Functional redundancy of acetylcholinesterase and neuroligin in mammalian neuritogenesis (original) (raw)
Abstract
Accumulated evidence attributes noncatalytic morphogenic activitie(s) to acetylcholinesterase (AChE). Despite sequence homologies, functional overlaps between AChE and catalytically inactive AChE-like cell surface adhesion proteins have been demonstrated only for the Drosophila protein neurotactin. Furthermore, no mechanism had been proposed to enable signal transduction by AChE, an extracellular enzyme. Here, we report impaired neurite outgrowth and loss of neurexin Iα mRNA under antisense suppression of AChE in PC12 cells (AS-ACHE cells). Neurite growth was partially rescued by addition of recombinant AChE to the solid substrate or by transfection with various catalytically active and inactive AChE variants. Moreover, overexpression of the homologous neurexin I ligand, neuroligin-1, restored both neurite extension and expression of neurexin Iα. Differential PCR display revealed expression of a novel gene, nitzin, in AS-ACHE cells. Nitzin displays 42% homology to the band 4.1 protein superfamily capable of linking integral membrane proteins to the cytoskeleton. Nitzin mRNA is high throughout the developing nervous system, is partially colocalized with AChE, and increases in rescued AS-ACHE cells. Our findings demonstrate redundant neurite growth-promoting activities for AChE and neuroligin and implicate interactions of AChE-like proteins and neurexins as potential mediators of cytoarchitectural changes supporting neuritogenesis.
Keywords: antisense/band 4.1 homologs/neuroligin–neurexin interactions/PC12/neurite
A strong body of evidence attributes morphogenic activities to the acetylcholine-hydrolyzing enzyme acetylcholinesterase (AChE), especially in association with neurite outgrowth (reviewed in ref. 1). An evolutionarily conserved capacity of AChE to promote process extension was observed in avian, amphibian, and mammalian primary neurons (2–4) and in rat glioma cells (5). In neuroblastoma cells, modulated expression of AChE revealed a direct correlation between AChE levels and neurite outgrowth (6). However, the molecular and cellular mechanism(s) by which AChE exerts its neuritogenic activities remain to be elucidated. Repeated observations that process-promoting activities of AChE are insensitive to certain active site inhibitors (2, 3) and to genetically engineered inactivation of its hydrolytic activity (4) demonstrated their noncatalytic nature. Together with the sequence homologies observed for AChE and several known cell-adhesion proteins, these findings hinted at a role for AChE in cell adhesion-related processes. Furthermore, they suggested that the neuritogenic function of AChE might be fulfilled, in some circumstances, by one of the catalytically inactive, AChE-homologous cell surface proteins.
Among the proteins carrying large AChE-like extracellular domains are Drosophila neurotactin (7), gliotactin (8), and the rat neuroligins (9). Unlike AChE, however, the cholinesterase-like proteins all possess a transmembrane region and a protruding cytoplasmic domain. As such, they are capable of transducing growth signals directly into the cell. In contrast, it is unclear how AChE might induce intracellular signals leading to neurite growth. We therefore considered the possibility that AChE may act by competing with members of the cholinesterase-like family for extracellular binding to common ligands such as neurexins. Neuroligins constitute a multigenic family of brain-specific AChE-homologous proteins that have been suggested to exert overlapping functions in mediating recognition processes between neurons (10). Neuroligins bind to a specific subset of neurexins, polymorphic neuronal cell surface proteins believed to play a role in neuronal differentiation and axogenesis (9, 11). Neurexin Iβ was shown to interact with rat neuroligin to induce heterotypic cell adhesion (12). Thus, neuroligin–neurexin interrelationships could be important in inter-neuronal recognition pathways regulating axon pathfinding. We previously reported that overexpressed transgenic AChE suppressed neurexin Iβ production in embryonic mouse motoneurons in vivo (13). These results were taken to indicate crosstalk between AChE and neurexins during development and strengthened the notion that AChE and neuroligin act on common elements.
To address the question of whether the cholinesterase-like proteins display overlapping functionality, we established a loss-of-function model in which AChE could be replaced by candidate substitutes. Here, we report that rat phaeochromocytoma (PC12) cells stably transfected with DNA-encoding antisense AChE cRNA (AS-ACHE cells) display severe AChE and neurexin Iα mRNA depletion. After nerve growth factor (NGF)-stimulated differentiation, AS-ACHE cells exhibit an aberrant phenotype characterized by attenuated neuritogenesis. Neuritogenesis was partially restored not only by AChE, but by neuroligin, which also rescued lost neurexin Iα expression. Using a nonbiased differential display analysis, we discovered a member of the band 4.1 protein superfamily the expression of which correlated with the ability of the rescued cells to extend neurites in response to NGF. Together, this work demonstrates functional redundancy for AChE and neuroligin and suggests that broad familial interactions between neurexins and AChE-like proteins mediate cell–cell recognition processes important for regulating neuronal cytoarchitecture.
MATERIALS AND METHODS
Vector Construction.
A fragment of rat AChE cDNA was amplified by reverse transcription (RT)-PCR, by using primers designed for the E6 exon of mouse AChE (positions 1728 and 1832, Genbank accession no. x56518). The amplification product was directly cloned into the pCR3 vector (Invitrogen) according to the manufacturer’s instructions. The orientation of the insert was determined by informative restriction analyses by using _Xmn_I and _Pst_I (New England Biolabs) and its nucleotide sequence confirmed in an ABI-377 automated sequencer (Perkin–Elmer). A pCR3 vector containing an unknown irrelevant DNA fragment served as a control.
Cell Lines and Transfections.
PC12 rat pheochromocytoma cells were grown as previously described (14). Tissue culture plates or coverslips were coated with 10 μg/ml collagen type IV (Sigma) and for rescue studies also with the same concentration of recombinant human AChE (Sigma). Transient transfections were performed with lipofectamine (GIBCO/BRL) according to the manufacturer’s instructions. For stable transfections, cells were incubated in medium containing 800 μg/ml G418 (GIBCO/BRL) for a period of 30 days and then maintained with 400 μg/ml G418.
PCR Analyses.
RNA extraction and semiquantitative RT-PCR amplifications were performed as previously described (14) by using the following specific primers: mE6 1832(+), 5′-TCT GGA CGA GGC GGA GCG CC-3′ and pCR3 759(−), 5′-AGA TGC ATG CTC GAG CGG CC-3′, to amplify the AS-E6 RNA. To amplify AChE mRNA, we used mAC 1361(+) 5′-CCG GGT CTA TGC CTA CAT CTT TGA A-3′ (upstream primer), mAC 1844(−) 5′-CAC AGG TCT GAG CAG CGC TCC TGC TTG CTA-3′, and mI4 74(−) 5′-TTG CCG CCT TGT GCA TTC CCT-3′, the downstream primers specific for E6-AChE and I4-AChE, respectively. Isoform-specific neurexin and neuroligin primers were as follows (numbers correspond to nucleotide position and Genbank accession no., respectively): αI(+) 4175–4198 and αI(−) 4667–4647, m96374; βI(+) 978–997 and βI(−) 1113–1090, m96375; αII(+) 2311–2330 and αII(−) 2503–2482, m96376; βII(+) 881–900 and βII(−) 1011–991, m96377; αIII(+) 4166–4185, αIII(−) 4390–4369, βIII(+) 1179–1198 and βIII(−) 1431–1411, L14851; neurolig1(−) 762–786 and neurolig1(−) 1497–1477, U22952; neurolig2(+) 2962–2981 and neurolig2(−) 3636–3615, U41662; neurolig3(+) 700–719 and neurolig3(−) 1169–1148, U41663.
The primers for specific amplification of differentially displayed transcripts were: collagentV 69(+), 5′-CTC CAG ATG ACA CAA ACA AAA C-3′; collagentV 301(−), 5′-CTC TCC TGT CTC CAG ATT GC-3′; and nitzin 57(+), 5′-AAT GCC AGG AAA GAC TTG AAG ACA C-3′; nitzin 498(−), 5′-CTT GAT GAG GGA AGA GTT TGC ATA C-3′. Neuronal cell adhesion molecule and actin primers were as described (5). Mouse β-tubulin and rat synaptophysin primers were as follows: βtub 197(+), 5′-CGG AGA GCA ACA TGA ATG AC-3′; βtub 365(−), 5′-AAA GAC CAA TGC TGG AGG AC-3′; rsyn 212(+), 5′-CCT TCA GGC TGC ACC AAG TGT ACT TTG ATG-3′; and rsyn 660(−), 5′-CAC GAA CCA TAA GTT GCC AAC CCA GAG CAC-3′.
Resultant PCR products obtained from similar amounts of RNA were removed at intervals of three PCR cycles, electrophoresed on agarose gels and stained with ethidium bromide.
Differential Display.
Differential display was performed by using a modification of the original Welsh protocol (15) as detailed elsewhere (16) by using the following primers: 410, 5′-AGG TGA CGT GGG ACA CT-3′; TATA, 5′-CCT CCG CGA GAT CAT CT-3′; 599(−), 5′-ACG ACT TTC ACA GAC GG-3′; R15, 5′-AGA GTG CAG GCC ATG-3′; and 17259, 5′-GGT GAA GTT AGC GCA AG-3′.
Biochemical AChE Analyses.
Protein extracts were obtained from analyzed cells by homogenization in PBS supplemented with 1% Triton and 10 μg/ml of leupeptin and aprotinin (Sigma). Homogenates were centrifuged at 13,000 × g in a microfuge and the supernatants kept frozen for subsequent use. AChE activity was determined as described (17).
In Situ Hybridization.
High resolution nonradioactive in situ hybridization was performed on 7-μm paraffin embedded sections from three individual whole newborn mice, using a previously reported AChEcRNA probe (13) or 10 μg/ml of the 50-mer 2′-O-methyl 5′-biotinylated nitzin cRNA probe, 5′-UGG CAG UGU CUU CAA GUC UUU CCU GGC AUU CUC AUC GCU GGU AUA AUC UC-3′, as previously described (13). Dewaxed sections were treated with proteinase K. Hybridization in 50% formamide and 5× SSC, pH 4.5, was at 52°C for 18 hr, and washes in 2× SSC were at 60°C.
RESULTS
AChE Suppression Is Associated with a Partially Reversible Neuritogenic Deficit.
To achieve potent long-term suppression of AChE production, we transfected PC12 cells with the pCRAS-E6 vector, which directs the expression of a 132-bp fragment of exon 6 from the rat ACHE gene in the antisense orientation. Eight independent stably transfected clones were selected, each displaying different expression levels of AChE cRNA. Four of these clones revealed significant reductions in the catalytic activity of AChE (of 12, 30, 70, and 80%). AChEcRNA thus suppressed AChE activity in PC12 cells considerably more effectively than similarly targeted antisense oligonucleotides (14). The latter clone, with maximal suppression of AChE activity, was termed AS-ACHE and was used for further analyses. RT-PCR analysis by using primers selective for exon 6 (E6) or pseudointron 4 (I4) in the ACHE gene revealed a reduction in AChE-E6 mRNA and the complete suppression of AChE-I4 mRNA transcripts in AS-ACHE cells (Fig. 1A and data not shown). Furthermore, AChE catalytic activity, which was suppressed by 80% in AS-ACHE cells as compared with the parental PC12 cell line, was not significantly enhanced by NGF-triggered differentiation. In contrast, the original PC12 cells display a 50% increase in AChE activity within 24 hr of NGF treatment (ref. 14 and Fig. 1B). When decorated with polyclonal anti-AChE antibodies, NGF-treated AS-ACHE cells revealed drastic reduction in AChE-immunofluorescent labeling as compared with the parental PC12 cells (data not shown).
Figure 1.

AChE suppression is associated with a partially rescuable neuritogenic deficiency. (A) Transcription levels. Total cellular RNA was extracted from PC12 or AS-ACHE cells and subjected to RT-PCR amplification with primers selective for antisense ACHE RNA or the alternative E6- or I4-ACHEmRNA transcripts. (A Left) Endpoint products of RT-PCR using antisense ACHE RNA-specific primers. Asterisk indicates expression of the antisense RNA exclusively in AS-ACHE cells. (A Center and Right) Kinetic RT-PCR analysis by using ACHE or actin mRNA-specific primers. Presented are samples of PCR products removed every third cycle from cycles 21–36 for ACHE mRNAs and cycles 15–30 for actin mRNA. Note the delayed appearance of ACHE-E6-derived products, the total absence of ACHE-I4 products, and the unmodified levels of actin PCR products in AS-ACHE cells as compared with the parental PC12 line. One of 3 reproducible tests. (B) Suppressed AChE catalytic activity in AS-ACHE cells. Presented are AChE specific activities in the original PC12 and antisense ACHE cells before (Ct) and after (+NGF) 3-days NGF-mediated differentiation. Data are expressed as a percent of the activity measured in undifferentiated PC12 cells. (C) Neuritogenic deficiency. Fibroblast-like appearance and paucity of neurites of AS-ACHE as compared with the parental PC12 cells 3 days after addition of NGF. Cells were stained by using the May–Grunwald stain (Sigma) followed by Gurr’s improved Giemsa stain (BDH). (D) Reversibility of the neuritogenic deficiency. Percentage of analyzed cells with various numbers of processes after growth in the absence (control) or presence (rhAChE) of 10 μg/ml catalytically active recombinant human AChE added to the collagen matrix substratum. Note the partial restoration of neurite outgrowth in AS-ACHE cells exposed to extracellular AChE (P < 0.01). The small neurite growth-promoting effect of AChE on the parental PC12 cell line was deemed statistically insignificant. A total of 50 cells from four different plates were analyzed for each group and neurite numbers determined by eye.
AS-ACHE cells displayed reduced neurite frequency after NGF treatment compared with the parent PC12 cells (2.2 ± 0.9 vs. 5.0 ± 1.0 neurites/cell; P < 0.01, two-tailed t test) (Fig. 1 C and D). To examine the reversibility of this phenotype, we coated the collagen matrix on which the cells were grown with highly purified and catalytically active recombinant human AChE (rhAChE). After 3 days in the presence of rhAChE and NGF, neurite frequency increased to 3.5 ± 1.1 (P < 0.01) (Fig. 1D), with cell bodies becoming more round. Recombinant rhAChE also had a small but not statistically significant neurite growth promoting effect on PC12 cells (5.2 ± 1.2 as compared with 5.0 ± 1.0 neurites/cell).
Transfected AChE and Neuroligin Rescue Neuritogenesis in AS-ACHE Cells.
The deficient neurite outgrowth displayed by AS-ACHE cells might be related to lost catalytic or noncatalytic properties of AChE, or both. To test these possibilities, we transiently transfected AS-ACHE cells with plasmids encoding the synaptic form of AChE (AChE-E6, 17), genetically inactivated AChE (AChE-In, 4), the catalytically inactive AChE homolog neuroligin-1 (9) or a plasmid encoding the mitochondrial protein StAR (18) as a control. The initial neurite frequency of AS-ACHE cells was 1.9 ± 0.7 neurites/cell in these experiments and remained low (2.4 ± 0.1 neurites/cell) after transient transfection with the irrelevant StAR plasmid. However, transfection with AChE-E6, AChE-E6-In, and neuroligin-1 enhanced neurite frequency to 3.3 ± 0.1, 3.3 ± 0.1, and 3.7 ± 0.5 neurites/cell, respectively (P < 0.01, Fig. 2). The similar extent of rescue achieved with ACHE-E6 and ACHE-In attested to the noncatalytic nature of AChE’s neuritogenic activity in PC12. Moreover, the partial rescue attained by transfection of neuroligin DNA demonstrated an overlapping functionality of AChE and neuroligin in promoting neuritogenesis in these cells.
Figure 2.

Catalytically inactive AChE and neuroligin-1 both rescue neurite outgrowth in AS-ACHE cells. AS-ACHE cells were transiently transfected with either control StAR plasmid (AS + ctrl) or with plasmids encoding AChE-E6 (AS + E6), catalytically inactive AChE (AS + In), or the AChE-homologous protein neuroligin-1 (AS + Nrl), stimulated with NGF, and assessed for number of neurites as compared with the parental PC12 or nontransfected AS-ACHE cells. Number of processes per cell is presented as in Fig. 1C. n = 50 cells for all groups. Note that both catalytically active and inactive AChE and neuroligin, but not StAR, partially rescue the neurogenetic deficiency of AS-ACHE cells.
The Neuritogenic Potential of PC12 Cells Is Associated with Production of Neurexin Iα.
Neurexins constitute a large polymorphic family of cell surface proteins derived from three genes, neurexins I, II, and III (19). Each gene may be transcribed from one of two alternative promoters to yield a large array of alternatively spliced longer (α) and shorter (β) transcripts. The observation that heterologous expression of neuroligin-1 could partially restore NGF-mediated neurite outgrowth indicated that ligands of neuroligins such as neurexins may be involved in sustaining the AS-ACHE phenotype. To test this hypothesis, we studied neuroligin and neurexin expression in AS-ACHE cells by RT-PCR. Of the three neuroligins, neuroligin-2 mRNA was the only one detected. It appeared in both the original PC12 cells and in AS-ACHE cells at similar levels (not shown). Of the six major neurexin isoforms, we detected mRNAs for neurexin Iα, Iβ, and IIβ in the original PC12 cells, with neurexin Iα mRNA being the most prominent (Fig. 3). In contrast, neurexin Iα mRNA was below detectable levels in AS-ACHE cells. This down-regulation did not involve an alternative choice of promoters because neurexin Iβ mRNA levels remained similarly low in both PC12 and AS-ACHE cells. Nevertheless, neurexin Iα mRNA levels increased considerably in neuroligin-1-transfected AS-ACHE cells, i.e., those in which NGF-induced neurite extension was most efficiently restored. The levels of neuronal cell adhesion molecule, actin, and β-tubulin mRNAs were similar in all the analyzed cell types (Fig. 3 and data not shown), demonstrating that the observed modulation in neurexin Iα mRNA levels was selective and suggesting that neurexin Iα is a prerequisite for neurite outgrowth in these cells. The increase we observed in neurexin Iα mRNA levels after neuroligin-1 transfection indicates a previously unknown relationship between neurexin Iα and neuroligin-1. Thus, reports that purified recombinant neuroligins bind only neurexin Iβ isoforms in vitro (12) do not necessarily exclude the possibility of additional interactions between other neurexins and neuroligins in the living cell.
Figure 3.

AS-AChE neurite deficiency is associated with suppressed neurexin production. Presented is kinetic follow-up of RT-PCR analysis of neurexin I mRNA levels in PC12 or AS-ACHE cells before or 2 days after NGF-mediated differentiation (+NGF) compared with that of AS-ACHE cells 4 days after transfection with a plasmid encoding neuroligin-1 and 2 days under NGF (AS + Nrl + NGF). Samples were taken every third cycle, starting with the first cycle of product appearance for each set of primers. Note the persistent loss of neurexin Iα mRNA from AS-ACHE cells and its restoration after neuroligin transfection. Neurexin Iβ and actin mRNA levels remained unchanged.
Increased Expression of Nitzin, a Band 4.1 Family Member Is Associated with AS-ACHE Neuritogenesis.
Additional protein(s) involved in AChE-dependent neuritogenesis were sought in AS-ACHE cells as compared with the parental cell line by differential PCR display (15, 16). Analysis of all of the differentially displayed PCR products (average length: 482 ± 191 bp) revealed seven transcripts that were prominently up- or down-regulated in AS-ACHE as compared with PC12 cells. The corresponding PCR fragments were cloned, sequenced, and sought in several databases. Five of these transcripts are yet unknown or presented weak homologies to known genes. The two others were rat procollagen type V-α2 and a protein, we termed nitzin, a putative member of the cytoarchitecture band 4.1 superfamily of proteins (20). RT-PCR amplification of procollagen type V-α2 and nitzin mRNAs, using specific primers, confirmed that these two mRNAs are detectable only in AS-ACHE cells (data not shown).
The band 4.1 superfamily includes the ezrin, radixin, moesin (ERM) family of proteins (20) involved in the intracellular anchoring of cell membrane proteins to the cytoskeleton. The nucleotide sequence of nitzin mRNA displays 55% homology to mammalian ERM mRNAs and 42% homology to band 4.1 genes. Moreover, amino acid motifs corresponding to consensus sequence blocks 3 and 4 (PROSITE accession nos. BL0060C and BL0060D, respectively) and motifs 4 and 5 of the ERM family (PR00661) are all preserved in nitzin. A search of expressed sequence tag collections revealed that the human nitzin gene is highly homologous to that from rat (Fig. 4A).
Figure 4.
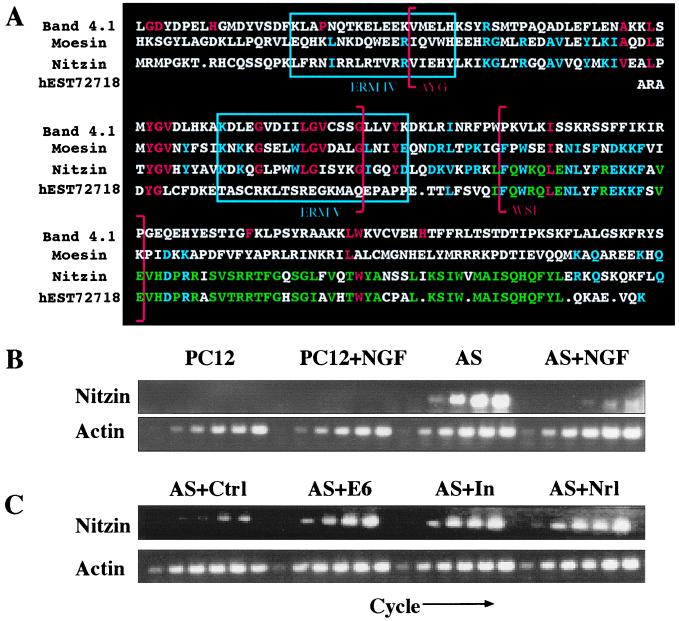
Expression of nitzin, a band 4.1 protein family member, is associated with AS-ACHE neuritogenesis. (A) Sequence alignment. Partial amino acid sequences from mouse band 4.1, human moesin, rat nitzin, and the corresponding human expressed sequence tag homolog to nitzin. Band 4.1 family consensus sequences are marked in red, ERM consensus sequences in blue, and nitzin/expressed sequence tag homologous residues in green. Band 4.1 signature no. 2 in sequence blocks 3 and 4 are marked by red brackets, and ERM family motifs IV and V are enclosed in blue boxes. (B) Transcriptional modulations. Nitzin mRNA levels were evaluated by RT-PCR in naive and NGF-differentiated PC12 and AS-ACHE cells, and in AS-ACHE cells transfected with a control plasmid (AS + ctrl), or plasmids encoding AChE-E6 (AS + E6), catalytically inactivated AChE (AS + In) or neuroligin-1 (AS + Nrl) as in Fig. 3. Samples were removed as described in the Fig. 3 legend. Note the pronounced expression of nitzin in AS-ACHE but not PC12 cells, its suppression under NGF, and the neuritogenic-associated increases in rescued AS-ACHE cells, under conditions in which actin mRNA levels remained unchanged.
The tentative identification of nitzin as a cytoarchitecture protein raised the possibility that its expression is modulated under NGF-induced neuritogenesis. Nitzin mRNA levels were below detection in PC12 cells before and after differentiation but high in naive AS-ACHE cells and down-regulated during the abortive neuritogenesis induced in these cells in the presence of NGF (Fig. 4B). Increased nitzin mRNA levels paralleled the efficiency of rescue observed after NGF-treatment of AS-ACHE cells transfected with AChE variants or neuroligin (Fig. 4C). This result indicated a role for nitzin in the cytoskeletal remodeling that provided for rescue of neurite extension in transfected AS-ACHE cells. To unravel the potential scope of such roles at the level of the developing organism in vivo, we analyzed nitzin’s pattern of expression in several tissues where various levels of AChE mRNA were detected in newborn mice by in situ hybridization and compared it to that of AChE. Nitzin and AChE mRNA signals were high and partially colocalized in the developing cerebellum, hippocampus, and retina, three tissues where a neuritogenic role for AChE was suggested (1). However, expression of nitzin also was observed in liver, thymus, and kidney tubules (Fig. 5 and data not shown), where no overlaps could be found with AChE expression and no developmental role(s) were suggested for AChE. These findings limit the predicted association between nitzin and AChE to the nervous system, yet suggest a more general role for nitzin in the cytoarchitectural changes involved with the development of several cells and organs.
Figure 5.
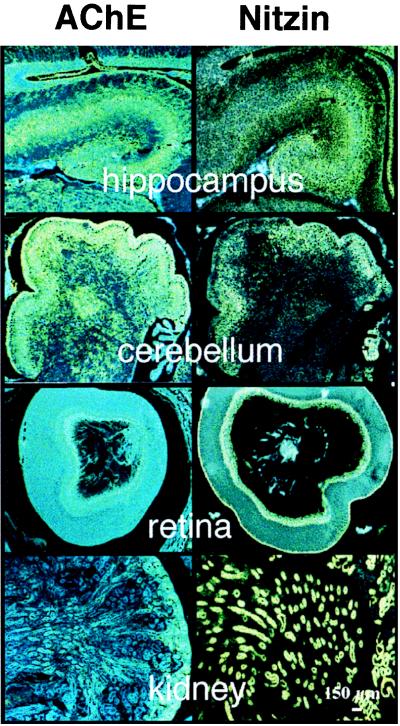
Partially overlapping expression patterns for nitzin and AChE mRNAs in developing mouse tissues. High resolution in situ hybridization results for various tissues from newborn FVB/N mice by using AChE (Left) and nitzin- (Right) specific probes (see Materials and Methods). From top to bottom: hippocampus, cerebellum, retina, and kidney. Note primarily overlapping expression patterns in both cholinergic (e.g., hippocampus) and noncholinergic brain regions (e.g., cerebellum), partial overlap in the retina, where AChE is notably involved with embryonic development (1) and no AChE expression in the kidney, which expresses high levels of nitzin.
DISCUSSION
We studied the neuritogenic activities of AChE by using a partially reversible AChE loss-of-function model in PC12 cells expressing antisense AChE cRNA. Stable, most effective suppression of AChE in PC12 cells imposed a block to normal NGF-mediated differentiation that was characterized by altered cytoarchitecture, a paucity of neurites, loss of neurexin Iα mRNA, and altered expression of a band 4.1 superfamily member, nitzin. Our findings that heterologous expression of neuroligin-1 restored morphological characteristics and gene expression patterns associated with normal differentiation demonstrate a functional redundancy of AChE and neuroligin in activating a critical morphogenetic pathway in these cells. Together with the sequence homologies shared by AChE, neuroligins, neurotactin, and gliotactin, our data therefore suggest that AChE and the various cholinesterase-like proteins interact with an overlapping set of heterotypic ligands such as neurexins and neurexin-like proteins.
Modest quantities of AChEcRNA transcripts suppressed AChE production in stably transfected PC12 cells more effectively than similarly targeted antisense oligodeoxynucleotides (14). This probably reflects more efficient formation of double stranded mRNA-cRNA hybrids by the longer cRNA chains, which reduced AChE levels below the amounts needed to support neuritogenesis. It was shown previously that the core domain of AChE could replace the homologous extracellular domain of neurotactin to generate a functional chimera (21). Thus, the cholinesterasic domain appears to play a conserved role in ligand recognition. Nevertheless, a membrane-associated form of the intact AChE polypeptide could not substitute for neurotactin in mediating heterotypic cell adhesion. Thus, the transmembrane and cytoplasmic elements present in the AChE-like proteins—but absent in AChE—appear indispensable in translating some ligand-binding interactions to changes in cytoarchitecture. In that case, competitive interactions between AChE and neurexins could serve a unique role in regulating growth processes associated with neuroligin–neurexin interactions.
As our cells were grown at low density on a collagen matrix, our observations in PC12 cells must reflect autologous interactions between substratum-bound or transgene-derived AChE, neuroligin, and a common ligand, possibly neurexin Iα. These and similar studies may therefore suggest that lateral cis membrane interactions between AChE, neuroligin, and neurexin mediate neuritogenic processes in some neuronal cell types. Yet, even if supported by additional in vitro studies, this hypothesis does not exclude in vivo situations in which heterotypic trans cell–cell interactions could predominate. Indeed, both AChE and neurexins are expressed in the developing nervous system and are considered to play central roles in establishing neuronal connectivity. Nevertheless, the partially reversible nature of the AS-ACHE phenotype demonstrates a degree of plasticity in AChE-mediated morphogenetic processes. As such, we might predict a role for noncatalytic AChE activities in neuronal remodeling in the adult nervous system as well. Consistent with this hypothesis, we recently have observed high expression of AChE to be associated with modulated dendrite branching in AChE transgenic mice (22). Long-term accumulation of AChE was further detected in mice subjected to acute psychological stress (23), also known to induce persistent changes in brain circuitry. In this light, the reduced AChE levels observed in the adrenal medulla of Alzheimer’s disease patients (24) may predict modified innervation of the medulla.
α-Neurexins contain cytoplasmic tails with sequences homologous to the glycophorin-C intracellular domain which binds the band 4.1 protein. In addition, they possess a conserved four-amino acid motif that functions as a recognition sequence for the PDZ domains of membrane-associated guanylate cyclase kinase (MAGUK) proteins (25). Complexes formed by the association of a glycophorin-C like domain, a band 4.1 protein and a MAGUK, were described in mammalian erythrocytes and Drosophila septate junctions (26, 27, and reviewed in ref. 19). In both cases, glycophorin-C, band 4.1, and MAGUK-related elements participate in mediating the cytoarchitectural organization that determines biological function. In Drosophila septate junctions, genomic knock-out of either the neurexin-like (27) or cholinesterase-like (8) component caused a similar disturbance in the integrity of the blood–nerve barrier. Thus, evolutionarily diverse interactions between AChE-like proteins and neurexin-like proteins can serve to mediate multiform intercellular recognition processes regulating cellular morphology. In the nervous system, modular compositions of neurexin-band 4.1-MAGUK-related complexes might similarly promote cytoskeletal arrangements/rearrangements in either autologous or heterologous fashions through interactions with members of the cholinesterase-related family of cell surface proteins. The apparently different band 4.1 elements in parental PC12 cells and “rescued” AS-ACHE cells, in which nitzin levels remain high, supports the notion of variable compositions of these complexes. In that case, AChE could regulate neurite outgrowth through competitive interactions with different neurexin complexes.
Currently approved drugs for the treatment of Alzheimer’s disease patients are all designed to suppress the catalytic activity of AChE (28). This is aimed at improving cholinergic neurotransmission under the acetylcholine deficiency caused by loss of cholinergic neurons. However, conventional AChE inhibitors induce increases in the amount of AChE protein (23). They may therefore facilitate the deleterious effects of noncatalytic morphogenic activities of AChE in the demented brain, such as the promotion of β-amyloid aggregation (23, 29). To reduce AChE protein levels in the brain, it is important to develop therapeutic antisense strategies targeted against ACHE mRNA (14). By preventing the production of AChE, antisense-ACHE agents, even if less effective than the transfection-delivered AChEcRNA, would improve cholinergic neurotransmission while simultaneously ameliorating the noncatalytic effects of this multifunctional protein (30). That the neuritogenic deficiencies induced by antisense AChE suppression can be reversed by AChE noncatalytic homologues supports the plausibility of antisense based therapies for the treatment of neurodegenerative diseases associated with cholinergic malfunction.
Acknowledgments
We are grateful to Dr. T. Sudhof (Dallas) for neuroligin 1 DNA, to Dr. I. Silman (Rehovot) for anti-AChE antibodies, and to M. Sternfeld and Dr. R. Broide (Jerusalem) for help with experiments. We also thank Prof. F. Eckstein and Dr. D. Glick for critical evaluation of this manuscript. This study was supported by grants from the German–Israeli Foundation (I-0512-206), the Israel Science Foundation (590/97), the United States Army (17.97.I.7007), the Zelman Cowen Foundation, and Ester Neurosciences, Ltd.
ABBREVIATIONS
AChE
acetylcholinesterase (protein)
ACHE
acetylcholinesterase (gene)
AS-ACHE
antisense oligodeoxynucleotide-treated PC12 cells
ERM
ezrin, radixin, moesin protein family
MAGUK
membrane-associated guanylate cylcase kinase
NGF
nerve growth factor
RT-PCR
reverse transcription–PCR
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF087945).
References
- 1.Layer P G, Willbold E. Prog Histochem Cytochem. 1995;29:1–99. doi: 10.1016/s0079-6336(11)80046-x. [DOI] [PubMed] [Google Scholar]
- 2.Small D H, Reed G, Whitefield B, Nurcombe V. J Neurosci. 1995;15:144–151. doi: 10.1523/JNEUROSCI.15-01-00144.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes C, Jones S A, Budd T C, Greenfield S A. J Neurosci Res. 1997;49:207–218. [PubMed] [Google Scholar]
- 4.Sternfeld M, Ming G-L, Song H-L, Sela K, Poo M-M, Soreq H. J Neurosci. 1998;18:1240–1249. doi: 10.1523/JNEUROSCI.18-04-01240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karpel R, Sternfeld M, Ginzberg D, Guhl E, Graessmann A, Soreq H. J Neurochem. 1996;66:114–123. doi: 10.1046/j.1471-4159.1996.66010114.x. [DOI] [PubMed] [Google Scholar]
- 6.Koenigsberger C, Chiappa S, Brimijoin S. J Neurochem. 1997;69:1389–1397. doi: 10.1046/j.1471-4159.1997.69041389.x. [DOI] [PubMed] [Google Scholar]
- 7.De la Escalera S, Bockamp E O, Moya F, Piovant M, Jimenez F. EMBO J. 1990;9:3593–3601. doi: 10.1002/j.1460-2075.1990.tb07570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auld V J, Fetter R D, Broadie K, Goodman C S. Cell. 1995;81:757–767. doi: 10.1016/0092-8674(95)90537-5. [DOI] [PubMed] [Google Scholar]
- 9.Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof T C. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 10.Ichtchenko K, Nguyen T, Sudhof T C. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 11.Puschel A W, Betz H. J Neurosci. 1995;15:2849–2856. doi: 10.1523/JNEUROSCI.15-04-02849.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen T, Sudhof T C. J Biol Chem. 1997;272:26032–26039. doi: 10.1074/jbc.272.41.26032. [DOI] [PubMed] [Google Scholar]
- 13.Andres C, Beeri R, Friedman A, Lev-Lehman E, Henis S, Timberg R, Shani M, Soreq H. Proc Natl Acad Sci USA. 1997;94:8173–8178. doi: 10.1073/pnas.94.15.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grifman M, Soreq H. Antisense Nucl Acid Drug Dev. 1997;7:351–359. doi: 10.1089/oli.1.1997.7.351. [DOI] [PubMed] [Google Scholar]
- 15.Welsh J, Chada K, Dalal S S, Cheng R, Ralph D, McClelland M. Nucleic Acids Res. 1992;20:4965–4970. doi: 10.1093/nar/20.19.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grifman M, Lev-Lehman E, El-Tamer A, Ginzberg D, Hanin I, Soreq H. Neuroscience Protocols. 1996;20:1–11. [Google Scholar]
- 17.Seidman S, Sternfeld M, Ben Aziz-Aloya R, Timberg R, Kaufer-Nachum D, Soreq H. Mol Cell Biol. 1995;15:2993–3002. doi: 10.1128/mcb.15.6.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark B J, Wells J, King S R, Stocco D M. J Biol Chem. 1994;269:28314–28322. [PubMed] [Google Scholar]
- 19.Missler M, Sudhoff T C. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- 20.Tsukita S, Yonemura S, Tsukita S. Trends Biochem Sci. 1997;22:53–58. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- 21.Darboux I, Barthalay Y, Piovant M, Hipeau-Jacquotte R. EMBO J. 1996;15:4835–4843. [PMC free article] [PubMed] [Google Scholar]
- 22.Beeri R, LeNovere N, Mervis R, Huberman T, Grauer E, Changeux J P, Soreq H. J Neurochem. 1997;69:2441–2451. doi: 10.1046/j.1471-4159.1997.69062441.x. [DOI] [PubMed] [Google Scholar]
- 23.Kaufer D, Friedman A, Seidman S, Soreq H. Nature (London) 1998;393:373–377. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- 24.Appleyard M E, McDonald B. Lancet. 1991;338:1085–1086. doi: 10.1016/0140-6736(91)91947-s. [DOI] [PubMed] [Google Scholar]
- 25.Songyang Z, Fanning A S, Fu C, Xu J, Marfatia S M, Chishti A H, Crompton A, Chan A C, Anderson J M, Cantley L C. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 26.Correas I, Leto T L, Speicher D W, Marchesi V T. J Biol Chem. 1986;261:3310–3315. [PubMed] [Google Scholar]
- 27.Baumgartner S, Littleton J T, Broadie K, Bhat M A, Harbecke R, Lengyel J A, Chiquet-Ehrismann R, Prokop A, Bellen H J. Cell. 1996;87:1059–1068. doi: 10.1016/s0092-8674(00)81800-0. [DOI] [PubMed] [Google Scholar]
- 28.Knapp M J, Knopman D S, Solomon P R, Pendlebury W W, Davis C S, Gracon S I. J Am Med Assoc. 1994;271:985–991. [PubMed] [Google Scholar]
- 29.Inestrosa N C, Alvarez A, Perez C A, Moreno R D, Vicente M, Linker C, Casanueva O I, Soto C, Garrido J. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 30.Grifman M, Lev-Lehman E, Ginzberg D, Eckstein F, Zakut H, Soreq H. In: Concepts in Gene Therapy. Strauss M, Barranger J A, editors. Berlin: de Gruyter; 1997. pp. 141–167. [Google Scholar]