Total Syntheses, Fragmentation Studies, and Antitumor/Antiproliferative Activities of FR901464 and Its Low Picomolar Analog (original) (raw)
. Author manuscript; available in PMC: 2008 Sep 5.
Published in final edited form as: J Am Chem Soc. 2007 Feb 6;129(9):2648–2659. doi: 10.1021/ja067870m
Abstract
FR901464 is a potent anticancer natural product that lowers the mRNA levels of oncogenes and tumor suppressor genes. In this article, we report a convergent enantioselective synthesis of FR901464, which was accomplished in 13 linear steps. Central to the synthetic approach was the diene-ene cross olefin metathesis reaction to generate the C6-C7 olefin without the use of protecting groups as the final step. Additional key reactions include a Zr/Ag-promoted alkynylation to set the C4 stereocenter, a mild and chemoselective Red-Al reduction, a reagent-controlled stereoselective Mislow-Evans-type [2,3]-sigmatropic rearrangement to install the C5 stereocenter, a Carreira asymmetric alkynylation to generate the C4′ stereocenter, and a highly efficient ring-closing metathesis-allylic oxidation sequence to form an unsaturated lactone. The decomposition pathways of FR901464's right fragment were studied under physiologically relevant conditions. Facile epoxide opening by β-elimination gave two enones, one of which could undergo dehydration via its hemiketal to form a furan. To prevent this decomposition pathway, a right fragment was then rationally designed and synthesized. This analog was 12 times more stable than the right fragment of the natural product. Using this more stable right fragment analog, an FR901464 analog, meayamycin, was then prepared in 13 linear steps. The inhibitions of human breast cancer MCF-7 cell proliferation by synthetic FR901464 and meayamycin were studied, and the GI50 values for these compounds were determined to be 1.1 nM and 10 pM, respectively. Thus, meayamycin is among the most potent anticancer small molecules that do not bind to either DNA or microtubule.
Keywords: FR901464, fragmentation, Meayamycin, hemiketal, NBS, Mislow-Evans rearrangement, olefin cross-metathesis, anticancer/antiproliferative activities, zirconium, alkynylation
Introduction
Controlled cellular processes such as signal transduction and gene expression are essential for homeostasis. When these processes become irregular, tumors can develop. Tumorigenesis can be accounted for by approximately 200 molecular mechanisms,1 many of which can in principle be targeted by drugs. However, currently available chemotherapeutic methods are limited to targeting DNA,2,3 nuclear hormone receptors,4 a tyrosine kinase,5 a proteasome,6,7 and the microtubule.8,9 Clearly, there is an urgent need to fill the gap between the molecular mechanisms of tumorigenesis and available pharmacological approaches.
In the quest for anticancer natural products possessing new modes of action, the Nakajima group at the Fujisawa Pharmaceutical Company employed a reporter assay in human breast adenocarcinoma MCF-7 cells using the simian virus 40 (SV40) promoter10 upstream of a reporter gene, which was stably transfected into the MCF-7 cell genome. Through their screening efforts, structurally unique FR901464 was isolated from the culture broth of a bacterium of Pseudomonas sp. No.2663, which activated the aforementioned reporter gene by 30-fold at 20 nM concentration in MCF-7 cells.11-13 The same group revealed that FR901464 possessed antitumor activity against MCF-7 cells, human lung adenocarcinoma A549 cells, colon cancer SW480 and HCT116 cells, and murine leukemia P388 cells with an IC50 of 0.6–3.4 nM.11 This natural product also exhibited a prominent effect at 0.056–0.18 mg/kg against human adenocarcinoma A549 and 0.18–1 mg/kg against murine Colon 38 and Meth A cells implanted in mice, indicating that FR901464 or its structurally related analogs have the potential for clinical application.13 FR901464 induced both G1 and G2/M phase arrest in MCF-7 cells.13,14 In contrast, adriamycin and camptothecin, both DNA synthesis inhibitors, induced S-phase arrest in the cell cycle, and Taxol®, a microtubule modulator, induced G2/M-phase arrest.13 Since FR901464 was originally discovered as an activator of SV40 promoter, the Nakajima group sought endogenous genes that were upregulated by the natural product. Their focused approach, prior to the invention of DNA microarray,15 showed that the mRNA levels of p53,16,17 p21 Cip-1,18,19 E2F-1,20,21 and c-Myc22,23 were downregulated while a housekeeping gene was intact. This result is both exciting and perplexing since p53 is a well-known tumor suppressor gene and c-Myc is an oncogene. These findings reported by the Nakajima group and our subsequent findings strongly suggest that the mode of action of FR901464 is different from that of clinically used anticancer drugs. After the discovery of FR901464, other pharmaceutical companies used the same SV40 promoter in reporter gene systems and found that the previously known natural product herboxidiene (a.k.a. GEX1)24-27 and the new natural product TMC-20528 exhibited closely related biological activity as FR901464, although these compounds were less potent.
Not surprisingly, the unique profile of FR901464 has intrigued many synthetic chemists,29 which culminated the first total synthesis of this natural product by the Jacobsen group30,31 and the second and third by the Kitahara group.32,33 The Jacobsen synthesis elegantly demonstrated the power of their asymmetric hetero-Diels-Alder reaction34 in their total synthesis, which consisted of 19 steps in the longest linear sequence and a total of 40 steps.
The subsequent total synthesis from the Kitahara group took advantage of the chiral pool to assemble their fragments. This total synthesis required 42 total steps with the longest linear sequence being 24 steps.35 The third total synthesis by the same group was the improvement of their earlier version with 41 steps in the longest linear sequence and 22 total steps.33 These syntheses revealed inefficient installation of the spiroepoxide at late stages and the latent instability of FR901464. Clearly, a more versatile and concise approach was desired for synthetic accessibility to analogs of FR901464 for biological studies. Moreover, more quantitative analysis of the instability of FR901464 was needed as part of our efforts to understand the mode of action of this natural product. This full account describes our synthetic efforts that eventually allowed us to develop a remarkably potent FR901464 analog via earlier and potentially risky installation of the spiroepoxide and ultimate convergency, namely the cross-coupling at the very end of the synthesis.36
Results and Discussion
First Generation Synthetic Studies
Scheme 1 shows our first generation retrosynthetic analysis of FR901464. We envisioned a Nozaki-Hiyama-Kishi reaction37,38 of 1 and 2 as the final coupling reaction because the reaction conditions are mild and it is generally in accordance with Felkin selectivity for additions into to α-chiral aldehydes. Vinyl iodide 1 would be prepared from acid 3 and amine 4 that were expected to be derived from propargylic alcohol 6 and the commercially available L-threonine derivative 7, respectively. Ketoaldehyde 2 could be prepared from allylic alcohol 5 by oxidative cleavages of both olefins. Finally, this alcohol would arise from epoxyalcohol 8.
Scheme 1.

First generation retrosynthetic analysis of FR901464
We envisioned that the first milestone of our synthetic studies would be the preparation of ketoaldehyde 2 (Scheme 2). The first step toward this end, a zinc-mediated coupling of propargyl alcohol and methallyl bromide39 afforded alcohol 9 in 93% yield. The subsequent Sharpless asymmetric epoxidation40 proceeded smoothly to generate epoxyalcohol 8 in 90% yield with >97:3 er. The volatile aldehyde 10 was obtained by the Dess-Martin oxidation41 of this alcohol in 81% yield. All of these steps could be executed in large scales (>20 g) without compromising yields.
Scheme 2.

Preparation of ketoaldehyde 2 a
_a_Conditions: (a) propargyl alcohol (1.1 equiv), allyl bromide (1.1 equiv), zinc dust (3.1 equiv); then methallyl bromide (1.0 equiv), THF, 23 °C, 93%; (b) (+)-DIPT (9 mol %), Ti(O_i_Pr)4 (8 mol %), _t_BuOOH (1.9–2.2 equiv), CH2Cl2, -20 °C, 90%, er >97:3; (c) Dess-Martin periodinane (1.5 equiv), CH2Cl2, 0→23 °C, 81%; (d) alkyne 11 or 12 (1.3 equiv), Zn(OTf)2 (1.2 equiv), (−)-_N_-methylephedrine (1.3 equiv), Et3N (1.3 equiv), toluene, 23→40 °C, 22 h; (e) see table below; (f) 4-O2NPhCO2H (3.0 equiv), diisopropyl azodicarboxylate (2.9 equiv), Ph3P (3.0 equiv), THF, 0→23 °C, 85%; (g) K2CO3 (2.5 equiv), MeOH, 0 °C, 88%; (h) TESCl (1.1 equiv), imidazole (1.4 equiv), THF, 0 °C, quant.; (i) OsO4 (0.05 equiv), NMO (1.0 equiv), THF, 0→23 °C, 58%; then NaIO4 (1.0), THF/H2O (1:1), 23 °C, quant.; (j) O3, CH2Cl2/MeOH (1:1), -78 °C; then Me2S (10 equiv), -78→23 °C, 54–82%.
With a large quantity of aldehyde 10 in hand, our next objective was to develop a stereoselective carbon-nucleophile addition to this aldehyde. Step d illustrates the two failed diastereomeric alkynylation reactions of aldehyde 10 using 11 and 12 as latent nucleophiles. We presumed that Carreira's asymmetric alkynylation conditions42 could be sufficiently mild to be compatible with aldehyde 10. To our disappointment, this aldehyde did not react at ambient temperature and decomposed at 40 °C.
In light of the poor selectivity in forming the C4 stereocenter described above, we turned our attention to substrate control (step e; Table 1). Vinylation of aldehyde 10 using CH2=CHMgBr in THF (entry 1) gave a 1:1 mixture of the desired alcohol 5 and the undesired alcohol 4-_epi_-5 in a combined 51% yield. The absolute configuration of these allylic alcohols was determined after further transformations as shown in Scheme 11. The addition of ZnCl2 to the reaction mixture with various stoichiometry did not improve the selectivity for the desired alcohol 5 (entries 2–4). Addition of hexanes to the reaction mixture finally improved the stereoselectivity for the desired alcohol (2:1) in 55% combined yield (entry 5). Treatment of 10 with vinyllithium, prepared in situ from CH2=CHSnBu3 (1.4 equiv) and _n_BuLi (1.3 equiv),43 gave a 1.2:1 mixture of 5 to 4-_epi_-5 in a combined 46% yield (entry 6). Alkynylation of 10 with HC≡CMgBr (3.5 equiv) at -78 °C (entry 7) gave an inseparable mixture of diastereomers in 32% yield and low stereoselectivity (dr = 1.7 :1), and higher reaction temperatures caused Payne rearrangements to occur. Partial hydrogenation of these propargylic alcohols gave 5 and 4-_epi_-5 in a combined yield of 93%, which enabled us to confirm the structures. After these and other even less fruitful attempts, high levels of stereocontrol in this transformation remained elusive.
Table 1.
Step e in Scheme 2 Optimization for the preparation of allylic alcohols 5 and 4-_epi_-5
| entry | Conditions | solvents | isolated yield (%)a | 5:4-_epi_-5b |
|---|---|---|---|---|
| 1 | 16 | THF | 51 | 1.0:1 |
| 2 | 16:ZnCl2 (2:1) | THF | 71 | 0.3:1 |
| 3 | 16:ZnCl2 (1:1) | THF | 42 | 1.2:1 |
| 4 | 16:ZnCl2 (1:2) | THF | 42 | 0.8:1 |
| 5 | 16:ZnCl2 (1:1) | THF:hexane (1:1) | 55 | 2.0:1 |
| 6 | 17 + _n_BuLi | THF | 46 | 1.2:1 |
| 7 a,c | 18 | THF | 32 | 1.7:1 |
Scheme 11.

Preparation of methyl glycoside 58 a
a Conditions: (a) NBS (1.1 equiv), THF, H2O, 0→23 °C, 75%; (b) PPTS (10 mol %), MeOH, CH2Cl2, 23 °C, 40%; (c) NBS (1.5 equiv), 3Å MS, MeOH, MeCN, 0→23 °C, 59%.
At this point, our priority was to examine the validity of the final coupling. With this in mind, we carried on with the result shown in entry 5, Table 1, and proceeded to prepare ketoaldehyde 2. To improve the overall yield for the formation of 5, the undesired 4-_epi_-5 was subjected to Mitsunobu conditions using 4-nitrobenzoic acid to invert the C4 stereocenter, and subsequent methanolysis of the resulting ester 13 with K2CO3 afforded the desired allylic alcohol 5 in 75% yield for the 2 steps. The resulting hydroxy group was protected as its triethylsilyl (TES) ether, 14, in quantitative yield. This compound was then subjected to oxidative cleavage conditions (OsO4, NMO; then NaIO4) to afford enone 15 in 58% yield, and its subsequent ozonolysis produced the unstable ketoaldehyde 2 in 54–82% yield. It should be mentioned that the direct ozonolysis of diene 14 to ketoaldehyde 2 was reproducibly low yielding (<10%). Although this synthetic route allowed us to prepare the target intermediate in a concise manner, we soon realized that chromatographic separation of alcohols 5 and 4-_epi_-5 in a large scale would be a daunting task.
With an established synthetic route to ketoaldehyde 2, we still desired to improve the C4 stereoselectivity (Scheme 3). Treatment of epoxyaldehyde 10 with methyl propiolate and _n_BuLi formed alcohol 19 and its C4-epimer in 50% combined yield, but with no diastereoselectivity (dr = 1:1). In contrast, it was gratifying to find that the alkynylation method developed in our laboratory afforded alcohol 19 in 84% with good stereocontrol (dr = 6:1),44 and the C4 stereochemistry was determined as described in our previous communication.36 After all the struggles, the solution was right under our noses! Unexpectedly, the partial hydrogenations of this alcohol and its TES ether 20 using Lindlar's catalyst failed. However, the reduction of 19 with Red-Al successfully produced 23 in 81% yield in a chemo- and stereoselective.45 Subsequent TES ether formation produced 24 in quantitative yield, and ozonolyses of both of the olefins of this compound produced ketoaldehyde 2 cleanly by crude 1H NMR analysis. Thus, access to this ketoaldehyde was attainable in a total of 7 steps. It is noteworthy that the ozonolysis of 24 was far more effective than that of 14.
Scheme 3.

Formation of C4 stereocenter with higher stereocontrol a
a Conditions: (a) Ag-C≡C-CO2Me (1.7 equiv), Cp2ZrCl2 (1.3 equiv), AgOTf (0.2 equiv), CH2Cl2, 23 °C, 84% (dr = 6:1); (b) H2 (1 atm), Lindlar's catalyst (1.2 –2.0 mol %), quinoline (0.94–1.5 equiv), EtOH, 23 °C, 7–17 h; (c) TESCl (1.1 equiv), imidazole (1.4 equiv), THF, 0 °C, 74%; (d) Red-Al (2.0 equiv), THF, -72 °C, 81%; (e) TESCl (1.4 equiv), imidazole (1.5 equiv), THF, 0 °C, quant.; (f) O3, CH2Cl2/MeOH (1:1), -78 °C; then Me2S (17 equiv), -78→23 °C, quant. (based on 1H NMR).
With the B-ring fragment 2 in hand, we embarked on the preparation of the A-ring fragment 1 as shown in Scheme 4. We chose the styryl unit to mask an aldehyde that would effectively suppress the volatility of otherwise low molecular weight intermediates. With this in mind, the preparation of acid 3 began by the homologation of cinnamaldehyde by the action of TMSCHN2 to give enyne 25 in 84% yield.46 The Corey-Fuchs method47 was less efficient, and the Seyforth-Ohira-Bestmann method48,49 gave 1-(1-methoxybut-3-ynyl)benzene. Subsequent Carreira asymmetric alkynylation42 of 25 with acetaldehyde generated propargylic alcohol 6 in 41% yield with 86:14 er. In this reaction, slow addition of acetaldehyde to the reaction mixture proved to be crucial since rapid addition resulted in the aldol condensation of acetaldehyde. Recrystallization of 6 further improved the er to 98:2. Unfortunately the catalytic version of the asymmetric alkynylation reaction50 was unsuccessful. After acetylation of propargylic alcohol 6, the resulting ester, 26, was converted to aldehyde 27 by ozonolysis in 89% yield. Ensuing oxidation of aldehyde 27 and partial reduction of the resulting alkyne using Lindlar's catalyst afforded acid 3 in 75% yield for the 2 steps, and in 6 total steps from cinnamaldehyde. More recently, we converted enyne 26 to acid 28 in 1 step via osmium tetroxide-mediated oxidative cleavage and subsequent partial reduction afforded 3 in 60% yield for the 2 steps. Therefore, acid 3 can now be prepared in 5 steps from cinnamaldehyde.
Scheme 4.

Preparation of acid 3
a Conditions: (a) cinnamaldehyde (1.1 equiv), TMSCHN2 (1.0 equiv), LDA (1.0 equiv), THF, -78→0 °C, 84%; (b) CH3CHO (2.3 equiv), Zn(OTf)2 (1.0 equiv), (−)-_N_-methylephedrine (1.0 equiv), Et3N (1.0 equiv), toluene, 23 °C, 41% (86:14 er); (c) Ac2O (5.0 equiv), pyridine, 23 °C, quant.; (d) O3, CH2Cl2, -78 °C; then Me2S (10 equiv), -78→23 °C, 89%; (e) NaClO2 (3.0 equiv), NaH2PO4 (2.0 equiv), 2-methyl-2-butene (15 equiv), _t_BuOH/H2O (1:1), 23 °C; (f) OsO4 (0.7 mol %), Oxone (4.0 equiv), DMF, 23 °C; (g) H2, Lindlar's catalyst (1 mol %), quinoline (10 mol %), EtOH, 23 °C, 75% (for steps e and g), 60% (for steps f and g).
Despite the successful preparation of acid 3, we wished to further improve the synthetic efficiency and scalability. More specifically, we deemed the use of TMSCHN2 less attractive in large scales due to potential safety concerns. Because other homologation methods to convert cinnamaldehyde to enyne 25 failed as described above, we decided to pursue a different synthetic route; tetrahydro-2-(2-propynoxy)-2_H_-pyran was coupled with _N_-acetylmorpholine to form ynone 29 in 78–93% yields (Scheme 5). This ynone was reduced using the CBS catalyst51 in EtNO252 to afford 30, which was acetylated to provide 31 in 97% yield for the 2 steps. Concurrent THP removal and oxidation using the Jones reagent afforded ynoic acid 28 in 74% yield and 86:14 er. Subsequent partial hydrogenation again gave acid 3. This scheme could provide acid 3 in 5 steps and should be more scalable than Scheme 4.
Scheme 5.

Alternative methods to prepare acid 3 a
a Conditions: (a) HC≡CCH2OTHP (3.0 equiv), _n_BuLi (3.0 equiv), THF, -78→0 °C, 78–93%; (b) (S)-2-methyl-CBS-oxazaborolidine (0.2 equiv), catecholborane (1.6 equiv), EtNO2, -78 °C; (c) Ac2O (4.6 equiv), pyridine (excess), 23 °C, 97% (2 steps); (d) Na2Cr2O7 (3.5 equiv), H2SO4, H2O, acetone, 0→23 °C, 74%; (e) see Scheme 4.
The last building block needed to test our final coupling strategy was intermediate 4, and our initial toward this intermediate is shown in Scheme 6 We chose to utilize the chiral pool as a source of material, since the commercially available L-threonine derivative 32 contained the C14 and C15 stereocenters identical to FR901464. Treatment of 32 with 2-methoxypropene and CSA afforded oxazolidine 7 in quantitative yield. Subsequent reduction with DIBALH, and immediate exposure of Garner aldehyde53 to Horner-Wadsworth-Emmons olefination conditions generated 33 in 84% yield. Hydrogenation of this unsaturated ester and lactonization afforded lactone 34, but revealed the poor diastereoselectivity (dr = 2:1) in the hydrogenation of 33. As it turned out later, the Kitahara group found similar results.32 Since we were unable to achieve stereoselectivity in forming the C12 stereocenter, we needed to alter our strategy.
Scheme 6.

Synthetic approach A-ring formation a
a Conditions: (a) 2-methoxypropene (2.0 equiv), CSA (1 mol %), CH2Cl2, 0 °C, quant.; (b) DIBALH (2.0 equiv), CH2Cl2, -78 °C; (c) (EtO)2P(O)CH(CH3)CO2Et (3.3 equiv), NaH (3.0 equiv), solvent, 23 °C, 84% (2 steps); (d) H2 (1 atm), PtO2 (1 mol %), EtOH, 23 °C, quant. (dr = 2:1); (e) AcOH, 58%; (f) DIBALH (2.0 equiv), CH2Cl2, -78 °C; then Ph3PCH3Br (2.1 equiv), KO_t_Bu (2.0 equiv), THF, -78→48 °C, 77%; (g) CSA (10 mol %), MeOH, 23 °C, 95%; (h) methacrylic acid (1.2 equiv), DCC (1.2 equiv), DMAP (0.1 equiv), CH2Cl2, 0 °C, 95%; (i) Ru-3 (10 mol %), ClCH2CH2Cl, 83 °C; (j) methallyl bromide (4.0 equiv), Ag2O (1.5 equiv), DMF, 23 °C, 86%; (k) Ru-1 (1 mol %), benzene, reflux, quant.; (l) PDC (6.0 equiv), ClCH2CH2Cl, reflux, 72%; (m) PCC (2.0 equiv), TBHP (4.0 equiv), Celite, benzene, 23 °C, 67% (n) H2, PtO2 (1 mol %), EtOH, 23 °C, quant. (dr = 10:1); (o) allyl-MgCl (2.0 equiv), THF, -78 °C, 96%.
Since the diastereoselectivity in the hydrogenation of 33 was poor, we planned to obtain better stereocontrol in a more rigid cyclic substrate. Oxazolidine 7 was transformed into 35 in a one-pot procedure (DIBALH; Ph3P=CH2)54 in 77% yield. Subsequent removal of the acetonide using catalytic CSA in MeOH afforded 36 in 95% yield. Methallylation of 36 by the action of Ag2O and methallyl bromide generated diene 37 in 86% yield, which was then subjected to ring-closing olefin metathesis conditions using 1 mol % of Ru-155 to form dihydropyran 38 in quantitative yield. Catalyst Ru-256 was also able to catalyze this reaction at 1 mol %, but we were unable to recover this catalyst. To prepare unsaturated lactone 39, we found PDC to be the most efficient and selective allylic oxidant for 38 (72%). PCC–TBHP gave similar results for the oxidation of 38, but gave peroxy-acetal 40 as a byproduct. Unfortunately, we were unable to effectively convert peroxy-acetal 40 to lactone 39 primarily due to epimerization at C14. A more direct approach to form the unsaturated lactone 39 was attempted by methacryl ester formation (step h) then ring-closing metathesis using catalyst Ru-3 (step i),57,58 but the second step was unsuccessful. Stereoselective hydrogenation of the unsaturated lactone 39 set the C-12 stereocenter of 34 in a 10:1 dr in quantitative yield. Allylation of this lactone afforded 41 as a mixture of hemiketal anomers and aminal anomers.
As shown in Scheme 7, subjection of 41 to reducing conditions (Et3SiH-BF3•OEt2) provided tetrahydropyran 42 in 20% as well as pyrrolidine 43 in 50% yield. The addition of CF3CH2OH to the reduction conditions improved the yield of tetrahydropyran 42 to 45% yield, possibly by establishing an equilibrium between the putative oxocarbenium and _N_-acyliminium ions. 12-_epi_-41 was subjected to the same reaction sequence, which gave tetrahydropyran 12-_epi_-42 stereo- and chemoselectively in 85% yield.
Scheme 7.

Preparation of 42 a
a Conditions: (a) Et3SiH (4.0 equiv), BF3•OEt2 (4.0 equiv), CH2Cl2, -78 °C, 42 (dr = 10:1); (b), Et3SiH (10 equiv), BF3•OEt2 (4.0 equiv), CF3CH2OH (8.0 equiv), CH2Cl2, -78 °C; then Boc2O (0.5 equiv), -78→23 °C, 42 (dr = 10:1).
Prior to the discovery of the reduction of 41 in the presence of CF3CH2OH, we explored other methods to improve the hemiketal reduction. To thwart the production of pyrrolidine 43-type compounds, we also tested other amine-protecting groups (Scheme 8). For example, we prepared picolinamide 44 from 38 in 82% yield. Subsequent oxidation and stereoselective reduction were comparable to using the Boc protecting group. Allylation of 46 was accomplished in 58% yield (not optimized), and reduction with Et3SiH-BF3•OEt2 afforded 50 in 44% yield (61% BORSM; dr = 8:1). Disappointingly, we failed to find deprotection conditions to give 50 after vigorous screening of reaction conditions including the literature procedure.59 Therefore, we turned our attention to other protecting groups.
Scheme 8.

Preparation of 41 using different amine protecting groups a
a Conditions: (a) TFA/CH2Cl2 (1:9), 23 °C; then pivaloyl chloride (3.0 equiv), DMAP (0.01 equiv), pyridine, 0→23 °C, 82% (38→44); (b) 6N HCl/THF (2:1), 23 °C; then TsCl (1.3 equiv), K2CO3, 23 °C, 86% (38→45); (c) PDC (4.0 equiv), _t_BuOOH (4.0 equiv), benzene, 23 °C, 70% (25) or 65% (26); (d) H2, PtO2 (3 mol %), EtOH, 23 °C, 97% (44→46) or 99% (45→47); (e) allyl-MgCl (3.0 equiv), THF, -98 °C, 58% (46→48), or -78 °C, 63% (47→49); (f) Et3SiH (10 equiv), BF3•OEt2 (4.0 equiv), CH2Cl2/MeCN (1:1), -20 °C, 44% (48→50, 61% BORSM; dr = 8:1), or THF, -20 °C, 68% (49→51, dr = 12:1); (g) (from 51) sodium naphthalenide, THF, -78 °C; then Boc2O (2.3 equiv), Et3N (4.0 equiv), DMAP (0.08 equiv), MeCN, 23 °C, 87% (51→42, 2 steps).
The undesired pyrrolidine formation proceeds via the putative highly electrophilic acyliminium ion, and the corresponding sulfonyliminium ion would be less electrophilic,60 suppressing the pyrrolidine formation. On the basis of this consideration, we decided to test a tosyl protecting group. Deprotection of the Boc group of 38 and subsequent protection of the resulting amine afforded sulfonamide 45 in 86% yield. Allylic oxidation and stereoselective hydrogenation were accomplished in 65 and 99% yields, respectively to afford lactone 47. Allylation of 47 gave 49 in 63% yield (not optimized), which was then subjected to reduction conditions (Et3SiH-BF3•OEt2) to afford 51 in 68% yield (dr = 12:1). Deprotection proceeded smoothly using sodium naphthalenide,61 which provided an alternative route to prepare amine 4 or carbamate 42.
The synthesis of vinyl iodide 1 is shown in Scheme 9. Compound 42 was converted to unsaturated aldehyde 52 using cross-olefin metathesis conditions employing a catalytic amount of Ru-3. Takai vinyl iodide formation62 generated iodoalkene 53 in 63% yield. Removal of the Boc group of this compound was accomplished in 1:9 TFA:CH2Cl2, and subsequent amide bond formation with acid 3 was accomplished by the action of HATU in 45% yield. Thus, access to vinyl iodide 1 was obtained in 10 longest linear steps.
Scheme 9.

Failed synthetic attempts using the NHK strategy a
a Conditions: (a) Ru-1 (5 mol %), methacrolein (10 equiv), ClCH2CH2Cl, 40 °C, 67%; (b) CHI3 (2 equiv), CrCl2 (6 equiv), THF, 0 °C, 63%; (c) TFA/CH2Cl2 (1:9), 23 °C; then 3 (1.7 equiv), HATU (1.7 equiv), _i_Pr2NEt (3.6 equiv), MeCN, 23 °C, 45%; (d) CrCl2 (7 equiv), NiCl2 (∼5 mol %), DMSO, 23 °C; (e) _n_BuLi (1–2 equiv), THF, -100→-78 °C.
With both coupling partners in hand, the stage was set to test the Nozaki-Hiyama-Kishi coupling. To our disappointment, treatment of vinyl iodide 1 and ketoaldehyde 2 under Nozaki-Hiyama-Kishi conditions37,38 caused proto-deiodination of 1 and decomposition of the ketoaldehyde. Similarly, treatment of vinyl iodide 53 and ketoaldehyde 2 to Nozaki-Hiyama-Kishi conditions gave diene 55 and decomposition of the fragile ketoaldehyde. Therefore, we opted to modify our strategy and attempted a lithium-halogen exchange between _n_BuLi and 53 to generate the corresponding alkenyllithium, but did not find any appreciable coupling to ketoaldehyde 2. Having realized the fragile nature of ketoaldehyde 2 and the difficulty of forming the C5–C6 bond in the late stages of a synthesis, we began to explore other coupling possibilities.
Second Generation Synthetic Studies
We envisioned the second retrosynthetic analysis featuring modified Julia coupling to form the C6–C7 bond as shown in Scheme 10. Julia olefinations63,64 are typically highly E-selective and compatible with various functional groups. Therefore, we decided to pursue this strategy, for which acid 3 and aldehyde 52 were already prepared, but the preparation of the B-ring fragment 56 from ketone 15 required new methodology.
Scheme 10.

Julia olefination coupling strategy
Displayed in Scheme 11 are our efforts focused on preparing the B-ring fragment 56. Typically, hemiketals are formed by the intramolecular attack of a hydroxy group onto a ketone, thus the hydroxy group is the origin of the ring oxygen atom. We questioned whether the ketone oxygen atom could be used as an alternative source for the ring oxygen atom. This would allow for the exciting possibility of trapping the putative intermediate oxocarbenium ion with water or other nucleophiles to obtain hemiketals and ketals, among others. To test this idea, enone 15 was treated with NBS in H2O/THF (1:8) to give 57 in 75% yield as a single diastereomer.65 Unambiguous structural determination was determined after conversion of this hemiketal to methyl glycoside 58 in 40% yield. Similarly, we could treat enone 15 with _N_-bromosuccinimide in the presence of MeOH and obtain methyl glycoside 58 in 59% in one step. However, the stereochemistry at C5 was incorrect thereby making preparation of sulfone 56 too difficult from this intermediate.
Since the stereochemistry in 57 and 58 was wrong, we thought that using 4-_epi_-15 for the cyclization could reverse the observed stereocontrol for C5. Towards this end, alcohol 4-_epi_-5 was protected as its TES ether, and subsequent regioselective oxidative cleavage (OsO4, NMO; NaIO4) gave enone 4-_epi_-15 in 60% yield (Scheme 12). Treatment of 4-_epi_-15 with NBS in H2O/acetone (1:8) gave hemiketal 59 in 95% yield as a single diastereomer. Methyl glycoside 60 could be prepared by the treatment of 59 with catalytic PPTS in MeOH/CH2Cl2 (1:3) in 75% yield. Alternatively, the same methyl glycoside could be formed in one step by treating 4-_epi_-15 with NBS in MeOH/MeCN (1:10) in 47% yield. The absolute stereochemistry of 60 was determined by X-ray crystallographic analysis, also revealing the diastereoselectivities for the vinylations of epoxyaldehyde 10 shown in Table 1 and the alkynylation of epoxyaldehyde 10 shown in Scheme 3.
Scheme 12.

Preparation of thioethers 61 and 63 a
a Conditions: (a) TESCl (1.2 equiv), imidazole (1.4 equiv), THF, 0 °C, quant.; (b) OsO4 (4.5 mol %), NMO (1.0 equiv), THF, H2O, 23 °C; then NaIO4 (1.0 equiv), Et2O, H2O, 23 °C, 60%; (c) NBS (2.0 equiv), acetone, H2O, 23 °C, 95%; (d) PPTS (10 mol %), MeOH, CH2Cl2, 23 °C, 75%; (e) NBS (1.5 equiv), 3Å MS, MeOH, MeCN, 0→23 °C, 47%; (f) 4-phenyltetrazole thiol (1.2 equiv), Et3N (2.0 equiv), MeCN, reflux, 60%; (g) TBAF (1.1 equiv), THF, 0 °C, 80%; (h) 4-phenyltetrazole thiol (1.0 equiv), Et3N (2.0 equiv), MeCN, 50 °C, 74%; (i) 4-O2NPhCO2H (2.1 equiv), DIAD (2.7 equiv), Ph3P (2.2 equiv), THF, 0→23 °C; then K2CO3 (1.0 equiv), MeOH, 23 °C.
To arrive at sulfone 56, the C-6 bromide needed to be substituted with a thiol, and the C4 stereocenter inverted. Treatment of methyl glycoside 60 with 4-phenyltetrazole thiol under various conditions only gave the undesired thioether 61. At this point, we were unsure whether the axial TES ether group was shielding the C6-Br group from SN2 reactions. To make the C6 position more sterically accessible, we removed the TES group from 60 to give 62 in 80% yield. We then treated this compound with 4-phenyltetrazole thiol under basic conditions but again only found epoxide opening with the thiol to form 63. A more severe problem is that Mitsunobu conditions failed to invert the C4 stereocenter presumably due to steric reasons. Due to the difficulty in inverting the C4-hyroxy group and sensitivity of the epoxide towards thiols, we abandoned the Julia olefination strategy. It is noteworthy that despite these synthetic problems, in 61 and 63 we observed couplings between the 3-hydroxy group and methylene protons adjacent to the sulfur atom in 1H NMR experiment in CDCl3, which supports the notion of a “strong” hydrogen bond.66,67
Third Generation Synthetic Studies
Disappointed by our two previous failed attempts, we had no choice but to employ a bolder approach to unite the two fully functionalized fragments. Such retrosynthetic analysis is shown in Scheme 13 (A), in which we continued to envisage forming the C6–C7 olefin as the final coupling. At the onset of this study, 1,3-diene–ene cross metatheses were poorly explored transformations68 and had not been used in natural product synthesis (Scheme 13, B). However, we believed that this would be a viable approach because: (1) a ruthenium catalyst would preferentially react with the olefin of monoene 70 rather than the conjugated olefins of diene 69; (2) the resulting ruthenium alkylidene 71 would preferentially react with 70 to form 67, but this is reversible at elevated temperature and could be minimized by adding 70 slowly to the reaction mixture; (3) eventually, alkylidene 71 would react with the electronically and sterically most accessible terminal olefin of 69 to form the thermodynamically favored 72; and (4) when ruthenium alkylidene 68 does form, this ruthenium species will react with 70 faster than 69 to form 72 and 66 respectively. This hypothesis was also corroborated by the Crimmins group in the crucial cross-coupling step.69
Scheme 13.

Diene-ene Cross Metathesis Coupling Strategy
To pursue this strategy, the first task was to prepare the right fragment 64. With ketoaldehyde 2 already in hand, a direct approach would be a chemoselective vinylation of this aldehyde (Table 2). Treatment of the fragile ketoaldehyde 2 with CH2=CHMgBr gave an inseparable mixture of 73 and 5-_epi_-73 with no diastereoselectivity (entry 1). Addition of HMPA or TMEDA (entries 2 and 3) slightly improved the Felkin selectivity and the overall yield of 73. Generation of vinyllithium in situ from _n_Bu3SnCH=CH2 and _n_BuLi, and subsequent addition of 2 afforded 73 and 5-_epi_-73 in 33% yield but again with no stereoselectivity (entry 4). Addition of HMPA gave better diastereoselectivity (dr = 2:1), but lower combined yield (16%) in producing 73 and 5-_epi_-73 (entry 5). Neither switching the solvent to CH2Cl2 or toluene nor making the organocerium reagent from CeCl3 and CH2=CHMgBr70,71 improved this difficult transformation. Additionally, ethynylation of ketoaldehyde 2 required higher temperatures (≥ -20 °C), leading to decomposition. We speculate that the low yields for these reactions are due to an intramolecular deprotonation-elimination pathway; the alkoxide generated after addition to the aldehyde can intramolecularly deprotonate a hydrogen atom at C2 and induce E1cb reaction. Although low yielding, we obtained the right coupling fragment 73 along with its inseparable diastereomer 5-_epi_-73 and decided to pursue preliminary coupling studies after preparing diene 54.
Table 2.
Optimization for the preparation of right fragment 73 a
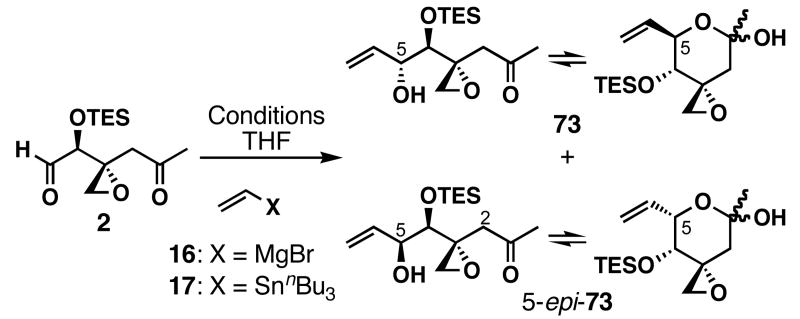 |
||||
|---|---|---|---|---|
| entry | reagent (2 equiv) | additives (equiv) | % yielda | 73:5-_epi_-73 b |
| 1 | 16 | --- | 40 | 1.0:1 |
| 2 | 16 | HMPA (2.5) | 46 | 1.3:1 |
| 3 | 16 | TMEDA (2.5) | 31 | 2.0:1 |
| 4 | 17 | _n_BuLi (1.9) | 33 | 1.0:1 |
| 5 | 17 | _n_BuLi (1.9), HMPA (2.5) | 16 | 2.0:1 |
Our attempts to prepare diene 54 are shown in Scheme 14. Aldehyde 52 was treated with ylide Ph3P=CH2 to form diene 55. Deprotection of the Boc protecting group in TFA/CH2Cl2 (1:9) and coupling with acid 3 gave 54 and 74 as an isomeric mixtures. Therefore, the Boc group removal should precede diene formation to avoid this acid-catalyzed isomerization. Removal of the Boc group in 42 (1:9 TFA/CH2Cl2) followed by coupling with acid 3 in one pot afforded 75 in 86% yield. Stereo- and regioselective cross metathesis of 75 with methacrolein using Ru-3 gave aldehyde 76 in 57% yield (67% BORSM), and subsequent Wittig reaction with ylide Ph3P=CH2 gave diene 54 in 86% yield.
Scheme 14.

Preparation of diene 54 a
a Conditions: (a) Ph3PCH3Br (1.4 equiv), _t_BuOK (1.3 equiv), THF, 0 °C, 80%; (b) TFA/CH2Cl2 (1:9), 23 °C; then 3 (1.7 equiv), HATU (1.7 equiv), _i_Pr2NEt (4.6 equiv), MeCN, 0 °C; (c) TFA/CH2Cl2 (1:9), 23 °C; then 3 (1.2 equiv), HATU (1.2 equiv), _i_Pr2NEt (4.0 equiv), MeCN, 23 °C, 86%; (d) Ru-3 (0.05 equiv), methacrolein (20 equiv), CH2Cl2, 23 °C, 57% (67% based on recovered 75); (e) Ph3PCH3Br (1.4 equiv), KO_t_Bu (1.2 equiv), THF, 0 °C, 86%.
With the fully functionalized intermediates 54 and 73 in hand, the stage was set to test our coupling strategy (Scheme 15). Treatment of diene 54 and the C6 epimeric mixture 73 gave the metathesis adduct 77 in 22% yield. Subsequent removal of the TES group was accomplished with HF•pyridine to furnish FR901464 (confirmed by HPLC analysis using the authentic FR901464) and presumably its C5-epimer, thereby supporting the viability of this strategy. We were very much surprised to find that when the reaction was neutralized with a pH 7.0 phosphate buffer, the natural product decomposed. This interesting observation prompted us to investigate the stability of FR901464 under physiological conditions (see Scheme 20).
Scheme 15.

Our first synthesis of FR901464 a
a Conditions: (a) Ru-1 (0.2 equiv), THF, 40 °C, 22%; (b) HF•pyridine, THF, 0 °C.
Scheme 20.
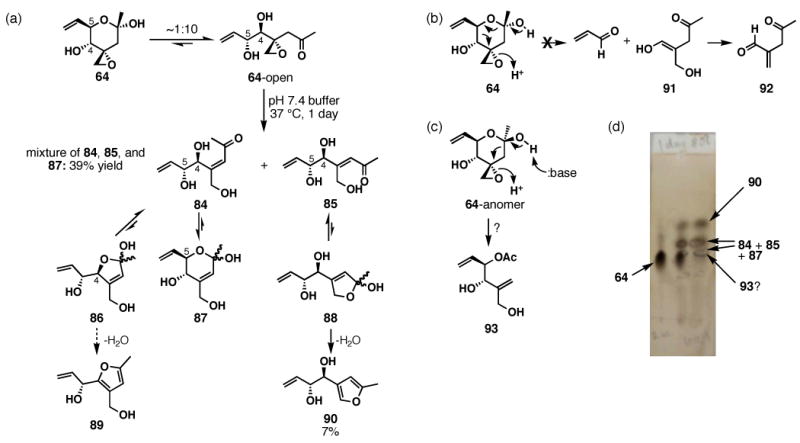
Decomposition of 64
Having realized that the final steps could potentially produce many byproducts due to instability, we strongly desired to prepare a right fragment in higher efficiency and with better stereocontrol of the C5 stereocenter. To test whether we could improve the C5 stereoselectivity, we switched to the TBS ether that might potentially favor the desired Felkin selectivity while preventing unwanted chelation of the α-alkoxy group (see Supporting Information). Preparation of this ketoaldehyde followed similarly as shown with the TES derivative (Scheme 2). However, subjection of the C4-OTBS-protected ketoaldehyde to various vinylation and alkynylation conditions formed the desired adduct in <5% yield and lacked the desired chemoselectivity. Therefore, we decided to develop a different approach to obtain the right fragment.
We turned our attention to the previously prepared enoate 24 (Scheme 3) since it contained all of the necessary carbons and the desired C4 stereochemistry. Up to this point, we were reluctant to pursue the following strategy because we could not predict the stereoselectivity of the [2,3]-sigmatropic rearrangement (Scheme 16).72 Regardless, we began to explore methods to prepare 81 from intermediate 24 (Table 3), hoping that we could somehow control the stereoselectivity in question. The DIBALH reduction of 24 afforded allylic alcohol 78 in 95% yield, which was subsequently transformed to selenide 79 or 80 in quantitative yields, setting us up to explore a Mislow-Evans–type [2,3]-sigmatropic rearrangement despite no closely related reported reactions.
Scheme 16.

Preparation of alcohols 81 and 5-_epi_-81 a
a Conditions: (a) DIBALH (3.0 equiv), THF, -78 °C, 95%; (b) ArSeCN (1.2 equiv), _n_Bu3P (1.4 equiv), THF, 0 °C, quant.
Table 3.
Step c in Scheme 16 Optimization of the [2,3]-sigmatropic rearrangements of 79 and 80
| entry | R | base (equiv) | solvent | temp. (°C) | % yield a | 81:5-_epi_-81 b |
|---|---|---|---|---|---|---|
| 1 | NO2 | pyridine (5) | THF | -20 | 94 | 4.0:1 |
| 2 | NO2 | pyridine (5) | EtOH | -44→-20 | 87 | 4.0:1 |
| 3 | NO2 | pyridine (5) | acetone | -20 | 96 | 3.5:1 |
| 4 | NO2 | pyridine (5) | CH2Cl2 | 0→20 | 67 | 3.5:1 |
| 5 | NO2 | 2,6-lutidine (5) | THF | -20→0 | 73 | 4.0:1 |
| 6 | NO2 | DMAP (5) | THF | -44→23 | 95 | 7.5:1 |
| 7 | NO2 | 4-pyrrolidinopyridine (5) | THF | -44→23 | 84 | 8.1:1 |
| 8 | NO2 | DMAP (3) | THF | -44→23 | 80 | 7.8:1 |
| 9 | NO2 | DMAP (1.5) | THF | -44→23 | 87 | 5.3:1 |
| 10 | NO2 | DMAP (0.5) | THF | -44→23 | 34 | 3.3:1 |
| 11 | H | DMAP (5) | THF | -44→23 | 91 | 7.0:1 |
Table 3 shows the optimization of the [2,3]-sigmatropic rearrangements of 79 and 80 to form the desired alcohol 81, whose stereochemistry was determined as shown in Scheme 17.36 While the transformation occurred rapidly in polar solvents (entries 1–3), it was sluggish in CH2Cl2 (entry 4). Surprisingly, the base employed in this reaction played a critical role as shown in entries 5–7. When 2,6-lutidine was used, the reaction was slower as compared to pyridine presumably due to the added steric bulk of the nucleophile, leading to no improved diastereoselectivity. When DMAP was used, the reaction was very slow at low temperatures and needed to be warmed to ambient temperature to proceed at an adequate rate. Gratifyingly, the diastereoselectivity of this reaction was improved to 7.5:1 in favor of alcohol 81. The more nucleophilic base, 4-pyrrolidinopyridine, gave the highest diastereoselectivity for this alcohol (dr = 8.1:1).73 Although DMAP showed slightly lower stereoselectivity, we were attracted by its lower cost. The effect of the DMAP stoichiometry are shown in entries 6, 8, 9, and 10; decreasing the amount of DMAP below 3 equivalents eroded selectivity presumably due to background hydrolysis reactions of the putative selenate. Finally, the nitro group on the aromatic ring was shown to be unnecessary for the high diastereoselectivity (entry 11).
Scheme 17.

Preparation of 64 a
a Conditions: (a) 2-O2NPhSeCN (1.2 equiv), _n_Bu3P (1.4 equiv), THF, 0 °C; then H2O2 (10 equiv), DMAP (4.0 equiv), -44→23 °C, 91% (dr = 7:1); (b) OsO4 (0.02 equiv), NaIO4 (2.5 equiv), 2,6-lutidine (1.6 equiv), dioxane/H2O (3:1), 0→23 °C, 27%; (c) TESCl (1.4 equiv), imidazole (1.6 equiv), THF, 0 °C, 95%; (d) OsO4 (0.01 equiv), NMO (0.96 equiv), THF/H2O (10:1), 0 to 23 °C; then Pb(OAc)4 (1.2 equiv), benzene, 0→23 °C, 71% (86% based on recovered 82); (e) AcOH/H2O/THF (3:1:3), 0→23 °C, 91%.
With the sufficiently stereoselective [2,3]-sigmatropic rearrangement in hand, we proceeded to prepare right fragment 64 (Scheme 17). Primary alcohol 78 was converted to the desired secondary alcohol 81 this time in one-pot procedure utilizing the selenide formation, [2,3]-rearrangement strategy in 91% combined yield (dr = 7:1). Oxidative cleavage of the 1,1-disubstituted olefin of 81 gave the previously prepared compound 73 in a diastereomerically pure form, but in only 27% yield. To improve the regioselectivity of the oxidative cleavage sequence, we prepared bis-TES ether 82 from 81 in 95% yield. Subsequent dihydroxylation and Pb(OAc)4-promoted diol cleavage afforded enone 83 in 71% yield. The removal of the TES ethers was found to occur best using AcOH/H2O/THF (3:1:3) to afford 64 in 91% yield. The diaxial nature of the C4 and C5 hydrogen atoms was confirmed by the 1H NMR analysis showing _J_H4-H5 = 9.8 Hz, similar to that found in the natural product (J = 10 Hz),12 thereby proving the relative stereochemistry of 64. We were now stereoselectively and efficiently form 64 in 11 linear steps. Thus, the only remaining question was the cross metathesis in the absence of any protecting groups.
As shown in Scheme 18, the catalyst employed in the cross-metathesis of 54 and 64 influenced the efficiency of the reaction. The desirable conditions found were to employ a 1:1.8 ratio of 54 to 64 in the presence of 12 mol % of Ru-3 in 1,2-dichloroethane to afford FR901464 in 40% yield after one recycle of unreacted starting materials (51% based on recovered 54 after one recycle). The fragile nature of 64 prevented more forcing reaction conditions, since facile fragmentation occurred at ≥47 °C in 1,2-dichloroethane. Additionally, right fragment 64 formed an unreactive homodimer under the reaction conditions, as determined by preparation of the right fragment homodimer and subjection of it to left fragment 54 and Ru-3.
Scheme 18.

Final stage of the total synthesis of FR901464 a
To recapitulate our total synthesis of FR901464, we completed the synthesis in 29 total steps with the longest linear sequence of 13 steps (Scheme 19). Highlights of the synthesis include a diene-ene cross metathesis as the final synthetic step in the absence of protecting groups, a Mislow-Evans type [2,3]-sigmatropic rearrangement of a selenoxide, an asymmetric Carreira alkynylation, formation of an unsaturated lactone by a ring-closing metathesis-regioselective allylic oxidation sequence, a mild, diastereoselective electron deficient alkynylation reaction, and a trans-selective Red-Al reduction. This total synthesis is the most concise to date, allowing for facile analog preparation and access to each fully functionalized fragment for biological and chemical studies.29
Scheme 19.

Summary of the total synthesis of FR901464
Stability of FR901464
Similar to the Jacobsen and Kitahara groups, we also noticed the instability of FR901464 toward acids, even as mild as silica gel.30,74 However, the sensitivity of FR901464 under physiologically relevant conditions remained unreported, and therefore we decided to determine the half-life of 64 as a means to examine its stability in various phosphate buffers at 37 °C as shown in Table 4. Alarmingly, the half-lives of 64 are only 8 and 4 hours at pH 7 and 7.4 respectively, possibly limiting FR901464's potential as a biological probe. Consequently, we became intrigued by the decomposition pathways and subjected 64 to pH 7.4 buffer at 37 °C for 1 day and observed >3 products by TLC and HPLC analyses, some of which were minor and not isolated (Scheme 20). Among the products that we were able to partially characterize were furan 90 and enones 84 and 85 (a). We did not observe acrolein (by 1H NMR or HPLC), 91, or 92 via Kitahara's decomposition pathway (b).74 We also looked for acetate 93 that would be formed by a Grob fragmentation (c),75,76 but have insufficient evidence to confirm this product formation. Hemiketal 64 exists as a 10:1 equilibrium mixture with linear ketoalcohol 64-open in both CD2Cl2 and D2O, in accordance with Jacobsen's observation.30 Therefore, enones 84 and 85 were presumably formed by the β-elimination via ketone 64-open, and furan 90 by the dehydration of hemiketal 88. Enone 84 can exist as its hemiketals 86 or 87, but 86 should be thermodynamically disfavored due to its ring strain, and therefore furan 89 is not a major byproduct of this reaction over the time period studied.
Table 4.
Instability of 64 in buffers
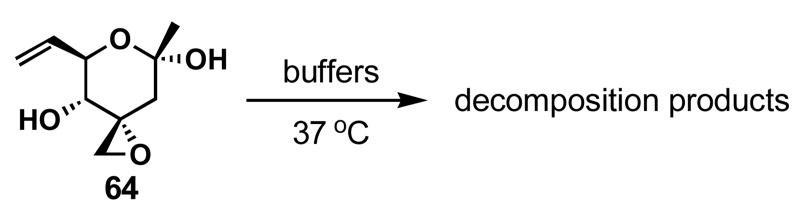 |
|
|---|---|
| pH | t1/2 (h) |
| 6 | 12 |
| 7 | 8 |
| 7.4 | 4 |
Tumor-specificity with pH sensitivity
It is intriguing to contemplate on why nature produces FR901464 with such subtle pH sensitivity. We speculate that under pressure for survival, the FR901464-producing organism may have had to find a way to inhibit the growth of enemies in acidic environments. Unfortunately this idea is difficult to test because the exact nature of the FR901464-producing organism has not yet been characterized. In the context of cancer medicine, we propose that the stability of the β-epoxy hemiketal under acidic environment can be exploited for the development of cancer-specific drugs because the pH within tumor cells is significantly lower than that of normal cells77,78 and therefore β-epoxy hemiketal-containing anticancer agents would decompose more rapidly in normal cells while remaining active in tumor cells longer. Although the acidity difference between cancer and normal cells is well-known, this difference has not yet been fully exploited. Thus this study may open the door for such an approach.
Although the pH-sensitivity could be exploited in therapeutic areas, the instability of FR901464 may limit its potential as a probe for biological study, requiring a more stable analog. The replacement of the C1-hydroxy group with either a hydrogen atom30 or a methoxy group74 only marginally improved the potency of FR901464. We hypothesized that such increased stability and more desirable van der Waals interaction with target proteins might improve the potency of FR901464. To test this hypothesis, we prepared right fragment 95 (Scheme 21) which should be more stable based on the fragmentation studies (Table 5). Treatment of alcohol 81 with Hg(OAc)2 followed by NaBH4 and Et3B79 to afford ether 94 in 76% yield. Deprotection of this tetrahydropyran was accomplished by the action of TBAF to give 95 in 97% yield. Interestingly, we found that deprotection of 94 with HF•pyridine caused a pinacol-like rearrangement to yield the ring-contracted aldehyde 96. We later found that treatment of 95 with HF•pyridine also caused this rearrangement to occur, indicating that the TES group is not essential for the rearrangement.
Scheme 21.

Preparation of the right fragment analog 95 a
a Conditions: (a) Hg(OAc)2 (1.1 equiv), THF, 0→23 °C; then NaBH4 (2.0 equiv), Et3B (1.0 equiv), -78→-44 °C, 76%; (b) TBAF (1.2 equiv), THF, 0 °C, 97%; (c) HF•pyr, THF, 0 °C, 73 %.
Table 5.
Decomposition of 95
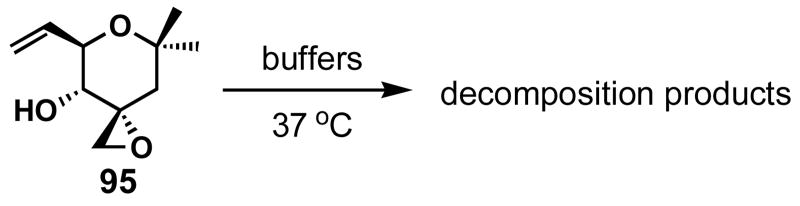 |
|
|---|---|
| pH | _t_1/2 (h) |
| 0.1 N H2SO4 | 3.5 |
| 3 | 24 |
| 4 | 40 |
| 5 | 48 |
| 6 | 40 |
| 7 | 48 |
| 7.4 | 48 |
The stability of 95 was then tested in various phosphate buffers at 37 °C as shown in Table 5. The _t_1/2 of 95 at pH 7 and 7.4 was now 48 hours, which is a dramatic increase from 64 (_t_1/2 = 4–8 h). Additionally, 95 did not appear to be significantly labile until 0.1N H2SO4 was employed as the solvent. It should also be mentioned that aldehyde 96 (Scheme 19) was not observed even in the presence of H2SO4, showing the pinacol-like rearrangement should not be biologically relevant. Replacement of the C1-OH with a methyl group enhanced the stability of the right fragment of FR901464, and therefore could provide a suitable biological probe to study the unique biology of FR901464.
Synthesis of a Stable FR901464 Analog
Encouraged by the chemical stability of 95, we proceeded to synthesize the more stable FR901464 analog meayamycin (97) in 59% yield after one recycling of recovered starting materials using the same diene-ene metathesis strategy (Scheme 22). By having a more stable right fragment, we were able to more efficiently produce the desired product. Therefore, we completed the synthesis of 97 in 28 total steps with the longest linear sequence of 13 steps. This synthetic scheme enabled us to produce 30 mg of this analog and should be scalable.
Scheme 22.

Cross metathesis of 54 and 95 a
a Conditions: (a) 54 (1.0 equiv), 95 (1.5 equiv), Ru-3 (0.10 equiv), ClCH2CH2Cl, 40 °C, 59% after 1 recycling of recovered 54 and 95.
Biological Assay
With synthetic FR901464 and its analog 97 in hand, we tested these compounds against MCF-7 breast cancer cells and measured the cell viability by using the MTS assay after the 7- and 10-day incubation periods.80 As Figure 1 shows, we observed the inhibition of cell growth in a concentration-dependent manner and determined the GI50 value of FR901464 to be 1.1 nM, which is in good agreement with the literature value of 1.8 nM.11 As we expected, the potency of meayamycin was better and exceeded our expectation: the GI50 value of this analog was 10 pM, giving approximately a 100-fold improvement in the antiproliferative activity. We also found that the mode of action of FR901464 did not involve either DNA binding or antimitotic activity.81
Figure 1.
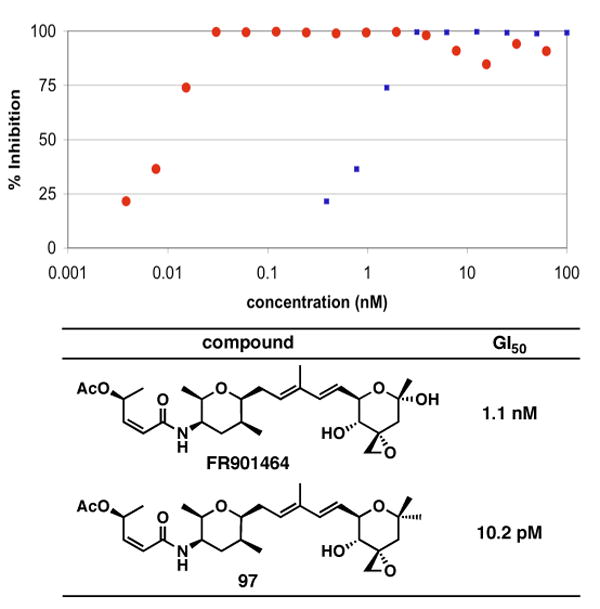
Growth inhibition of MCF-7 cells by FR901464 (blue) and meayamycin (97; red)
Conclusion
We completed the most concise total synthesis of FR901464 to date, in a total of 29 steps. We then performed degradation studies on the fully functionalized right fragment of FR901464, and used this insight to rationally design a more stable right fragment analog. Enabled by our total synthesis, we completed the synthesis of a FR901464 analog (meayamycin) using the more stable right fragment in a total of 28 steps. The GI50 value of this analog was a remarkable 10 pM in MCF-7 cells, making it one of the most potent anticancer agents known to date. With the successes of FR901464 in the xenograft models13 and impressive potency of meayamycin, we are currently working toward preparing this analog on a larger scale. The potency and stability of this analog should facilitate our efforts to isolate cellular targets of FR901464. We are also studying how the stability of meayamycin would affect the tumor specificity.
Supplementary Material
si20061103_105. Supporting Information Available.
Experimental procedures and spectroscopic data for all the new compounds and FR901464. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
B.J.A. is thankful for a Graduate Excellence Fellowship. N.L.C. is a recipient of the Arnold and Mabel Beckman Scholar Award, the Lilly Summer Research Fellowship, and the Averill Scholarship. This work was supported by the University of Pittsburgh, the American Chemical Society (PRF No. 38542-G1), The American Cancer Society George Heckman Institutional Research Grant, The Competitive Medical Research Fund, and the National Cancer Institute (1R01CA120792-01).
References
- 1.Service RF. Science. 2005;310:1132–1134. doi: 10.1126/science.310.5751.1132. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Burger RM. Chem Rev. 1998;98:1153–1169. doi: 10.1021/cr960438a. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Leonessa F, Welch JN, Skaar TC. Pharmacol Rev. 2001;53:25–71. [PubMed] [Google Scholar]
- 5.Druker BJ. Trends Mol Med. 2002;8:S14–S18. doi: 10.1016/s1471-4914(02)02305-5. [DOI] [PubMed] [Google Scholar]
- 6.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J Clin Oncol. 2005;23:630–639. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 7.Adams J. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 8.Jordan MA, Wilson L. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Giannakakou P. Curr Med Chem Anti-Cancer Agents. 2005;5:65–71. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]
- 10.Jones NC, Rigby PWJ, Ziff EB. Genes Dev. 1988;2:267–281. doi: 10.1101/gad.2.3.267. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. J Antibiot. 1996;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 12.Nakajima H, Takase S, Terano H, Tanaka H. J Antibiot. 1997;50:96–99. doi: 10.7164/antibiotics.50.96. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. J Antibiot. 1996;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 14.Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. J Antibiot. 1996;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 15.Schena M, Shalon D, Davies RW, Brown PO. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 16.Resnick-Silverman L, Manfredi JJ. J Cell Biochem. 2006;99:679–689. doi: 10.1002/jcb.20925. [DOI] [PubMed] [Google Scholar]
- 17.Braithwaite AW, Prives CL. Cell Death and Differentiation. 2006;13:877–880. doi: 10.1038/sj.cdd.4401938. [DOI] [PubMed] [Google Scholar]
- 18.Kumar R, Gururaj AE, Barnes CJ. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 19.Gartel AL, Radhakrishnan SK. Cancer Res. 2005;65:3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 20.Stevens C, La Thangue NB. DNA Repair. 2004;3:1071–1079. doi: 10.1016/j.dnarep.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 21.Bell LA, Ryan KM. Cell Death and Differentiation. 2004;11:137–142. doi: 10.1038/sj.cdd.4401324. [DOI] [PubMed] [Google Scholar]
- 22.Dominguez-Sola D, Dalla-Favera R. Nat Cell Biol. 2004;6:288–289. doi: 10.1038/ncb0404-288. [DOI] [PubMed] [Google Scholar]
- 23.Janz S. Oncogene. 2005;24:3541–3543. doi: 10.1038/sj.onc.1208473. [DOI] [PubMed] [Google Scholar]
- 24.Sakai Y, Yoshida T, Ochiai K, Uosaki Y, Saitoh Y, Tanaka F, Akiyama T, Akinaga S, Mizukami T. J Antibiot. 2002;55:855–862. doi: 10.7164/antibiotics.55.855. [DOI] [PubMed] [Google Scholar]
- 25.Isaac BG, Ayer SW, Elliott RC, Stonard RJ. J Org Chem. 1992;57:7220–7226. [Google Scholar]
- 26.Millerwideman M, Makkar N, Tran M, Isaac B, Biest N, Stonard R. J Antibiot. 1992;45:914–921. doi: 10.7164/antibiotics.45.914. [DOI] [PubMed] [Google Scholar]
- 27.Sakai Y, Tsujita T, Akiyama T, Yoshida T, Mizukami T, Akinaga S, Horinouchi S, Yoshida M, Yoshida T. J Antibiot. 2002;55:863–872. doi: 10.7164/antibiotics.55.863. [DOI] [PubMed] [Google Scholar]
- 28.Sakurai M, Kohno J, Nishio M, Yamamoto K, Okuda T, Kawano K, Nakanishi N. J Antibiot. 2001;54:628–634. doi: 10.7164/antibiotics.54.628. [DOI] [PubMed] [Google Scholar]
- 29.Koide K, Albert BJ. J Synth Org Chem Jpn. 2007 Accepted for publication. [Google Scholar]
- 30.Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2001;123:9974–9983. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 31.Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2000;122:10482–10483. [Google Scholar]
- 32.Horigome M, Motoyoshi H, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8207–8210. [Google Scholar]
- 33.Motoyoshi H, Horigome M, Watanabe H, Kitahara T. Tetrahedron. 2006;62:1378–1389. [Google Scholar]
- 34.Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem Int Ed. 1999;38:2398–2400. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 35.Horigome M, Motoyoshi H, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8207–8210. [Google Scholar]
- 36.Albert BJ, Sivaramakrishnan A, Naka T, Koide K. J Am Chem Soc. 2006;128:2792–2793. doi: 10.1021/ja058216u. [DOI] [PubMed] [Google Scholar]
- 37.Haolun Jin JiU, Christ William J, Kishi Yoshito. J Am Chem Soc. 1986;108:5644–5646. [Google Scholar]
- 38.Kazuhiko Takai KK, Kuroda Tooru, Hiyama Tamejiro, Nozaki Hitosi. Tetrahedron Lett. 1983;24:5281–5284. [Google Scholar]
- 39.Frangin Y, Gaudemar M. J Organomet Chem. 1977:9–22. [Google Scholar]
- 40.Katsuki T, Sharpless KB. J Am Chem Soc. 1980:5974–5976. [Google Scholar]
- 41.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 42.Frantz DE, Fassler R, Carreira EM. J Am Chem Soc. 2000;122:1806–1807. [Google Scholar]
- 43.Seyferth D, Weiner MA. J Am Chem Soc. 1962;84:361–363. [Google Scholar]
- 44.Shahi SP, Koide K. Angew Chem Int Ed. 2004;43:2525–2527. doi: 10.1002/anie.200353400. [DOI] [PubMed] [Google Scholar]
- 45.Meta CT, Koide K. Org Lett. 2004;6:1785–1787. doi: 10.1021/ol0495366. [DOI] [PubMed] [Google Scholar]
- 46.Miwa K, Aoyama T, Shioiri T. Synlett. 1994:107–108. [Google Scholar]
- 47.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769–3772. [Google Scholar]
- 48.Ohira S. Synth Commun. 1989;19:561. [Google Scholar]
- 49.Muller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521–522. [Google Scholar]
- 50.Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 51.Corey EJ, Raman KB, Shibata S, Chen CP, Singh VK. J Am Chem Soc. 1987;109:7925–7926. [Google Scholar]
- 52.Yu CM, Kim C, Kweon JH. Chem Comm. 2004:2494–2495. doi: 10.1039/b407387h. [DOI] [PubMed] [Google Scholar]
- 53.Garner P, Park JM. J Org Chem. 1987;52:2361–2364. [Google Scholar]
- 54.Wei ZY, Knaus EE. Synthesis. 1994:1463–1466. [Google Scholar]
- 55.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 56.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J Am Chem Soc. 1999;121:791–799. [Google Scholar]
- 57.Grela K, Harutyunyan S, Michrowska A. Angew Chem Int Ed. 2002;41:4038–4040. doi: 10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 58.Michrowska A, Bujok R, Harutyunyan S, Sashuk V, Dolgonos G, Grela K. J Am Chem Soc. 2004;126:9318–9325. doi: 10.1021/ja048794v. [DOI] [PubMed] [Google Scholar]
- 59.Koul AK, Prashad B, Bachawat JM, Ramegowda NS, Mathur NK. Synth Commun. 1972;2:383–388. [Google Scholar]
- 60.Royer J, Bonin M, Micouin L. Chem Rev. 2004;104:2311–2352. doi: 10.1021/cr020083x. [DOI] [PubMed] [Google Scholar]
- 61.Ji S, Gortler LB, Waring A, Battisti A, Bank S, Closson WD. J Am Chem Soc. 1967;89:5311–5312. [Google Scholar]
- 62.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408–7410. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 63.Julia M, Paris JM. Tetrahedron Lett. 1973;14:4833–4836. [Google Scholar]
- 64.Kocienski PJ, Lythgoe B, Ruston S. J Chem Soc Perkin Trans. 1978;1:829–834. [Google Scholar]
- 65.Albert BJ, Koide K. Org Lett. 2004;6:3655–3658. doi: 10.1021/ol049160w. [DOI] [PubMed] [Google Scholar]
- 66.Alkorta I, Rozas I, Elguero J. Chem Soc Rev. 1998;27:163–170. [Google Scholar]
- 67.Perrin CL, Nielson JB. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 68.Randall ML, Tallarico JA, Snapper ML. J Am Chem Soc. 1995;117:9610–9611. [Google Scholar]
- 69.Crimmins MT, Christie HS, Chaudhary K, Long A. J Am Chem Soc. 2005;127:13810–13812. doi: 10.1021/ja0549289. [DOI] [PubMed] [Google Scholar]
- 70.Dimitrov V, Bratovanov S, Simova S, Kostova K. Tetrahedron Lett. 1994;35:6713–6716. [Google Scholar]
- 71.Dimitrov V, Kostova K, Genov M. Tetrahedron Lett. 1996;37:6787–6790. [Google Scholar]
- 72.Wirth T. Angew Chem Int Ed. 2000;39:4189–4189. [Google Scholar]
- 73.Hassner A, Krepski LR, Alexanian V. Tetrahedron. 1978;34:2069–2076. [Google Scholar]
- 74.Motoyoshi H, Horigome M, Ishigami K, Yoshida T, Horinouchi S, Yoshida M, Watanabe H, Kitahara T. Biosci Biotechnol Biochem. 2004;68:2178–2182. doi: 10.1271/bbb.68.2178. [DOI] [PubMed] [Google Scholar]
- 75.Grob CA. Angew Chem Int Ed. 1969;8:535–546. [Google Scholar]
- 76.Grob CA, Schiess PW. Angew Chem Int Ed. 1967;6:1–15. [Google Scholar]
- 77.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 78.Simon SM, Schindler M. Proc Natl Acad Sci USA. 1994;91:3497–3504. doi: 10.1073/pnas.91.9.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang SH, Lee JH, Lee SB. Tetrahedron Lett. 1998;39:59–62. [Google Scholar]
- 80.Buttke TM, Mccubrey JA, Owen TC. J Immunol Methods. 1993;157:233–240. doi: 10.1016/0022-1759(93)90092-l. [DOI] [PubMed] [Google Scholar]
- 81.Pommier, Y.; Vogt, A.; McPherson, P.; Koide, K., Unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
si20061103_105. Supporting Information Available.
Experimental procedures and spectroscopic data for all the new compounds and FR901464. This material is available free of charge via the Internet at http://pubs.acs.org.