containing epitope tags (Figure 1). The tags allowed protein expression to be confirmed in all of our experiments and similar expressionlevels to be demonstrated when activity of several different molecules was compared (e.g. Figure 5D and data not shown). None of the Frizzled derivatives we expressed in 293T cells was able to stabilize β-catenin to a significant extent (Figure 3A and data not shown). An example of this is shown in Figure 3A. Fzd1 has previously been associated with canonical Wnt/β-catenin signaling and has been shown to synergize with Wnt1 to activate TCF-dependent transcription (Gazit et al., 1999; Mao et al., 2001a). However, in our experiments, neither full-length Fzd1 nor a truncated derivative with the first extracellular domain deleted (ΔN-Fzd1) was able to increase β-catenin levels when expressed in the absence of Wnt1 (Figure 3A). To pursue this further we assayed β-catenin/TCF-dependent transcription using the luciferase reporter plasmid pTOPFLASH (van de Wetering et al., 1997). Expression of Wnt1 gave a >10-fold increase in luciferase activity in this assay, but neither full-length Fzd1 nor ΔN-Fzd1 gave more than a 2-fold increase, even at much higher concentrations of plasmid DNA (Figure 3B). While this suggested that Fzd1 might have weak signaling potential, other experiments showed that high level expression of Fzd1 was actually inhibitory to signaling mediated by Wnt1 (Figure 3C).
Figure 5. Signaling by ΔN-LRP6 requires intracellular PPSP motifs.
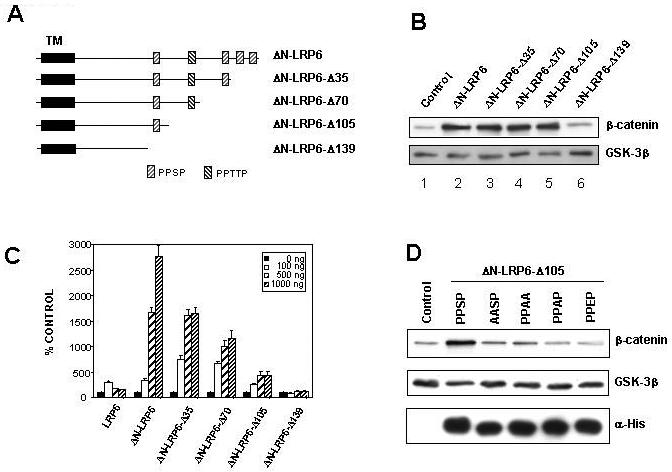
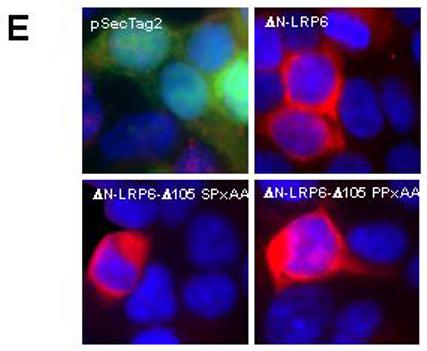
(A) Diagram of ΔN-LRP6 and C-terminal deletions thereof. The repeated motifs PPSP and PPTTP are indicated by hatched boxes and the transmembrane domain (TM) with a black rectangle. (B) Effect of ΔN-LRP6 C-terminal deletions on β-catenin stabilization. 293T cells were transfected with empty vector (lane 1) or vectors encoding the indicated ΔN-LRP6 derivatives (lanes 2-6). Cytosolic β-catenin levels were analyzed as in Figure 2 and GSK-3β was used as a loading control. Removal of 105 amino acids does not reduce activity in this assay (lane 5) but removal of 139 residues eliminates signaling (lane 6). (C) Effect of ΔN-LRP6 C-terminal deletions on TCF/β-catenin-dependent transcription. 293T cells were transfected with the indicated amounts of LRP6 or ΔN-LRP6 derivatives, together with pTOPFLASH and pRL-TK. Luciferase assays were performed as in Figure 3. Results are the mean + S.D. of six replicates. Note the progressive loss of activity with increasing C-terminal truncation. (D) Mutations in the sole PPSP motif of ΔN-LRP6-Δ105 abolish signaling to β-catenin. Cells were transfected with His-tagged ΔN-LRP6-Δ105 constructs carrying the indicated point mutations. Cytosolic β-catenin and GSK-3β levels were analyzed by Western blotting and membrane fractions were probed with anti-His antibody to verify expression of the mutant LRP6 proteins. (E) Subcellular localization of inactive ΔN-LRP6-Δ105 mutants by immunofluorescence. Cells transfected with the indicated Myc-tagged ΔN-LRP6 proteins or empty vector were fixed and immunostained (red) with anti-Myc antibody in the absence of detergents. The cells were co-transfected with pEGFP-C3 (Clontech) to identify transfected cells (green) independently of LRP6 expression.
Figure 3. Overexpression of Frizzled1 does not activate Wnt/β-catenin signaling.
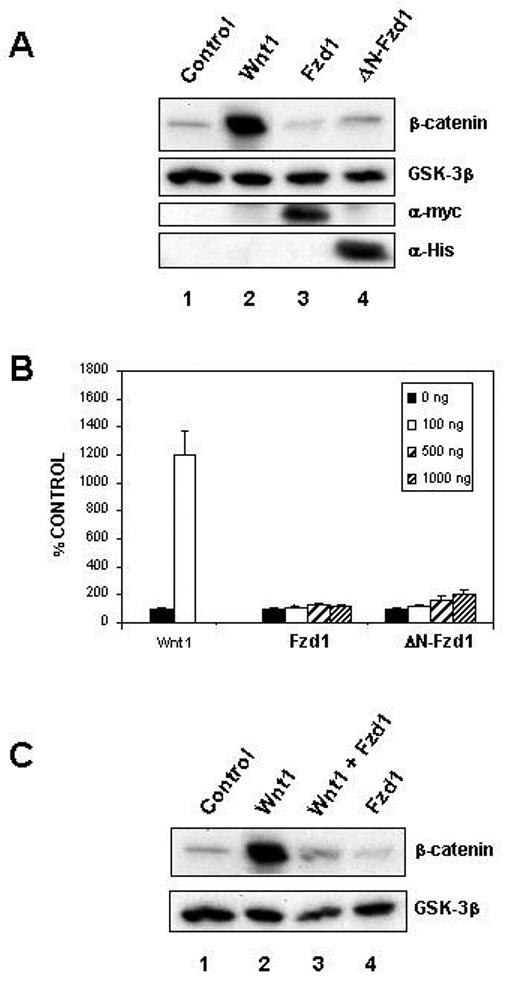
(A) Full length Fzd1 and the deletion mutant ΔN-Fzd1 do not stabilize β-catenin. 293T cells were transfected with vectors encoding either Wnt1 (lane 2), full-length Fzd1 (lane 3), ΔN-Fzd1 (lane 4), or with empty vector (lane 1) and cytosolic β-catenin levels were determined by Western blotting. GSK-3β served as a loading control. Expression of the epitope-tagged Fzd proteins was confirmed using anti-Myc and anti-His antibodies (lanes 3 and 4). Only Wnt1 caused significant stabilization of β-catenin. (B) Overexpression of full length Fzd1 or ΔN-Fzd1 does not significantly activate TCF/β-catenin-dependent transcription. 293T cells were transfected with the indicated amounts of plasmid encoding Wnt1, full length Fzd1 or ΔN-Fzd1, together with the TCF/β-catenin responsive reporter pTOPFLASH. The cells were also transfected with the Renilla luciferase expression plasmid pRL-TK as an internal control. Luciferase activities were measured and TOPFLASH values normalized to Renilla values. Results shown are the means + S.D. of six replicates. (C) Overexpression of full length Fzd1 can block Wnt1 signaling. 293T cells were transfected with empty vector (lanes 1 and 4) or Wnt1 (lanes 2 and 3) in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of full-length Fzd1. Cytosolic β-catenin levels were analyzed as in Figure 2 and GSK-3β was used as a loading control.
LRP6 overexpression mimics Wnt1 signaling
In contrast to the behavior of Frizzled proteins in the above assays, we found that overexpression of LRP6 was sufficient to stabilize β-catenin (Figure 4A, lane 5). Having observed signaling activity with full-length LRP6, we tested deletion derivatives missing specific regions of the extracellular domain. These included: ΔLDLR-LRP6, missing the LDLR repeats conserved among the LDL receptor-related proteins (Brown et al., 1998; Hussain et al., 1999); ΔEGF-LRP6, missing the large domain containing four EGF-like repeats; and ΔN-LRP6, a derivative with virtually all of the extracellular domain deleted (Figure 1). All three of these mutants were clearly capable of stabilizing cytosolic β-catenin to an extent similar to that achieved by Wnt1 (Figure 4A). We also tested an N-terminally deleted form of the Drosophila LRP5/6 homolog, Arrow (ΔN-Arr), and found that this was capable of causing similar increases in mammalian β-catenin levels (Figure 4A, lane 10).
Figure 4. Overexpression of full-length LRP6, or extracellular domain deletion derivatives, mimics Wnt1 signaling.
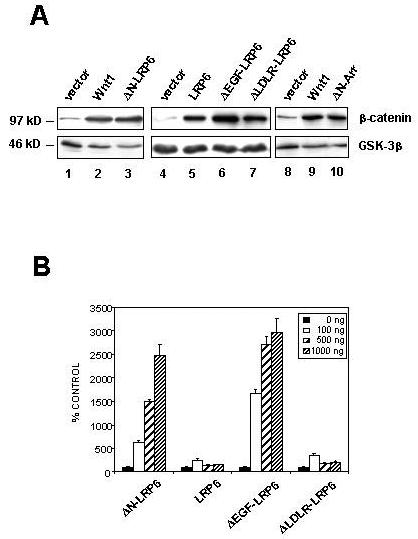
(A) LRP6 derivatives stabilize β-catenin. Western analysis of cytosolic β-catenin in 293T cells transfected with empty vector (lanes 1, 4 and 8), Wnt1 (lanes 2 and 9), full-length LRP6 (lane 5), the indicated LRP6 constructs (lanes 3, 6, and 7), or ΔN-Arr (lane 10). GSK-3β was used as a loading control. (B) LRP6 derivatives lacking the EGF-like repeat domain potently activate TCF/β-catenin-dependent transcription. 293T cells were transfected with varying amounts of plasmid encoding the indicated LRP6 constructs, together with pTOPFLASH and pRL-TK. Luciferase activities were measured and TOPFLASH values normalized to Renilla values. Results shown are the means + S.D. of six replicates. LRP6 and ΔLDLR-LRP6 have significant activity above background, but ΔN-LRP6 and ΔEGF-LRP6 have markedly stronger activity.
LRP6 and the three N-terminal deletion mutants also activated β-catenin/TCF-dependent transcription in TOPFLASH assays (Figure 4B). However, in contrast to the cytosolic β-catenin assays, in which all four LRP6 constructs showed similar levels of signaling, the transcriptional reporter assay revealed quantitative differences. Thus full-length LRP6 and ΔLDLR-LRP gave maximal increases of 2 to 3 fold in reporter activity, while ΔEGF-LRP6 and ΔN-LRP6 increased activity by >25-fold although the four proteins were expressed at similar levels. These results indicate that truncation mutations of LRP6 are potent activators of β-catenin/TCF signaling in this assay. They also illustrate that measurement of cytosolic β-catenin levels provides a highly sensitive assay for Wnt signaling, but that transcriptional reporter assays provide a greater range of quantification.
Identification of a minimal domain and sequence motif required for ΔN-LRP6 signaling
Having established that ΔN-LRP6 is capable of signaling in these assays, we made a series of progressive C-terminal deletions of ΔN-LRP6 (Figure 5A) in order to define a minimal region sufficient for activity. Cytosolic β-catenin and TOPFLASH assays using these constructs are illustrated in Figures 5B and 5C, respectively. The cytosolic β-catenin results clearly fell into two qualitatively distinct groups indicating that deletion of up to 105 C-terminal amino acids left signaling intact but deletion of 139 amino acids abolished it (Figure 5B). In contrast, the TOPFLASH transcription assays showed quantitative differences, with stepwise reductions in activity as amino acids were removed. Again, however, removal of 139 amino acids from the C-terminus resulted in no detectable activity (Figure 5C). Both assays therefore identify ΔN-LRP6-Δ105 as the shortest of these constructs capable of eliciting a signal, and define a 34 amino acid region between positions 1474 and 1508 that contains one or more sequences necessary for this activity.
The intracellular domain of LRP6 contains five copies of a short amino acid motif, present as either PPSP or PPTTP. The 34 aa region implicated in signaling above contains only a single copy of PPSP. This motif is conserved between LRP5, LRP6, and their Drosophila homolog, Arrow, suggesting that it is functionally important. Interestingly, the progressive loss of activity in our deletion series appeared to correlate with the removal of PPSP/PPTTP sequence elements. This trend was further supported by analysis of protein levels in the transfections since in general the shorter mutant proteins were expressed more abundantly than the longer ones (data not shown). A similar deletion analysis of LRP5 has also lead to the identification of a region containing PPSP/PPTTP motifs that is required for the signaling activity (Mao et al., 2001b). However in that analysis at least three PPSP/PPTTP motifs were required for the activity, making it difficult to prove their importance. Therefore, to perform a definitive test of the functional significance of the PPSP/PPTTP motifs, we generated site-specific mutations converting the single PPSP sequence in ΔN-LRP6-Δ105 to AASP, PPAA, PPAP, or PPEP. In each case the mutant ΔN-LRP6-Δ105 derivatives showed no significant signaling ability, yet the proteins were expressed as abundantly as the parental ΔN-LRP6-Δ105 (Figure 5D) and with similar subcellular localization (Figure 5E). These data therefore define the PPSP motif in ΔN-LRP6-Δ105 as a sequence necessary for signaling.
LRP6 can signal in a ligand-independent manner
ΔN-LRP6 contains only 7 amino acids of the LRP6 extracellular domain, and thus is lacking the region reported to bind Wnt ligands (Mao et al., 2001a). The ability of this construct to stabilize β-catenin therefore suggested that it does so in a ligand-independent manner. However, in view of the possibility of endogenous Wnt proteins in the cell culture, we wished to test this notion more rigorously. As described above, truncated forms of either Fzd8 or LRP6 can act in a dominant negative manner to prevent Wnt1-mediated signaling in 293T cells (Figure 2). In other experiments we have also observed that Fzd8 Ex inhibits signaling by several other Wnts, (data not shown) and there is evidence suggesting that it does so by sequestering Wnt ligands (Hsieh et al., 1999; Uren et al., 2000). We therefore challenged LRP6-induced and ΔN-LRP6-induced signaling by co-expressing Fzd8 Ex. While Fzd8 Ex was a potent inhibitor of Wnt1-mediated signaling (Figure 6A, lanes 2 and 3), its expression at similar levels had no inhibitory effect on β-catenin stabilization by LRP6 or ΔN-LRP6 (Figure 6B, lanes 2, 3, 8, 9 and data not shown). Similarly, the inhibitory LRP6 construct LRP6-Δ173 had little or no effect on β-catenin levels induced by full-length LRP6 or ΔN-LRP6 (Figure 6). Furthermore, expression of Fzd8 Ex and LRP6-Δ173 with LRP6 and ΔN-LRP6 did not significantly alter the signaling of these molecules in TOPFLASH transcription assays (Figure 6C). These results support the conclusion that expression of these LRP6 constructs can initiate signaling to β-catenin in the absence of functional Wnt ligands.
Figure 6. LRP6 can signal in a ligand-independent manner.
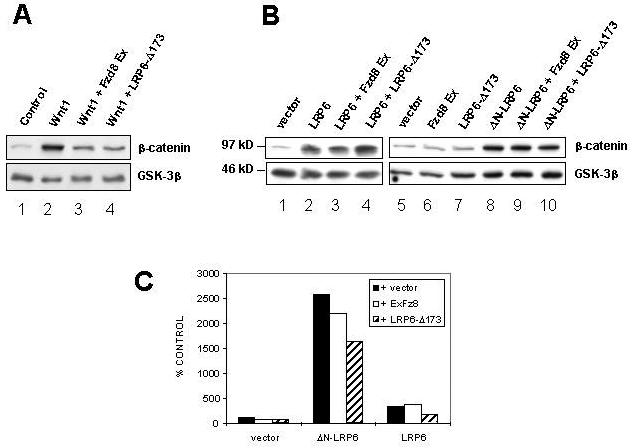
(A) Fzd8 Ex and LRP6-Δ173 both inhibit Wnt-mediated accumulation of β-catenin, as in Figure 2. (B) The same Fzd8 Ex and LRP-Δ173 constructs fail to inhibit β-catenin stabilization mediated by LRP6 or ΔN-LRP6, implying that signaling by the LRP6 cytoplasmic domain in these experiments is ligand-independent. Cytosolic β-catenin levels in co-transfected 293T cells were analyzed by Western blotting as in Figure 2. (C) Effect of Fzd8 Ex and LRP6-Δ173 expression on TCF/β-catenin-dependent transcription induced by LRP6 or ΔN-LRP6. 293T cells were transfected with the empty vector, LRP6 or ΔN-LRP6, together with pTOPFLASH and pRL-TK, in the presence or absence of Fzd8 Ex and LRP6-Δ173. Luciferase assays were performed as in Figure 3. Results are the mean + S.D. of six replicates. The expression of either dominant negative protein does not significantly alter LRP6 or ΔN-LRP6 signaling.
LRP6 derivatives can signal in the absence of functional Frizzled proteins
A particularly important question was to ask whether Frizzled molecules were required for signaling activated by expression of LRP6 derivatives. Since members of the Frizzled protein family are endogenously expressed in several mammalian cell lines, it is difficult to test their contribution to the observed Wnt/β-catenin signaling in such cells. Consequently, we made use of Drosophila S2 cells as these have previously been reported not to express the Drosophila Fz proteins fz, Dfz2 and Dfz3 (Bhanot et al., 1996; Nusse et al., 1997; Sato et al., 1999). In addition, it has been shown that Wingless (the Drosophila Wnt1 homolog) does not induce accumulation of the β-catenin homolog, Armadillo, in S2 cells unless they are transfected with exogenous fz or Dfz2 (Bhanot et al., 1996; Nusse et al., 1997). This finding is recapitulated in Figure 7A and demonstrates that S2 cells do not express endogenous Frizzleds that can support Wingless-mediated signaling. To determine whether the S2 cells used in these experiments express any of the four fz genes, we conducted an RT-PCR analysis (Figure 7B and data not shown). Our analysis showed that S2 cells contain transcripts for all four fz genes. However quantification of their expression indicated that transcripts for all four genes were present at significantly less than one transcript per cell. The low abundance of these transcripts therefore suggests that none of the Fz proteins is expressed at a significant level.
Figure 7. A truncated form of Drosophila Arrow can signal independently of Frizzled proteins.
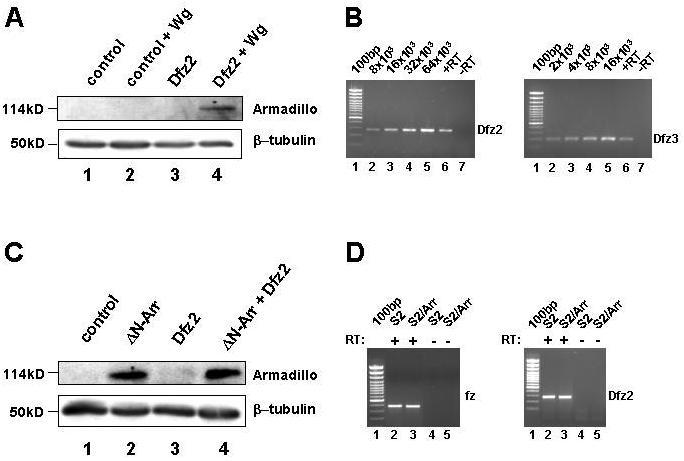
(A) Drosophila S2 cells do not respond to Wingless unless transfected with Dfz2. S2 cells were transfected with empty vector (lanes 1 and 3) or vector encoding the Wnt1 homolog Wingless (Wg; lanes 2 and 4) in the presence (lanes 3 and 4) or absence (lanes 1 and 2) of Dfrizzled2 (Dfz2). Cytosolic levels of Armadillo (the Drosophila homolog of β-catenin) were analyzed by Western blot and β-tubulin was used as a loading control. (B) Quantitative RT-PCR analysis of Dfz2 and Dfz3 transcripts in S2 cells. Analysis of S2 cells (lane 6 and 7) indicated that Dfz2 and Dfz3 transcripts were present, but comparison to reactions containing known numbers of template molecules (lanes 2-5) demonstrated that the transcripts were at very low abundance. From this comparison it is possible to estimate the number of transcripts per cell. Given an average yield of 40μg of total RNA from 8×106 S2 cells and the use of 0.5μg of total RNA in each RT-PCR, these results indicated that there were approximately 0.16 and 0.04 transcripts per cell for Dfz2 and Dfz3 respectively. (C) Expression of ΔN-Arr is sufficient to cause stabilization of Armadillo, independently of Dfz2 expression. S2 cells were transfected with empty vector (lane 1), or vectors encoding ΔN-Arr, an N-terminally truncated form of the LRP6 homolog Arrow (lane 2), Dfz2 (lane 3), or both ΔN-Arr and Dfz2 (lane 4). Armadillo and β-tubulin levels were analyzed by Western blot as in (A). (D) Overexpression of ΔN-Arr in S2 cells does not induce fz or Dfz2 expression. RT-PCR analysis of S2 cells (lanes 2 and 4) and S2 cells expressing ΔN-Arr (lanes 3 and 5) demonstrated that the numbers of fz and Dfz2 transcripts were the same or similar in the two cell types.
To establish whether LRP6 derivatives can signal in the absence of significant Fzd protein expression, we expressed an N-terminally truncated form of Arrow, ΔN-Arr, in S2 cells. This construct was shown above to induce β-catenin stabilization when expressed in 293T cells (Figure 4A). As shown in Figure 7C, expression of ΔN-Arr in S2 cells caused robust stabilization of Armadillo, irrespective of whether Dfz2 was co-expressed. Furthermore, expression of ΔN-Arr did not induce the expression of any of the four Drosophila Fz transcripts (Figure 7D and data not shown). Together these data indicate that ΔN-Arr can signal in a Frizzled-independent manner to cause stabilization of β-catenin/Armadillo.
DISCUSSION
Despite the widespread importance of Wnt signaling in development and cancer, the molecular mechanisms of Wnt signal transduction across the plasma membrane are not yet well characterized. Powerful genetic evidence has implicated both Frizzled proteins and Arrow/LRP5/6 as candidate Wnt receptors, but their respective contributions to signal transduction have not been defined. Here we have shown that the canonical Wnt/β-catenin signaling pathway can be activated by overexpression of full length LRP6, or expression of truncated forms of LRP6 or Arrow that are missing their extracellular domain, and we have identified sequences within the intracellular domain of LRP6 that are required for this. Importantly, we have demonstrated that truncated forms of Arrow/LRP6 activate the signaling pathway in both a ligand-independent and Frizzled-independent manner. In contrast, we did not observe activation of the intracellular Wnt signaling pathway upon expression of full-length Frizzled proteins or truncated Fzd mutants. Instead we found that overexpression of a full length Frizzled protein can be inhibitory to ligand-induced Wnt/β-catenin signaling. Since LRP6/Arrow has the capacity to initiate Fzd-independent signaling to β-catenin, LRP5/6 proteins are the leading candidates to be the signal-generating components of Wnt receptors in the canonical Wnt/β-catenin pathway.
Overexpression of LRP6 is sufficient to activate the Wnt signaling pathway
Our results indicate that LRP6 proteins bearing extracellular deletions of different lengths are all able to activate the canonical Wnt signaling pathway as monitored by the accumulation of β-catenin (see Figure 4A). However, these same proteins induced β-catenin-dependent transcription from the TOPFLASH reporter to very different extents (Figure 4B). Specifically, ΔN-LRP6 and ΔEGF-LRP6 induced much higher levels of luciferase expression than ΔLDLR-LRP6; similar results have been reported by others (Mao et al., 2001a; Mao et al., 2001b). This suggests that there are sequences within the large EGF-like repeat domain that restrain signaling by LRP6. Consistent with this, it has been reported recently that the EGF-like repeat domain causes oligomerization of LRP6 and that the oligomer is inactive in signaling (Liu et al., 2003). We also found that a derivative of the Drosophila LRP5/6 homolog Arrow, deleted for its extracellular domain (ΔN-Arr), could activate Wnt/β-catenin signaling in both human and Drosophila cells (Figures 4A and 7C). This implies that extracellular deletions in LRP5/6/Arrow proteins can generate activated receptors in widely different species. The result appears to contrast with a report that a myristoylated form of the Arrow intracellular domain was unable to activate Wnt/β-catenin signaling in Drosophila (Tolwinski et al., 2003). However, this may simply reflect differences in protein expression levels in the two systems, or possibly differences in membrane targeting of the relevant constructs.
In addition to the effects of N-terminal truncations, our data show that overexpression of full-length LRP6 is sufficient to induce accumulation of cytosolic β-catenin to levels similar to that achieved by Wnt1. However, endogenous LRP5/6 protein levels in the cell are apparently insufficient to generate a canonical Wnt/β-catenin signal. Interestingly, Dickkopf-1, an inhibitor of Wnt signaling, has been shown to interact with LRP6 (Bafico et al., 2001; Mao et al., 2001a; Semenov et al., 2001) and, together with Kremen proteins, to promote endocytosis of LRP6 (Mao et al., 2002). It is therefore possible that endogenous Dickkopf proteins produced by 293T cells limit the levels and signaling activity of endogenous full-length LRP6. However when LRP6 and ΔLDLR-LRP6 are over expressed this degradation system may become saturated, leading to accumulation of the exogenous proteins and hence signaling. In addition, it is possible that Dickkopf and Kremen limit LRP5/6 protein levels to prevent spontaneous signaling in vivo. In this regard it is interesting that a familial mutation has been identified in human LRP5 which alters its interaction with Dickkopf-1 and results in increased Wnt signaling in certain tissues (Boyden et al., 2002; Little et al., 2002).
LRP6 signaling requires a repeated intracellular motif
Our deletion analysis of the intracellular domain of ΔN-LRP6 indicated that C-terminal sequences downstream of amino acids 1474 were necessary for ΔN-LRP6 to activate the canonical Wnt pathway, and that ΔN-LRP6-Δ105 (terminating at amino acid 1508) was the shortest construct capable of generating a signal (Figure 5). This defines a 34 amino acid region containing essential sequences. However this region was not sufficient for the full signaling capacity of LRP6. We observed an inverse correlation between the magnitude of TOPFLASH activation by the C-terminally deleted ΔN-LRP6 derivatives and the extent of their C-terminal truncation (figure 5D). Sequences within the C-terminal 105 amino acids are thus also important. This segment contains several copies of the short conserved motif PPSP/PPTTP and the TOPFLASH activity of the deletion constructs loosely correlated with the number of motifs present. One such PPSP motif is retained in ΔN-LRP6-Δ105 and lies within the 34 amino acid region defined above. A previous analysis of the related molecule LRP5 also identified an intracellular region containing three PPSP motifs as being important for signaling, and for the interaction of Axin with LRP5 (Mao et al., 2001b), but those experiments did not specifically demonstrate that the PPSP motifs were functionally important. The presence of a single PPSP motif within ΔN-LRP6-Δ105 allowed us to test readily the functional significance of the motif by generating point mutants. Conversion of either the first pair or second pair of residues to alanines abolished activity, as did mutation of the serine to either alanine or glutamic acid. These results demonstrate that the PPSP motif is absolutely required for signaling (Figure 5D). It seems likely that the four additional copies of PPSP/PPTTP in the full-length cytoplasmic domain contribute to the strong signaling potential we observed for ΔN-LRP6 as compared to ΔN-LRP6-Δ105. Consistent with these findings, Tamai et al. (2004) have very recently reported that the single PPSP motif in a similar ΔN-LRP6 construct lacking the last 107 amino acids is required for signaling. They also showed that the PPSP motif is sufficient to mediate a physical interaction between LRP6 and Axin in a phosphorylation-dependent manner, emphasizing the key role of this motif in Wnt receptor signaling.
Overexpression of Fzd alone in 293T cells does not activate Wnt/β-catenin signaling
Our efforts to activate Wnt/β-catenin signaling by expression of Fzd constructs were consistently unsuccessful. These findings stand in apparent contrast to previous reports that the overexpression of Frizzled molecules can be sufficient to activate the Wnt/β-catenin signaling pathway in some circumstances. For example, ectopic expression of XFzd3 in animal cap explants from Xenopus embryos induced transcription of the Wnt target genes Siamois and Xnr3 (Umbhauer et al., 2000). In addition, it has been reported that expression of rFzd9 in 293T cells caused modest activation of Wnt/β-catenin signaling and an approximately 2-fold increase in TOPFLASH activity (Karasawa et al., 2002). XFzd3 and rFzd9 were not among the Fzd family members that we tested and this provides a possible explanation for the apparent discrepancy. However, the studies of XFzd3 and rFzd9 did not address whether these proteins could activate Wnt/β-catenin signaling independently of endogenous Wnt ligands or LRP5/6. Thus it is possible that overexpression of certain Fzd proteins permits an endogenous Wnt ligand to signal through Wnt-Fzd-LRP complexes. This notion is consistent with evidence of cooperative signaling when both Wnt and Fzd are provided exogenously (Gazit et al., 1999; Mao et al., 2001a). Similarly, a recent study indicates that certain Wnt-Frizzled fusion proteins can activate Wnt/β-catenin signaling in mammalian cells, but that they are unable to do so in the presence of a dominant negative LRP6 protein (Holmen et al., 2002). This implies that signaling under these circumstances is dependent on LRP6. Further evidence against the notion that Fzd proteins can independently generate downstream signals comes from our own data (Figure 3C) and those of Bhanot et al. (1996), indicating that overexpression of full-length Fzd proteins can be antagonistic to Wnt-mediated signaling to β-catenin or Armadillo. Thus we suggest that when activation of Wnt/β-catenin signaling occurs from overexpression of Frizzled proteins it involves formation of a complex with a Wnt ligand and/or LRP5/6. If the Fzd component is in large excess of LRP5/6 it may instead sequester Wnt ligand in inactive complexes.
LRP6 signaling can be ligand- and Frizzled-independent
In contrast to the behavior of Fzd proteins, the ability of full length or N-terminally deleted LRP6 to generate signals in the presence of an inhibitory form of Fzd8 that sequesters multiple Wnt ligands indicates that LRP6 proteins can signal in a ligand-independent manner (Figure 6). In addition, the ability of ΔN-Arr to stabilize Armadillo in S2 cells is a particularly telling result (Figure 7). In contrast to commonly used mammalian cell lines, Drosophila S2 cells express no endogenous Fz that can support canonical Wnt signaling (Bhanot et al., 1996) (Figure 7). We were able to detect transcripts for all four Drosophila fz genes by RT-PCR (Figure 7 and data not shown), but quantification of their expression indicated that all four transcripts were present at significantly less than one transcript per cell. This suggests that infrequent transcripts may have arisen from “illegitimate transcription” events which can be detected by RT-PCR but lead to transcripts that are not translated (Chelly et al., 1989; Kaplan et al., 1992). Others have also detected Dfz2 and Dfz3 transcripts in S2 cells, but also by using highly sensitive RT-PCR methods (Schweizer & Varmus, 2003). The low abundance of these transcripts in our assays implies that none of the Fz proteins is expressed at functionally significant levels in S2 cells. Our data therefore indicate that LRP6/Arrow derivatives can signal in the absence of Frizzled proteins. A recent demonstration that an LRP6 derivative similar to ΔN-LRP6 can signal in S2 cells in the presence of dsRNAi against Dfz2 also supports this possibility (Schweizer & Varmus, 2003). Together these data suggest that LRP5/6/Arrow functions downstream of Fzd in the Wnt/β-catenin signaling cascade. However, since ligand binding to both Frizzled and LRP5/6 has been reported (Tamai et al., 2000), current models suggest that both proteins are components of a Wnt receptor complex that signals to β-catenin. In light of these models, our data imply that LRP5/6/Arrow proteins constitute the distal signal-generating component of these receptors.
The ability of the N-terminally deleted LRP6 proteins to signal in a Fzd-independent manner raises questions about the role Fzd proteins play in Wnt signal transduction. One possible interpretation of our data is that the Fzd proteins bind ligand and present it to LRP/Arrow but play no other role in initiating the intracellular signal. This is unlikely, however, as sequences within the C-terminal domain of Fzd proteins are known to be important for signal (Umbhauer et al., 2000), and switching the C-terminal intracellular domains of Fz and Dfz2 exchanges the respective abilities of these proteins to activate planar cell polarity and Wnt/β-catenin pathways (Boutros et al., 2000). Thus there is evidence that the intracellular domain of Fzd plays a role in activating the Wnt/β-catenin pathway. The recent findings of Liu et al. (2003) provide possible insights into this role. These authors show that full length LRP6 can be present at the cell surface as a non-functional oligomer and that, in response to a Wnt signal, there is separation of the intracellular domains of LRP6 coincident with signaling (Liu et al., 2003). It is possible that the C-terminal domain of Frizzled facilitates this separation of the LRP6 intracellular domains in response to Wnt ligands. If so, this function of Fzd would be predicted to be unnecessary for signaling by LRP6/Arrow derivatives lacking their extracellular domain since such constructs form monomers at the cell surface and so signal constitutively (Liu et al., 2003). This prediction is borne out by our data showing that ΔN-Arr can signal in a Fzd-independent manner.
In summary, it is clear that under normal circumstances Wnt/Wingless cannot signal via endogenous LRP/Arr in the absence of Fzd proteins (Figure 7)(Bhanot et al., 1996). However, our experiments demonstrate that truncated mutants of LRP6/Arrow can stabilize β-catenin in a ligand-independent and Frizzled-independent manner. The PPSP motifs within the intracellular domain of LRP6 are required to initiate this signaling. These data, and the inability of overexpressed Frizzled proteins to cause ligand-independent signals, lead us to conclude that LRP5/6 proteins are downstream of Frizzled in the sense that they initiate distal intracellular signals to β-catenin. The N-terminally truncated forms of LRP/Arr differ from their full-length counterparts in several important respects: (a) they fail to form oligomers that would otherwise attenuate their signaling, (b) they are resistant to endocytosis and degradation mediated by Dkk and Kremen, and (c) they signal in a ligand- and Fzd-independent manner. This combination of properties highlights the possibility that LRP5 and 6 may be proto-oncogenes whose mutation in cancer cells might result in constitutive Wnt/β-catenin signaling. It is therefore appropriate to seek evidence of truncated forms of these proteins in cancer cells.
Materials and Methods
Plasmid constructs
Mouse LRP6 cDNA was kindly provided by Dr. Fred Hess (Brown et al., 1998). LRP6 Ex is a secreted form of the LRP6 extracellular domain (amino acids 1-1372), while LRP6-Δ173 is a transmembrane protein (amino acids 1-1440) from which has the majority of the intracellular domain is deleted. cDNAs encoding these proteins were sub-cloned into pcDNA3.1/Myc-His (Invitrogen), thus introducing Myc and poly-histidine tags at the C-termini. LRP6 constructs bearing deletions within the extracellular domain were as follows. ΔLDLR-LRP6 lacks amino acids 1245-1364, which include all 3 low density lipoprotein receptor (LDLR) repeats; ΔEGF-LRP6 lacks amino acids 20-1244, which include all 4 epidermal growth factor (EGF)-like repeats and the 20 YWTD repeats; ΔN-LRP6 lacks amino acids 20-1365 and thus all but 7 residues of the extracellular domain. The signal peptides of these mutant LRP6 proteins, and of full-length LRP6, were replaced by the Ig κ-chain signal peptide by sub-cloning the cDNAs into the expression vector pSecTag2 (Invitrogen) and c-Myc and poly-histidine epitopes were introduced immediately downstream of the signal cleavage site. A further series of constructs derived from ΔN-LRP6, designated ΔN-LRP6-Δ35, ΔN-LRP6-Δ70, ΔN-LRP6-Δ105, and ΔN-LRP6-Δ139, were obtained by progressive C-terminal deletion of the indicated number of amino acids. The single PPSP motif at position 1488-91 in ΔN-LRP6-Δ105 was converted to AASP, PPAA, PPAP, or PPEP by site-directed mutagenesis.
A partial cDNA encoding Drosophila Arrow was obtained from Research Genetics, Inc. ΔN-Arrow, comprising amino acids 1445-1678, was expressed from the plasmid pMT/BIP/V5-His (Invitrogen), which contains a Drosophila metallothionein promoter and N-terminal BiP signal peptide. c-Myc and poly-histidine epitopes were introduced immediately after the signal cleavage site to permit detection of the ΔN-Arrow protein.
Murine Fzd8 cDNA was kindly provided by Dr. Jeremy Nathans (Wang et al., 1996) and rat Fzd1 by Dr. Robert Nissenson (Chan et al., 1992). Fzd8 Ex, a secreted form of the extracellular domain of Fzd8 (amino acids 1-270) carrying a single Myc tag at its C-terminus, was expressed from pcDNA3 (Invitrogen). ΔN-Fzd1, composed of amino acids 309-642, was expressed from a pSecTag2 vector. c-Myc and polyhistidine tags were introduced after the signal peptide. A tagged form of full-length rat Fzd1 was obtained by introducing a Myc epitope at amino acid 310 and sub-cloning the resultant cDNA into pcDNA3.1(+) (Invitrogen). Full length Dfz2 cDNA carrying an N-terminal Myc epitope tag and signal peptide from human CD33 was obtained from Dr. Vincent Zecchini (University of Cambridge). For expression in Schneider2 cells, this cDNA was sub-cloned into the Drosophila heat-shock vector pPHygroHS (K. Brennan, unpublished). Full-length Drosophila wingless cDNA was cloned into the AKSII vector (generously provided by Dr. Simon Kidd, The Rockefeller University), such that it is expressed from the Drosophila actin5C promoter.
Cell culture and transfection conditions
Human embryonic kidney cell line 293T was maintained in Dulbecco's modified Eagle's medium (DMEM; 4.5g/liter D-glucose) containing 10% fetal bovine serum (Life Technologies, Inc.), 100 units/ml penicillin, and 100μg/ml streptomycin at 37°C and 7.5% CO2. The Drosophila cell line Schneider2 was maintained in Schneider's medium (Gibco-BRL) containing 15% heat-inactivated fetal bovine serum (LabTech), 100 units/ml penicillin, and 100μg/ml streptomycin at 22°C and atmospheric CO2. For luciferase assays, 293T cells were transfected using LipofectAMINE (Invitrogen) according to the manufacturer's instructions. Other transfections , of 293T and S2 cells, were by the calcium phosphate co-precipitation method.
Determination of cytoplasmic β-catenin levels
Immunoblot analysis of cytosolic β-catenin was carried out essentially as previously described (Giarre et al., 1998). 5×105 293T cells seeded in 60mm plates were transfected with 4μg of DNA. 48 hrs after transfection, the cells were lysed with a Dounce homogenizer and cytosol and membrane fractions prepared by high-speed centrifugation. The protein concentration of cytosolic fractions was determined by Bio-Rad protein assay (Bio-Rad Laboratories) and 5μg of protein was analyzed for β-catenin levels by Western blotting with a mouse monoclonal antibody (Transduction Laboratories). Equal loading of the samples was confirmed by probing the Western blots with mouse anti-GSK-3β antibody (Transduction Laboratories). Expression of LRP6, Arrow, Fzd1, and Fzd8 proteins was confirmed by loading equivalent amounts of the membrane fraction and probing with mouse anti-Myc antibody (9E10, Santa Cruz) and/or mouse anti-His antibody (Qiagen).
Determination of cytoplasmic Armadillo levels
Drosophila S2 cells seeded in 6 well plates at 3×106/well were transfected with 20 μg of DNA. 48 hrs later, the cells were washed twice with PBS (100mM Na2HPO4, 18mM KH2PO4, 140mM NaCl, 10mM KCl) and lysed in boiling SDS lysis buffer (10mM Tris-HCl, pH7.4, 2%SDS). Genomic DNA was sheared by passing the lysate 10 times through a 23-gauge syringe needle. Protein concentrations were then determined by BCA assay (Pierce). To evaluate Armadillo levels, 50 μg of protein was analyzed by Western blotting using a mouse monoclonal anti-Armadillo antibody (N2 7A1, Developmental Studies Hybridoma Bank). Equal loading of the samples was confirmed by probing with mouse anti-tubulin antibody (generous gift of K. Gull, University of Manchester, UK). Expression of Arrow and Dfz2 proteins was confirmed by using anti-Myc (Santa Cruz) and anti-His antibodies (Sigma).
Luciferase assays
Luciferase assays were carried out as described previously (Howe et al., 2001). Briefly, 293T cells were seeded in 24-well plates at 1×105 cells/well. 20 hrs later, a transfection cocktail containing 1.2 ml of serum-free DMEM, 3.6 μl of LipofectAMINE (Invitrogen), and 2.2 μg of DNA (including 0.5 μg pTOPFLASH and 0.2 μg pRL-TK) was divided equally between six wells and the cells were incubated with cocktail for 4 hrs. Lysates were prepared 48 hrs after transfection, and Firefly and Renilla luciferase activities in 5 μl of lysate were measured using a Dual Luciferase Reagent kit (Promega) and Monolight 2010 luminometer (Analytical Luminescence Laboratory).
RT-PCR analysis
Total RNA was isolated from S2 cells using Stat-60 (Tel-Test, Inc.) and contaminating genomic DNA removed using an RNeasy miniprep kit (Qiagen). The average yield of total RNA from 8×106 cells was 40μg. cDNA was prepared from 5μg of total RNA by random priming using Superscript II reverse transcriptase (Invitrogen) and RNA/DNA hybrids were removed by incubation with RNase H (Invitrogen). One tenth of the prepared cDNA was used in the subsequent PCRs. The presence of fz, Dfz2, Dfz3 and Dfz4 cDNA was established using the primers below and following cycle conditions: 94°C for 30secs, 62°C for 30secs and 72°C for 1 minute. fz transcripts were detected after 35 cycles, Dfz2 and Dfz3 after 31 cycles, and Dfz4 after 29 cycles.
Fz: 5'-GCG TCC CTG ATC CTT CCA CT-3'; 5'-CCA GCA TGG AGC AAA GGT AC-3'
Dfz2: 5'-ATC ACC TCG GGC GTG TGG AT-3'; 5'-AAC GTG GTG GTG CAG GTG GT-3'
Dfz3: 5'-TCC AGG TCT CTG CTC TCC CT-3'; 5'-GTG GGT GAG AGG CAG TCA TC-3'
Dfz4: 5'-GGA GCA GCA ACT GGA CAC CT-3'; 5'-TGC GAG GAT GTG GAG TGA TG-3'
Immunofluorescence
Cells transfected with LRP6 constructs were washed twice with PBS and fixed for 10 minutes with 4% paraformaldehyde in PBS at room temperature. The cells were blocked for 30minutes with 1% horse serum in PBS and incubated for 1 hour with anti-Myc antibody, diluted 1:200 in PBS + 1% horse serum, at room temperature. The cells were washed extensively and blocked again before incubating with rhodamine-tagged anti-mouse antibody (Jackson), diluted 1:400 in PBS + 1% horse serum, and 0.12μg/ml Hoescht for 30 minutes at room temperature. After further washing the cells were mounted in DAKO fluorescent mounting medium and photographed under a Zeiss Axiophot2 microscope.
ACKNOWLEDGEMENTS
We are grateful to Drs. Fred Hess, Hans Clevers, Vincent Zecchini, Simon Kidd, Robert Nissenson and Jeremy Nathans for supplying plasmids. We also thank C. Dong, M. Semenov, and M. Goll, for contributions to the early phases of analyzing Frizzled constructs, and K. Tolle for technical assistance. The mouse anti-Armadillo antibody N2 7A1, was produced in the laboratory of Dr. Eric Wieschaus and was obtained from the Developmental Studies Hybridoma Bank (DSHB). The DSHB was developed under the auspices of the NICHD and is maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242.
This work was supported by US Army Medical Research and Materiel Command fellowships DAMD17-99-1-9388 (to K.B.) and DAMD17-02-1-0359 (to L.A.C.S.), by NIH MSTP grant GM67739, by a fellowship from the Ministerio de Educacion, Cultura y Deportes of Spain (to J.M.G.S.), and by NIH grant CA47207 (to A.M.C.B.).
Abbreviations
APC
Adenomatous Polyposis Coli
Arr
Arrow
CRD
Cysteine Rich Domain
EGF
Epidermal Growth Factor
Fzd/fz
Frizzled
GSK-3β
Glycogen Synthase Kinase-3β
LDL
Low Density Lipoprotein
LDLR
LDL Receptor
LRP
LDLR Related Protein
S2
Schneider 2
TCF
T-cell Factor
REFERENCES
- Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Nature Cell Biology. 2001;3:683–686. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Harryman Samos C, Hsieh J-C, Wang Y-S, Macke JP, Andrew D, Nathans J, Nusse R. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- Bhanot P, Fish M, Jemison JA, Nusse R, Nathans J, Cadigan KM. Development. 1999;126:4175–4186. doi: 10.1242/dev.126.18.4175. [DOI] [PubMed] [Google Scholar]
- Binari RC, Staveley BE, Johnson WA, Godavarti R, Sasisekharan R, Manoukian AS. Development. 1997;124:2623–2632. doi: 10.1242/dev.124.13.2623. [DOI] [PubMed] [Google Scholar]
- Boutros M, Mihaly J, Bouwmeester T, Mlodzik M. Science. 2000;288:1825–8. doi: 10.1126/science.288.5472.1825. [DOI] [PubMed] [Google Scholar]
- Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- Bradley RS, Brown AMC. Mol. Cell. Biol. 1995;15:4616–4622. doi: 10.1128/mcb.15.8.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Twells RCJ, Hey PJ, Cox RD, Levy ER, Soderman AR, Metzker ML, Caskey CT, Todd JA, Hess JF. Biochemical and Biophysical Research Communications. 1998;248:879–888. doi: 10.1006/bbrc.1998.9061. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Genes and Development. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Chan SDH, Karpf DB, Fowlkes ME, Hooks M, Bradley MS, Vuong V, Bambino T, Liu MYC, Arnaud CD, Strewler GJ, Nissenson RA. J. Biol. Chem. 1992;267:25202–25207. [PubMed] [Google Scholar]
- Chelly J, Concordet JP, Kaplan JC, Kahn A. Proc Natl Acad Sci U S A. 1989;86:2617–21. doi: 10.1073/pnas.86.8.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Struhl G. Development. 1999;126:5441–52. doi: 10.1242/dev.126.23.5441. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Tan C, Conrad LJ, Klein PS. Development. 1998;125:2687–2700. doi: 10.1242/dev.125.14.2687. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Tan C, Saint-Jeannet JP, Klein PS. Development. 2001;128:3655–3663. doi: 10.1242/dev.128.19.3655. [DOI] [PubMed] [Google Scholar]
- Eastman Q, Grosschedl R. Curr Opin Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- Gazit A, Yaniv A, Bafico A, Pramila T, Igarashi M, Kitajewski J, Aaronson SA. Oncogene. 1999;18:5959–5966. doi: 10.1038/sj.onc.1202985. [DOI] [PubMed] [Google Scholar]
- Giarre M, Semenov MV, Brown AMC. Ann N Y Acad Sci. 1998;857:43–55. doi: 10.1111/j.1749-6632.1998.tb10106.x. [DOI] [PubMed] [Google Scholar]
- He X, Saint Jeannet J-P, Wang Y, Nathans J, Dawid I, Varmus H. Science. 1997;275:1652–1654. doi: 10.1126/science.275.5306.1652. [DOI] [PubMed] [Google Scholar]
- Hecht A, Kemler R. EMBO Rep. 2000;1:24–8. doi: 10.1093/embo-reports/kvd012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmen SL, Salic A, Zylstra CR, Kirschner MW, Williams BO. Journal of Biological Chemistry. 2002;277:34727–34735. doi: 10.1074/jbc.M204989200. [DOI] [PubMed] [Google Scholar]
- Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AMC. J Biol Chem. 2001;276:20108–20115. doi: 10.1074/jbc.M010692200. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Rattner A, Smallwood PM, Nathans J. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3546–51. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J, Behrens J. Journal of Cell Science. 2002;115:3977–3978. doi: 10.1242/jcs.00089. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Birchmeier W. Current Opinion in Genetics and Development. 2001;11:547–553. doi: 10.1016/s0959-437x(00)00231-8. [DOI] [PubMed] [Google Scholar]
- Hussain MM, Strickland DK, Bakillah A. Annu Rev Nutr. 1999;19:141–172. doi: 10.1146/annurev.nutr.19.1.141. [DOI] [PubMed] [Google Scholar]
- Kaplan JC, Kahn A, Chelly J. Hum Mutat. 1992;1:357–60. doi: 10.1002/humu.1380010502. [DOI] [PubMed] [Google Scholar]
- Karasawa T, Yokokura H, Kitajewski J, Lombroso PJ. Journal of Biological Chemistry. 2002;277:37479–37486. doi: 10.1074/jbc.M205658200. [DOI] [PubMed] [Google Scholar]
- Leyns L, Bouwmeester T, Kim SH, Piccolo S, DeRobertis EM. Cell. 1997;88:747–756. doi: 10.1016/s0092-8674(00)81921-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Perrimon N. Nature. 1999;400:281–284. doi: 10.1038/22343. [DOI] [PubMed] [Google Scholar]
- Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. Am J Hum Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Bafico A, Harris VK, Aaronson SA. Molecular and Cellular Biology. 2003;23:5825–5835. doi: 10.1128/MCB.23.16.5825-5835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo KA, Kennell JA, Ochocinska MJ, Ross SE, Wright WS, MacDougald OA. J Biol Chem. 2002;277:38239–38244. doi: 10.1074/jbc.M206402200. [DOI] [PubMed] [Google Scholar]
- Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Nature. 2002;417:664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. Nature. 2001a;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH, 3rd, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D. Mol Cell. 2001b;7:801–9. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- Mumm JS, Kopan R. Dev Biol. 2000;228:151–65. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- Nusse R, Samos CH, Brink M, Willert K, Cadigan KM, Wodarz A, Fish M, Rulifson E. Cold Spring Harb Symp Quant Biol. 1997;62:185–90. [PubMed] [Google Scholar]
- Nusse R, Varmus HE. Cell. 1992;69:1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. Nature. 2000;407:535–8. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Polakis P. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Sato A, Kojima T, Ui-Tei K, Miyata Y, Saigo K. Development. 1999;126:4421–30. doi: 10.1242/dev.126.20.4421. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schweizer L, Varmus H. BMC Cell Biol. 2003;4:4. doi: 10.1186/1471-2121-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Current Biology. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Julius MA, Zheng Z, Giarre M, Brown AMC, Kitajewski J. Cell Growth Diff. 1997;8:1349–1358. [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. Nature. 2000;407:530–5. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, He X. Mol Cell. 2004;13:149–56. doi: 10.1016/s1097-2765(03)00484-2. [DOI] [PubMed] [Google Scholar]
- Tolwinski NS, Wehrli M, Rives A, Erdeniz N, DiNardo S, Wieschaus E. Developmental Cell. 2003;4:407–418. doi: 10.1016/s1534-5807(03)00063-7. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Hong Z, Kuzuya T, Tada M, Hori M. J Cell Biol. 2000;150:225–241. doi: 10.1083/jcb.150.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Kamimura K, Nakato H, Archer M, Staatz W, Fox B, Humphrey M, Olson S, Futch T, Kaluza V, Siegfried E, Stam L, Selleck SB. Nature. 1999;400:276–280. doi: 10.1038/22336. [DOI] [PubMed] [Google Scholar]
- Umbhauer M, Djiane A, Goisset C, Penzo-Mendez A, Riou J-F, Boucaut J-C, Shi D-L. EMBO Journal. 2000;19:4944–4954. doi: 10.1093/emboj/19.18.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren A, Reichsman F, Anest V, Taylor WG, Muraiso K, Bottaro DP, Cumberledge S, Rubin JS. J Biol Chem. 2000;275:4374–4382. doi: 10.1074/jbc.275.6.4374. [DOI] [PubMed] [Google Scholar]
- Uusitalo M, Heikkila M, Vainio S. Experimental Cell Research. 1999;253:336–348. doi: 10.1006/excr.1999.4710. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, Peifer M, Mortin M, Clevers H. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Wang SW, Krinks M, Lin KM, Luyten FP, Moos M. Cell. 1997;88:757–766. doi: 10.1016/s0092-8674(00)81922-4. [DOI] [PubMed] [Google Scholar]
- Wang YS, Macke JP, Abella BS, Andreasson K, Worley P, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Journal of Biological Chemistry. 1996;271:4468–4476. doi: 10.1074/jbc.271.8.4468. [DOI] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Cancer Cell. 2002;1:279–288. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. Nature. 2000;407:527–30. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Yang-Snyder J, Miller JR, Brown JD, Lai CJ, Moon RT. Current Biology. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]
- You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, MacDougald OA, Brown AMC, Evan G, Kitajewski J, Wang CY. J Cell Biol. 2002;157:429–440. doi: 10.1083/jcb.200201110. [DOI] [PMC free article] [PubMed] [Google Scholar]