Metabolomic and Mass Isotopomer Analysis of Liver Gluconeogenesis and Citric Acid Cycle: II. HETEROGENEITY OF METABOLITE LABELING PATTERN (original) (raw)
Abstract
In this second of two companion articles, we compare the mass isotopomer distribution of metabolites of liver gluconeogenesis and citric acid cycle labeled from NaH13CO3 or dimethyl [1,4-13C2]succinate. The mass isotopomer distribution of intermediates reveals the reversibility of the isocitrate dehydrogenase + aconitase reactions, even in the absence of a source of α-ketoglutarate. In addition, in many cases, a number of labeling incompatibilities were found as follows: (i) glucose versus triose phosphates and phosphoenolpyruvate; (ii) differences in the labeling ratios C-4/C-3 of glucose versus (glyceraldehyde 3-phosphate)/(dihydroxyacetone phosphate); and (iii) labeling of citric acid cycle intermediates in tissue_versus_ effluent perfusate. Overall, our data show that gluconeogenic and citric acid cycle intermediates cannot be considered as sets of homogeneously labeled pools. This probably results from the zonation of hepatic metabolism and, in some cases, from differences in the labeling pattern of mitochondrial versus extramitochondrial metabolites. Our data have implications for the use of labeling patterns for the calculation of metabolic rates or fractional syntheses in liver, as well as for modeling liver intermediary metabolism.
This second of two companion articles concentrates on a comparison of the mass isotopomer distributions of metabolites of gluconeogenesis and the citric acid cycle in livers perfused with precursors of [1-13C]PEP.2 One substrate was NaH13CO3 that labels liver GNG from lactate or pyruvate via carboxylation and isotopic exchange reactions (1). The second substrate was dimethyl [1,4-13C2]succinate that labels PEP via reactions of the citric acid cycle and PEPCK. We modulated the rates of GNG from lactate, pyruvate, or [1,4-13C2]succinate using mercaptopicolinate (MPA), an inhibitor of PEPCK (2,3), or aminooxyacetate (AOA), an inhibitor of the glutamate-aspartate shuttle (4–6). Our data reveal major incompatibilities in the labeling of gluconeogenic intermediates extracted from the whole rat liver.
EXPERIMENTAL PROCEDURES
_Materials_—The materials and rat liver perfusion experiments are described in detail in the accompanying article (28). Briefly, livers from 18-h fasted rats (180–220 g) were perfused (7) with nonrecirculating bicarbonate buffer (40 ml/min) containing the following: (i) 40% enriched NaH13CO3 and 5 mm lactate, or 2 mm pyruvate ± 0.3 mm MPA, or 0.5 mm AOA (protocol I), or (ii) dimethyl [1,4-13C2]succinate ± 0.3 mm MPA (protocol II). In orientation experiments, we found that the labeling of gluconeogenic and CAC intermediates as well as glucose production were two to four times greater with dimethyl [1,4-13C2]succinate than with [1,4-13C2]succinate (not shown). Similar ratios in glucose production from dimethyl succinate and succinate were reported by Rognstad (8). Therefore, we conducted all the experiments of this group with 0.5 mm dimethyl [1,4-13C2]succinate ± 0.3 mm MPA.
_Sample Preparation_—Powdered frozen tissue (1.5 g), spiked with internal standards (150 nmol of [13C6]citrate, 100 nmol of [13C4]succinate, and 50 nmol of (RS)-3-hydroxy-[2H5]glutarate) was extracted with 19 ml of chloroform/methanol, 2:1, pre-cooled at –25 °C, using a Polytron homogenizer. During the 5-min extraction, the tube was partially immersed in acetone kept at –25 °C by periodic addition of dry ice. Then, 6 ml of ice-cold water was added to the tube, and the extraction (Folch wash (9)) was continued for 5 min. The slurry was centrifuged at 670 × g for 20 min at 4 °C. The upper methanol/water phase was collected and treated with 200 μmol of methoxylamine-HCl to protect ketoacids. The lower chloroform phase was vortexed for 5 min with 10 ml of methanol/water, 3:2, pre-cooled at –20 °C. After 20 min of centrifugation, the two upper methanol/water phases were combined, adjusted to pH 8.0 with NaOH, and evaporated in a Savant vacuum centrifuge. The residue was reacted with 100 μl of_N,O_-bis(trimethylsilyl)trifluoroacetamide with 10% trimethylchlorosilane (Regisil) at 70 °C for 50 min to form the TMS and methoxamate/TMS derivatives of the analytes. The MID of CAC intermediates in the effluent perfusate was assayed by the following: (i) acidifying 2 ml of effluent spiked with 5 nmol of [13C6]citrate internal standard; (ii) extracting with ethyl ether; (iii) preparing TMS derivatives; and (iv) GC-MS analysis.
For the assay of aspartate, glutamate, and glutamine in effluent perfusate, 2-ml samples were acidified with HCl and then loaded on a 2-ml AG-1–50 H+ resin column. The column was rinsed with 8 ml of H2O, with 8 ml of 3 m NH4OH, and 8 ml of H2O. The ammonia and second water washes were combined, evaporated, and treated to form TMS derivatives.
GC-MS Assays_—Analyses were carried out on an Agilent 5973 mass spectrometer, linked to a model 6890 gas chromatograph equipped with an autosampler, a Varian VF-5MS capillary column (60 m, 0.25 mm inner diameter), and an EZ guard column (10 m). The carrier gas was helium (1 ml/min) with a pulse pressure of 40 p.s.i. The injection was either 1 μl split 10:1 or 2 μl splitless. The injector temperature was set at 270 °C and the transfer line at 280 °C. The GC temperature program was as follows: start at 80 °C, hold for 1 min, increase by 10 °C/min to 320 °C, hold at 320 °C for 5 min. The ion source and the quadrupole were set at 150 °C. The ammonia pressure was adjusted to optimize peak areas. For each analyte, we monitored the signals at the nominal m/z (M0) and at all detectable naturally labeled mass isotopomers (M1, M2, and M3). The MID of each analyte was compared with the theoretical distribution. The electron ionization fragmentation pattern of analytes was introduced in the National Institute of Science and Technology software to help with the identification and to detect interferences. The relative concentrations and MIDs of compounds of interest, identified during electron ionization runs, were assayed under ammonia-positive chemical ionization conditions. Retention times and_m/z monitored are listed in Ref.10. To measure the enrichment of C-6 of citrate,3 the TMS derivative was assayed with electron ionization, monitoring_m/z_ 375–381 (C-1–C-6) and 273–278 (C-1–C-5).
Because of an interference with the assay of PEP by the above procedure, the 13C-enrichment of PEP was determined by an assay that involves the following: (i) perchloric acid extraction of a liver sample; (ii) reduction of extant pyruvate in the neutralized extract by NaBH4; (iii) acidification to destroy excess NaBH4 and re-neutralization; (iv) hydrolysis of PEP with alkaline phosphatase; (v) derivatization of pyruvate with methoxylamine and pentafluorobenzyl bromide; and (vi) ammonia negative chemical ionization GC-MS (11).
The concentration and total labeling of glucose in the effluent perfusate were assayed by the following: (i) isotope dilution with [13C6]glucose; (ii) evaporating the sample; (iii) reacting the residue with CH3I and NaOH to form permethylglucose; and (iv) ammonia-positive chemical ionization GC-MS (12). The 13C labeling of individual glucose carbons was assayed using the methoxamate-TMS and the aldonitrile pentaacetate derivatives (12).
_Calculations_—The relative concentrations of metabolites (inhibitor versus control) were calculated (10) as shown inEquation 1,
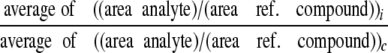 |
(Eq. 1) |
|---|
where subscripts i and c represent the intervention and control group, respectively. Equation 1 was used to calculate relative concentrations (inhibitor_versus_ control group). When metabolites were m1- and m2-labeled,4 their total labeling was calculated as shown inEquation 2,
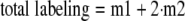 |
(Eq. 2) |
|---|
The theoretical mass isotopomer distribution of glucose, based on the measured m1 fractional enrichment of PEP, was calculated as shown in Equations3 and4,
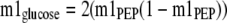 |
(Eq. 3) |
|---|
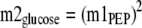 |
(Eq. 4) |
|---|
The theoretical mass isotopomer distribution of glucose, based on the measured m1 fractional enrichment of the two triose phosphates, was calculated as shown in Equations 5 and6,
 |
(Eq. 5) |
|---|
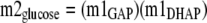 |
(Eq. 6) |
|---|
We used four formulas to calculate fractional GNG (_f_GNG) from the labeling of glucose and gluconeogenic intermediates. First, we used a modification of the formula by Rossetti et al. (13) adapted for a stable isotopic tracer as indicated in Equation 7,
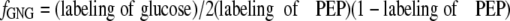 |
(Eq. 7) |
|---|
Second, we used the two formulas by Hellerstein (14) to calculate (i) the theoretical average enrichment of triose phosphates (p) from the m2/m1 labeling ratio of glucose, and (ii) _f_GNG as given in Equations 8 and9,
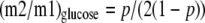 |
(Eq. 8) |
|---|
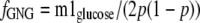 |
(Eq. 9) |
|---|
Third, we modified Equation 9 by replacing the theoretical average enrichment of triose phosphates (p) by the average of the measured enrichments of triose phosphates (TP) and achieved Equation 10,
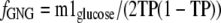 |
(Eq. 10) |
|---|
Fourth, we modified Equation 10 to take into account the actual labeling of each triose phosphate andEquation 11 shows the result,
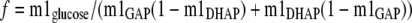 |
(Eq. 11) |
|---|
_Statistics_—Data are presented as mean ± S.E. Significance was tested by independent sample t test with SPSS software. Statistical significance was set for p < 0.05.
RESULTS AND DISCUSSION
The design of the metabolomic assays allowed the measurement of the MID of metabolites, in addition to their relative concentrations (calculated fromEquation 1) presented in the accompanying article (28). The MIDs, presented in Tables 1,2,3, reveal unexpected features of the labeling of intermediates.
TABLE 1.
Comparison of the labeling on C4 and C3 of effluent glucose with labeling of GAP and DHAP in liver
Each group of livers was perfused with the substrate + label ± inhibitor indicated in the left column. Glucose was labeled only on C4 and C3. Labeling data are expressed in mole percent enrichments (mean ± SE,n = 4–6).

TABLE 2.
Labeling from 40% NaH13CO3 of liver CAC and gluconeogenic intermediates
Livers were perfused with buffer containing 40% NaH13CO3 + 5 mm lactate or 2 mm pyruvate ± 0.3 mm MPA or 0.5 mm AOA. Labeling data are expressed in mole percent enrichments (mean ± S.E., n = 4–7). Most metabolites are only M1-labeled, except for citrate and glucose that are M1- and M2-labeled. The total labeling of citrate and glucose was calculated using Equation 2.

TABLE 3.
Labeling from dimethyl [1,4-13C2]succinate of CAC and GNG intermediates
Livers were perfused with buffer containing 0.5 mm dimethyl [1,4-13C2]succinate ± 0.3 mm MPA.n =4–6.

_Comparisons of Labeling Patterns of Glucose, Triose Phosphates, and PEP_—In the presence of NaH13CO3, glucose becomes labeled only on C-3 and C-4, as shown by others and us (1,12). In the presence of [1,4-13C2]succinate, glucose should also be labeled only on C-3 and C-4 because [1,4-13C2]succinate yields [1,4-13C2]OAA, which is converted to [1-13C]PEP by PEPCK. Indeed, in all cases, label was found only on C-3 and C-4 of glucose (Table 1). The sum of the C-3 and C-4 labeling was close to the total labeling of glucose (compare with Table 2, row p1). In seven of eight cases, C-4 was significantly more labeled than C-3 (Table 1). In the 1950s, it was reported that, when GNG was investigated with tracers that label glucose via C-1 of PEP, C-4 of glucose was slightly, but significantly, more labeled than C-3 (15). This was ascribed to the combination of (i) dilution of triose phosphates by unlabeled endogenous glycerol, and (ii) incomplete isotopic equilibration of GAP and DHAP by triose-phosphate isomerase (16). In all our perfusions with lactate or pyruvate + NaH13CO3, the labeling sequences were GAP > DHAP > α-glycerophosphate > unlabeled glycerol (Table 2, rows l, m, and o). In perfusions with [1,4-13C2]succinate dimethyl ester (Table 3; rows l, m, and o), the labeling sequences were GAP·DHAP > α-glycerophosphate > unlabeled glycerol. Our data clearly show that the labeling of α-glycerophosphate is much lower than those of triose phosphates, thus supporting the conclusion by Landau et al. (15) that the asymmetry of glucose labeling appears to depend on the availability on unlabeled trioses. This dilution is present even in isolated livers perfused with buffer containing no glycerol. The diluting glycerol is most likely derived from the action of hepatic lipases (17).
Because the labeling of C-4 and C-3 of glucose is derived from those of GAP and DHAP, one would expect the following: (i) the labeling ratios (C-4 of glucose)/GAP and (C-3 of glucose)/DHAP to be to be close to 1.0, and (ii) the labeling ratio C-4/C-3 in glucose to be identical to the labeling ratio GAP/DHAP. This was not the case (Table 1). In five of the six groups of livers perfused with 40% NaH13CO3, the GAP/DHAP labeling ratio was significantly greater than the C-4/C-3 labeling ratio of glucose. In contrast, in the two groups of livers perfused with [1,4-13C2]succinate, the GAP/DHAP labeling ratio was significantly lower than the C-4/C-3 labeling ratio of glucose.
To test for the compatibility of the labeling of glucose with those of triose phosphates and PEP, we calculated the theoretical labeling of glucose based on the measured labeling of GAP and DHAP (Equations5 and6; line q of Tables2 and3), and on the measured labeling of PEP (Equations 3 and4; line r of Tables2 and3). Comparison of these theoretical labeling with the actual labeling of glucose (line p of Tables2 and3) reveals significant differences in 13 of 20 comparisons (9 theoretical labelings underestimate actual labeling and 4 overestimate). Therefore, the labeling of glucose via GNG in the intact liver cannot be computed by a simple combinatorial analysis of the measured labeling of triose phosphates or PEP. This conclusion will be further developed when we consider estimates of fractional GNG.
Other incompatibilities show up between the labeling of GAP and PEP. In five of six groups of livers perfused with lactate or pyruvate + 40% NaH13CO3, GAP was significantly more labeled than PEP (Table 2, compare rows l and i), except in the lactate control group. In contrast, in the two groups of livers perfused with [1,4-13C2]succinate, GAP was significantly less labeled than PEP (Table 3, compare rows l and i). Note that in all groups, the enrichments of 2- and 3-phosphoglycerate were close to that of PEP. So the labeling of GAP appears to be influenced by other factor(s) than the known processes that label GAP from either NaH13CO3 or [1,4-13C2]succinate. Besides the effect of metabolic zonation of the liver on the labeling patterns of intermediates (to be dealt with below), the data might suggest the existence of an unknown CO2 fixating reaction, the product of which is GAP or a compound converted to GAP. In perfusions with 40% NaH13CO3, such13CO2 fixation would explain a GAP/PEP labeling ratio greater than 1.0. In perfusions with [1,4-13C2]succinate, the fixation of unlabeled CO2 (used to gas the perfusate) would explain a GAP/PEP labeling ratio smaller than 1.0. If such CO2 fixating reaction existed, the GAP thus formed would not mix with the GAP used for glucose synthesis. However, no such CO2 fixating process has been described in any organism.
Could some reactions of the pentose phosphate pathway (PPP) result in the over-labeling or under-labeling of GAP from NaH13CO3 or [1,4-13C2]succinate, respectively? This can be excluded because (i) both label sources are channeled to glucose via [1-13C]PEP, and (ii) we found no labeling on C-2 of glucose. The incompatibilities of labeling of metabolites presented in this section are reflected in the varying estimates of fractional GNG (_f_GNG) computed from the labeling patterns.
_Estimates of Fractional Gluconeogenesis_—Two types of equations have been presented to calculate _f_GNG from the measured or calculated labeling patterns of glucose and/or gluconeogenic intermediates. Rossetti et al. (13) have used the specific activity ratio (UDP-glucose)/2(PEP) to calculate _f_GNG in livers from animals infused with [U-14C]lactate. In these experiments, the labeling of UDP-glucose was taken as a proxy of the labeling of glucose-6-P. Hellerstein (14) presented two equations, the first of which calculates the enrichment of triose phosphates (p) from the m2/m1 labeling ratio of glucose. A given m2/m1 enrichment ratio can result from many combinations of m2 and m1 enrichments. However, the second equation by Hellerstein (14) calculates _f_GNG by selecting the m2/m1 combination corresponding to the measured m1 enrichment of glucose. So the two equations yield single values of p and f. For example, for m2 = 4% and m1 = 16%, p = 33% and f = 70%. The main assumption of the Rossetti and Hellerstein equations is that the enrichment of gluconeogenic intermediates is the same in all cells that synthesize glucose. In Tables2 and3, we present four calculations of _f_GNG. The first calculation (Tables2 and3, row t, andEquation 7) is similar to that of Rossetti et al. (13), except that (i) Equation 7 is adapted to stable isotope technology, and (ii) it uses the labeling of effluent glucose instead of that of UDP-glucose. This is appropriate for data from isolated liver experiments where the inflowing perfusate does not contain glucose. The second calculation (Tables2 and3, row u, and Equations8 and9), which uses the equations by Hellerstein (14), is based on the measured m2 and m1 enrichments of glucose. The third calculation (Tables2 and3, row v, andEquation 10) uses a modification of the second equation by Hellerstein (Equation 9) with the measured average enrichments of triose phosphates (Tables2 and3, row n). Finally, the fourth calculation (Tables 2 and3, row w, andEquation 11) derives from the third calculation (Equation 10) by taking into account the actual enrichment of each triose phosphate (Tables2 and3, rows l and m).
In livers from 18 to 22-h fasted rats, perfused with nonrecirculating buffer containing no glucose, but lactate, pyruvate, or succinate,_f_GNG should be close to 100%, about 90–100%. A computed _f_GNG, lower than 90%, must represent an underestimation of GNG. A computed _f_GNG greater than 100% is a biological impossibility. Values of _f_GNG calculated from the labeling of PEP and glucose usingEquation 7 (Tables2 and3, row t) are reasonable in only three of nine groups, i.e. in livers perfused with pyruvate. In the six other groups, _f_GNG is impossibly high (142–223%). These are the same groups where the mass isotopomer distribution of glucose was not compatible with that of PEP (Tables2 and3, compare rows p and r). In the accompanying article (28), Fig. 5, A–C, and Fig. 6 show that in the groups with impossibly high _f_GNG, PEP concentrations were much lower than in the corresponding control groups with reasonable _f_GNG. One possible explanation of the high _f_GNG would be that in the livers with low PEP concentrations, the PEP in the low-GNG perivenous area has a lower labeling than in the high-GNG periportal area. This would cause the labeling of PEP in the total liver extract to be incompatible with that of glucose.
In livers perfused with lactate or pyruvate + 40% NaH13CO3, the measured average triose phosphate enrichment matches within 20% the triose phosphate enrichment calculated from the MID of glucose (Equation 8 and Table 2, compare rows n and s). However, in livers perfused with [1,4-13C2]succinate, the measured average triose phosphate enrichment is much lower than the triose phosphate enrichment calculated from the MID of glucose (Equation 8, Table 3, compare rows n and s). In the presence of dimethyl [1,4-13C2]succinate and no MPA, the calculated triose phosphate enrichment (Table 3, line s) is compatible with the following: (i) the enrichment of PEP (Table 3, line i), (ii) the measured MID of glucose (line p), and (iii) the theoretical MID of glucose calculated from the enrichment of PEP (line r). In this case, the labeling incompatibility is limited to the measured enrichment of triose phosphates. However, in the presence of dimethyl [1,4-13C2]succinate + MPA, the calculated triose phosphate enrichment (Table 3, line s) is not compatible with the following: (i) the enrichment of PEP (Table 3, line i), (ii) the measured MID of glucose (line p), and (iii) the theoretical MID of glucose calculated from the enrichment of PEP (line r).
Also, in most cases, the average of the measured enrichment of the triose phosphates was very close to the enrichment calculated fromEquation 8 (identical toEquation 1 by Hellerstein and Neese (18)) (Table 2, compare rows r and s). So it would appear that the enrichment of triose phosphates was similar in all cells that synthesized glucose from lactate or pyruvate in the presence of 40% NaH13CO3. In livers perfused with erythrocyte-free 25 mm bicarbonate buffer at 4 ml·min–1·g–1, the endogenous production of CO2 (about 1 μmol·min–1·g–1 (7)) decreased the enrichment of the bicarbonate buffer by a negligible amount. The constancy of the bicarbonate enrichment between the periportal and perivenous areas of the liver lobule resulted in compatible labeling of triose phosphates and glucose.
In perfusions with dimethyl [1,4-13C2]succinate,_f_GNG calculated fromEquation 9 was reasonable for the control and MPA conditions (91 and 82%,Table 3, row u). However, the average of the measured enrichment of the triose phosphates was 2- and 4-fold lower than the enrichment calculated fromEquation 8 (Table 3, compare rows n and s). To explain this discrepancy, one would have to assume the following: (i) that most GNG is periportal (19); (ii) the computed values of triose phosphate enrichment and _f_GNG shown inTable 3, rows s and u, reflect what occurs in the periportal area; and (iii) the enrichment of triose phosphates from dimethyl [1,4-13C2]succinate is very low in the pericentral area. So when the whole liver was extracted, the enrichment of triose phosphates in the liver extract was lower than in the area where glucose was produced from dimethyl [1,4-13C2]succinate.
In conclusion, the compatibility of the labeling of triose phosphates extracted from the liver with that of glucose can neither be assumed nor rejected. It varies with both the substrate and the tracer, which are processed through the gluconeogenic pathway.
Reversibility of Isocitrate Dehydrogenase Revealed from the Labeling Pattern of Citrate and α_-Ketoglutarate_—In our previous study in livers perfused with 0.5 mm [13C5]glutamate or [13C5]glutamine, the presence of M5 citrate in the tissue extract demonstrated the reversibility of the reactions catalyzed by isocitrate dehydrogenase and aconitase (20,21). However, this reversibility could have resulted from an increase in the αKG pool induced by exogenous glutamate or glutamine. In this study conducted without αKG precursor, we were able to demonstrate the reversibility of ICDH, using the relative labeling of C-1 and C-6 of citrate.
In perfusions with lactate or pyruvate + 40% NaH13CO3, all metabolites shown inTable 2 were M1-labeled, except for citrate and glucose, which were M1- and M2-labeled. The total labeling of citrate and glucose was calculated fromEquation 2. Let us compare the enrichments of citrate and αKG (Table 2, rows a and c). The labeling of αKG is substantially lower than one-half that of the total labeling of citrate. This is unexpected because, in perfusions with lactate or pyruvate, (i) only carbons 1 and 6 of citrate become labeled from NaH13CO3 (via pyruvate carboxylase and citrate synthase), and (ii) randomization of label between C-1 and C-4 of OAA (via the reversal of the malate dehydrogenase and fumarase reactions) would result in the labeling of C-6 of citrate to be at most equal to that of C-1. We hypothesized that the labeling of C-6 of citrate occurred in part via reversal of isocitrate dehydrogenase (ICDH) and aconitase. To test this hypothesis, we needed to measure the labeling of C-6 and C-1 of citrate. The electron ionization mass spectrum of the TMS derivative of citrate includes a fragment at m/z 273 corresponding to the loss of C-6 from the parent molecular ion at m/z 375. This was assessed using standards of unlabeled, [13C6]-, [1,5-13C2]-, and [6-13C]citrate (not shown). Because in the presence of NaH13CO3, citrate is labeled only on C-1 and C-6, the enrichments of C-1 and of the C-1–5 fragment are identical. Also, the enrichment of αKG (Table 2, row c) must be equal to that of C-1 of citrate. This allowed us to calculate the labeling of C-1 and C-6 of liver citrate (Table 2, row b). In all experiments with 40% NaH13CO3, C-6 was 1.7–3.6 times more labeled than C-1 (p < 0.05). The labeling of α-ketoglutarate was compatible with the difference (total labeling of citrate) minus (labeling of C-6 of citrate). Thus, the labeling pattern of citrate reflects the reversibility of ICDH.
Additional evidence of ICDH reversibility was found in perfusions with dimethyl [1,4-13C2]succinate where all CAC intermediates (except αKG) were M1- and M2-labeled (Table 3). Starting from [1,4-13C2]succinate, citrate should be equally labeled (and only labeled) on C-1 and C-6. Because the labeling of α-ketoglutarate reflects that of C-1 of citrate, comparison of rows b and c of Table 3 shows that C-6 of citrate is three times less labeled than C-1. The lower labeling of C-6 reflects a dilution by unlabeled CO2 via the reversal of ICDH. This unlabeled CO2 is that used to equilibrate the bicarbonate buffer of the perfusate. Thus, the opposite labeling ratios C-1/C-6 of citrate in perfusions with NaH13CO3 versus dimethyl [1,4-13C2]succinate reflect the reversal of the ICDH reaction. In conclusion, in the intact liver, the reaction catalyzed by isocitrate dehydrogenase is reversible even when the αKG pool is not increased by exogenous glutamate or glutamine.
_Labeling Patterns of Citric Acid Cycle Intermediates in Liver_—Because αKG is only labeled on C-1 from NaH13CO3, the sequence α-ketoglutarate → succinyl-CoA → succinate should yield unlabeled succinate. The low, but easily measurable, labeling of succinate (Table 2, row d) must result from partial equilibration with fumarate which, as well as malate, becomes labeled from OAA by the reversible reactions of malate dehydrogenase and fumarase.
The measured labeling of whole tissue OAA (Table 2, row g) should be compatible with the combination of the labeling of the following: (i) a mitochondrial pool with a labeling equal to twice that of C-1 of citrate (assuming complete randomization of label between C-1 and C-4 of OAA before it enters the citrate synthase reaction), and (ii) a cytosolic pool with a labeling equal to twice that of PEP (because one-half of the label of OAA labeled from NaH13CO3 is lost in the PEPCK reaction). The labeling of OAA should be between twice the labeling of C-1 of citrate and twice the labeling of PEP. In fact, in five of six cases, the labeling of OAA is much lower than twice the labeling of C-1 of citrate or twice the labeling of PEP. In the sixth case (lactate + MPA), the labeling of OAA is greater than twice the labeling of C-1 of citrate or twice the labeling of PEP. We recognize that the comparison between the labeling of OAA, C-1 of citrate and PEP assumes the constancy of labeling of the three compounds across the liver lobule. This condition is obviously not fulfilled. Because one cannot account for the labeling of OAA, the interpretation on the labeling of aspartate should not be based on the labeling of OAA, although aspartate can only become labeled from OAA. In perfusions with lactate, the labeling of aspartate was decreased 9 times by AOA, whereas in perfusions with pyruvate the labeling of aspartate was not affected by AOA (Table 2, row h). This probably results from the 2.8-fold higher concentration of OAA in perfusions with pyruvate + AOA versus lactate + AOA (see Fig. 4_C_ in the accompanying article (28)). The higher OAA concentration allows isotopic equilibration of OAA and aspartate in the presence of AOA.
In perfusions with M2 dimethyl [1,4-13C2]succinate, the MIDs of fumarate, malate, and OAA include both M1 and M2 isotopomers (Table 3, rows e–g). Because all label from [1,4-13C2]succinate should be lost after one turn of the CAC, the presence of M1 isotopomers reflects extramitochondrial processes such as the conversion of M2 to M1 malate via reversible malic enzyme, resulting in loss of label from C-4 of malate, and the loss of label from M2 OAA via the OAA ↔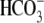 exchange catalyzed by the first step of the PEPCK reaction, resulting in loss of label from C-4 of OAA (22). The presence of M1 citrate is unlikely to result from the return of M1 malate to the mitochondria, because GNG from succinate involves the export of reducing equivalents to the cytosol via malate. A more likely explanation is that M2 citrate (labeled on C-1 and C-6) undergoes a partial loss of label from C-6 via the reversibility of mitochondrial isocitrate dehydrogenase, replacing13Cby 12C from unlabeled CO2 used to gas the perfusate.
exchange catalyzed by the first step of the PEPCK reaction, resulting in loss of label from C-4 of OAA (22). The presence of M1 citrate is unlikely to result from the return of M1 malate to the mitochondria, because GNG from succinate involves the export of reducing equivalents to the cytosol via malate. A more likely explanation is that M2 citrate (labeled on C-1 and C-6) undergoes a partial loss of label from C-6 via the reversibility of mitochondrial isocitrate dehydrogenase, replacing13Cby 12C from unlabeled CO2 used to gas the perfusate.
Addition of MPA increased the labeling of CAC intermediates from fumarate to α-ketoglutarate. This reflects the decreased transfer of label to GNG and, probably, a decreased rate of CAC operation (resulting from the inhibition of GNG).
_Differences in Labeling of CAC Intermediates in Effluent Versus Liver_—Fig. 1, A and_B,_ compares the labeling of CAC intermediates in liver tissue and effluent perfusate for groups 1 (livers perfused with 5 mm lactate + 40% NaH13CO3) and 7 (livers perfused with 0.5 mm [1,4-13C2]succinate). In both cases, the labeling of fumarate and malate was lower in the effluent perfusate than in liver. In the presence of dimethyl [1,4-13C2]succinate, the labeling of effluent pyruvate was five times lower than that of tissue pyruvate. Similar differences were observed in groups 2–6 and 8 (data not shown). These differences probably result from cytosolic reactions that alter the labeling of mitochondrial CAC intermediates and pyruvate before they leave the cell. Such reactions might include the transamination of aspartate and alanine (derived from proteolysis) forming unlabeled OAA and pyruvate. Unlabeled OAA would decrease the enrichment of malate and fumarate via cytosolic malate dehydrogenase and fumarase. Reversal of the reaction of isocitrate dehydrogenase in cytosol can result in loss of label on C-6 of citrate. Exchange reaction sequences such as malate → pyruvate → malate, catalyzed by malic enzyme, and the first of the PEPCK sub-reactions,i.e. the OAA-bicarbonate exchange (22,23), would convert M2 malate to M1 and can explain the differences in labeling of malate and pyruvate in effluent versus liver. In the presence of dimethyl [1,4-13C2]succinate, there is a decrease in labeling from PEP (48%) to tissue pyruvate (27%) to effluent pyruvate (4%) (Table 3, rows i and h2;Fig. 1_B_). Metabolic zonation can also contribute to the release of low-labeled pyruvate from the glycolytic pericentral area of the lobule. In all perfusions with 40% NaH13CO3 (groups 1–6), the labeling of α-ketoglutarate was slightly, but significantly, higher in the effluent perfusate than in liver (Fig. 1_A_). The cause of this difference is not clear. In the presence of MPA, there was a decrease in the difference in labeling between metabolites in liver and in effluent perfusate (not shown). This probably results from the increase in the concentration of CAC intermediates induced by MPA (see Fig. 5, A and C, in accompanying article (28)). Also, inhibition of PEPCK decreased the productions of M1 PEP and M1 pyruvate. In conclusion, the differences in labeling pattern of tissue versus effluent metabolites probably result from the following: (i) modification of the labeling of CAC intermediates in the cytosol, (ii) dilution by amino acids derived from proteolysis, and (iii) inverse zonation of GNG versus glycolysis. These processes contribute to the lack of homogeneity of metabolite labeling.
FIGURE 1.

Comparison of the mass isotopomer distribution of citric acid cycle intermediates and pyruvate in liver tissue versus the effluent perfusate. A, livers perfused with 5 mm lactate + 40% NaH13CO3. B, livers perfused with 0.5 mm [1,4-13C2]succinate dimethyl ester.SUC, succinate; FUM, fumarate; MAL, malate;CIT, citrate; α_KG_, α-ketoglutarate;PYR, pyruvate; MPE, molar percent enrichment.
In conclusion of the second part of our study, our data reveal a number of incompatibilities between the labeling of gluconeogenic intermediates and glucose. In addition, we show some incompatibilities between the measured and calculated enrichments of triose phosphates. These incompatibilities are not systematic, but they vary with the metabolic conditions of the livers and the nature of labeled substrates added to the perfusate. The heterogeneity of labeling of intermediates is probably caused by the zonation of liver metabolism (19), resulting in gradients of concentration and labeling of intermediates across the liver lobules. Our data reinforce earlier findings that demonstrated the existence of gradients of labeling of triose phosphates and of lipogenic acetyl-CoA, from the mass isotopomer analysis of newly synthesized glucose, fatty acids, and sterols (24–26).
What are the implications of our findings? First, the reversibility of liver ICDH under conditions where the pool of αKG is not increased by the supply of glutamate or glutamine decreases the number of irreversible reactions of the CAC to those catalyzed by citrate synthase and α-ketoglutarate dehydrogenase. Second, the multiple incompatibilities observed between the labeling of glucose and of key gluconeogenic intermediates (PEP, triose phosphates) probably result from the zonation of enzyme activities and of metabolite concentrations across the liver. Although there is a fair amount of knowledge on the zonation of enzyme activities, there is essentially no knowledge on the distribution of metabolite concentrations across the liver lobule. Note that the labeling incompatibilities we observed in perfused livers probably underestimate those occurring in the liver in vivo. In vivo, the liver is perfused with blood at about 1 ml·g·min–1. However, the isolated liver is perfused with a high flow rate of nonrecirculating buffer (4 ml·g·min–1) to compensate for the low oxygen content of red blood cell-free perfusate. As a result, there is very little or no decrease in the concentration and labeling of substrates as perfusate passes through the liver. In contrast, in vivo, there is a clear decrease in concentration and labeling of substrates across the liver (27). Thus, in vivo, the incompatibilities of metabolite labeling are probably greater than those we observed in perfused livers.
A final implication of our data relates to the modeling of metabolic processes in the liver, especially in experiments using labeled tracers or labeled substrates. We previously showed that the labeling of fatty acids and sterols from [13C]acetate can be reasonably explained by the existence of decreasing gradients of enrichment of lipogenic acetyl-CoA across the liver lobule (26). It will be very challenging to build a comprehensive model of hepatic GNG and CAC based on measured labeling profiles of intermediates, because these profiles vary across the liver lobule, and between cytosol and mitochondria.
*
This work was supported, in whole or in part, by National Institutes of Health Metabolomics Roadmap Initiative Grant R33DK070291. This work was also supported by the Cleveland Mt. Sinai Health Care Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “_advertisement_”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
2
The abbreviations used are: PEP, phosphoenolpyruvate; AOA, aminooxyacetate; DHAP, dihydroxyacetone phosphate; CAC, citric acid cycle; GAP, glyceraldehyde 3-phosphate; GNG, gluconeogenesis; MID, mass isotopomer distribution; MPA, mercaptopicolinate; OAA, oxaloacetate; PEPCK, phosphoenolpyruvate carboxykinase; TMS, trimethylsilyl; GC-MS, gas chromatography-mass spectrometry.
3
Carbons 1, 2, 3, and 6 of citrate derive from carbons 4, 3, 2, and 1 of oxaloacetate, respectively.
4
Mass isotopomers are designated as Mi, where i is the number of atomic mass units above the molecular weight of the monoisotopic mass M. The mol fraction of labeled mass isotopomers, after correction for natural enrichment, are designated as m1 and m2.
References
- 1.Esenmo, E., Chandramouli, V., Schumann, W. C., Kumaran, K., Wahren, J., and Landau, B. R. (1992) Am. J. Physiol. 263 E36–E41 [DOI] [PubMed] [Google Scholar]
- 2.Blackshear, P. J., Holloway, P. A., and Alberti, K. G. (1975) Biochem. J. 148 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jomain-Baum, M., Schramm, V. L., and Hanson, R. W. (1976) J. Biol. Chem. 251 37–44 [PubMed] [Google Scholar]
- 4.Rognstad, R., and Clark, D. G. (1974) Arch. Biochem. Biophys. 161 638–646 [DOI] [PubMed] [Google Scholar]
- 5.Ochs, R. S., and Harris, R. A. (1980) Biochim. Biophys. Acta 632 260–269 [DOI] [PubMed] [Google Scholar]
- 6.Cornell, N. W., Zuurendonk, P. F., Kerich, M. J., and Straight, C. B. (1984) Biochem. J. 220 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunengraber, H., Boutry, M., and Lowenstein, J. M. (1973) J. Biol. Chem. 248 2656–2669 [PubMed] [Google Scholar]
- 8.Rognstad, R. (1984) Arch. Biochem. Biophys. 230 605–609 [DOI] [PubMed] [Google Scholar]
- 9.Folch, J., Arsove, S., and Meath, J. A. (1951) J. Biol. Chem. 191 819–831 [PubMed] [Google Scholar]
- 10.Yang, L., Kasumov, T., Yu, L., Jobbins, K. A., David, F., Previs, S. F., Kelleher, J. K., and Brunengraber, H. (2006) Metabolomics 2 85–94 [Google Scholar]
- 11.Previs, S. F., Des Rosiers, C., Beylot, M., David, F., and Brunengraber, H. (1996) J. Mass Spectrom. 31 643–648 [DOI] [PubMed] [Google Scholar]
- 12.Beylot, M., Previs, S. F., David, F., and Brunengraber, H. (1993) Anal. Biochem. 212 526–531 [DOI] [PubMed] [Google Scholar]
- 13.Rossetti, L., Giaccari, A., Barzilai, N., Howard, K., Sebel, G., and Hu, M. (1993) J. Clin. Investig. 92 1126–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellerstein, M. K. (1991) J. Biol. Chem. 266 10920–10924 [PubMed] [Google Scholar]
- 15.Landau, B. R., Hastings, A. B., and Nesbett, F. B. (1955) J. Biol. Chem. 214 525–535 [PubMed] [Google Scholar]
- 16.Rose, I. A., Kellermeyer, R., Stjernholm, R., and Wood, H. G. (1962) J. Biol. Chem. 237 3325–3331 [Google Scholar]
- 17.Debeer, L. J., Beynen, A. C., Mannaerts, G. P., and Geelen, M. J. (1982) FEBS Lett. 140 159–164 [DOI] [PubMed] [Google Scholar]
- 18.Hellerstein, M. K., and Neese, R. A. (1992) Am. J. Physiol. 263 E988–E1001 [DOI] [PubMed] [Google Scholar]
- 19.Jungermann, K. (1987) Diabetes Metab. Rev. 3 269–293 [DOI] [PubMed] [Google Scholar]
- 20.Des Rosiers, C., Fernandez, C. A., David, F., and Brunengraber, H. (1994) J. Biol. Chem. 269 27179–27182 [PubMed] [Google Scholar]
- 21.Des Rosiers, C., Di Donato, L., Comte, B., Laplante, A., Marcoux, C., David, F., Fernandez, C. A., and Brunengraber, H. (1995) J. Biol. Chem. 270 10027–10036 [DOI] [PubMed] [Google Scholar]
- 22.Chang, H. C., Maruyama, H., Miller, R. S., and Lane, M. D. (1966) J. Biol. Chem. 241 2421–2430 [PubMed] [Google Scholar]
- 23.Rognstad, R. (1982) J. Biol. Chem. 257 11486–11488 [PubMed] [Google Scholar]
- 24.Landau, B. R., Fernandez, C. A., Previs, S. F., Ekberg, K., Chandramouli, V., Wahren, J., Kalhan, S. C., and Brunengraber, H. (1995) Am. J. Physiol. 269 E18–E26 [DOI] [PubMed] [Google Scholar]
- 25.Previs, S. F., Fernandez, C. A., Yang, D., Soloviev, M. V., David, F., and Brunengraber, H. (1995) J. Biol. Chem. 270 19806–19815 [DOI] [PubMed] [Google Scholar]
- 26.Bederman, I. R., Reszko, A. E., Kasumov, T., David, F., Wasserman, D. H., Kelleher, J. K., and Brunengraber, H. (2004) J. Biol. Chem. 279 43207–43216 [DOI] [PubMed] [Google Scholar]
- 27.Puchowicz, M. A., Bederman, I. R., Comte, B., Yang, D., David, F., Stone, E., Jabbour, K., Wasserman, D. H., and Brunengraber, H. (1999) Am. J. Physiol. 277 E1022–E1027 [DOI] [PubMed] [Google Scholar]
- 28.Yang, L., Kombu, R. S., Kasumov, T., Zhu, S.-H., Cendrowski, A. V., David, F., Anderson, V. E., Kelleher, J. K., and Brunengraber, H. (2008) J. Biol. Chem. 283 21978–21987 [DOI] [PMC free article] [PubMed] [Google Scholar]