Autophagy in neuroprotection and neurodegeneration: A question of balance (original) (raw)
. Author manuscript; available in PMC: 2008 Sep 19.
Published in final edited form as: Future Neurol. 2008 May;3(3):309–323. doi: 10.2217/14796708.3.3.309
Summary
A central issue in developing therapies for neurodegenerative diseases involves understanding why adaptive responses to stress or injury fail to prevent synaptic dysfunction and neuronal cell death. Macroautophagy is a major, evolutionarily conserved response to nutrient and bioenergetic stresses, which has the capacity to remove aggregated proteins and damaged organelles such as mitochondria. This has prompted intense interest in autophagy-related therapies for Huntington’s, Alzheimer’s, Parkinson’s, stroke and other neurological diseases. However, excessive or imbalanced induction of autophagic recycling can actively contribute to neuronal atrophy, neurite degeneration and cell death. Oxidative-, aging- and disease-related increase in demand for autophagy, coupled with declining axonal trafficking, lysosomal degradation or biosynthetic efficiencies promote increased susceptibility to a harmful state of autophagic stress. A more complete understanding of dysfunction along the entire spectrum of autophagic recycling, from autophagosome formation through clearance and regeneration of new cellular components is necessary to restore balance to the system, promote neuronal health and maximize therapeutic potentials.
Keywords: autophagy, protein aggregation, neurite degeneration, neuronal cell death, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, lysosomal storage disease, hypoxic-ischemic brain injury, aging
INTRODUCTION
Cytoplasmic and nuclear inclusions composed of aggregated, often polyubiquitinated proteins comprise key pathologic hallmarks of numerous neurodegenerative diseases. These include neuritic plaques and neurofibrillary tangles of Alzheimer’s disease, the Lewy bodies and Lewy neurites of Parkinson’s disease and Lewy body dementias, aggregated polyglutamine proteins in trinucleotide repeat disorders, and ubiquitinated inclusions in frontotemporal dementias. While the mechanisms by which protein aggregation contributes to synaptic dysfunction and neuronal degeneration remain to be fully elucidated, recent evidence suggests that pathologic alterations in autophagy-related pathways contribute to neurodegeneration [1, 2]. New therapeutic approaches based on manipulating lysosomal degradation systems may thus be broadly applicable to a wide range of neurodegenerative disorders [3].
Autophagy is the orchestrated bulk degradation of intracellular proteins or whole organelles. There are at three major types of autophagy (Figure 1). Chaperone mediated autophagy involves direct import of proteins containing a targeting amino acid motif across the lysosomal membrane [4]. Substrates for microautophagy bud directly into invaginations of the lysosomal membrane [5]. Macroautophagy involves the formation of membranous vacuoles, which are transported along microtubules for eventual fusion with the lysosome [6]. Because macroautophagy is likely the major pathway for aggregated proteins, it forms the focus of this review and the terms “autophagy” and “autophagic” refer to macroautophagy unless otherwise indicated.
Figure 1. Major Phases of Autophagic Recycling.
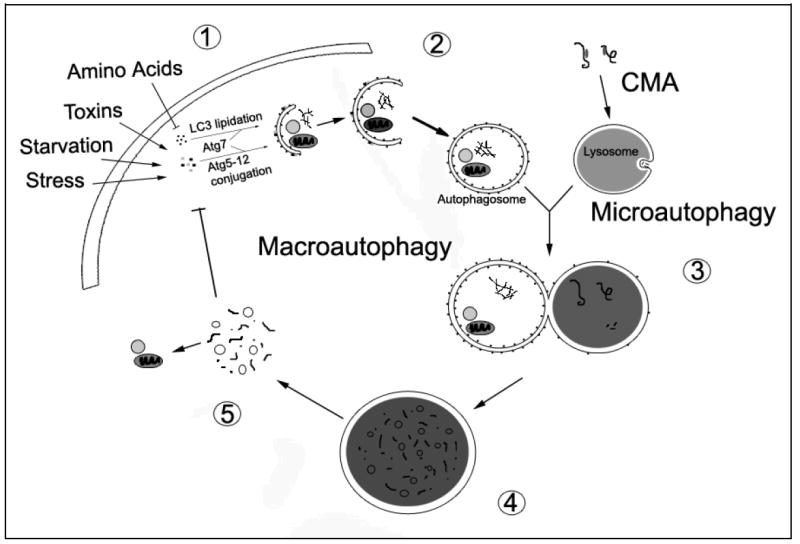
The cell induces macroautophagy in response to a number of physiological and pathological signals. A series of Atg proteins transduce these signals for the execution of macroautophagy, resulting in the conjugation of Atg5 to Atg12 and the lipidation of LC3 (conversion of LC3-I to LC3-II) by Atg7 (1). While many other molecules are involved in autophagy regulation [90], these conjugation reactions are now widely used to monitor and manipulate autophagy induction. Sequestration of cytosolic components into autophagosomes follows Atg5-12 conjugation and LC3 lipidation. The autophagic cargo may include long-lived proteins, misfolded or damaged proteins, and dysfunctional organelles (2). The maturing autophagosome, which has lost the Atg5-12 complex, fuses with the lysosome (3). Successful fusion may depend upon microtubule-dependent trafficking, intermediate fusion with acidified endocytic vesicles to form amphisomes or multivesicular bodies, lysosomal pH or other unknown lysosomal membrane properties (not illustrated). Material delivered to the lysosome is degraded by lysosomal hydrolases that function at acidic pH (4). The degraded contents of the lysosome are released into the cytosol for reutilization in the biosynthesis of new proteins and organelles (5). The released biomolecules (i.e. amino acids) can also regulate macroautophagy through effects on protein synthesis [103] or through modulation of signaling pathways. The processes of chaperone-mediated autophagy (CMA) and microautophagy converge with macroautophagy at the lysosome. In CMA, molecular chaperones select proteins by their KFERQ-like motif and shuttle them to the lysosome for degradation. In microautophagy, the lysosome invaginates to remove cytosolic components for degradation.
The phases of autophagic recycling (Figure 1)
Autophagic recycling of long-lived proteins, organelles and unneeded, effete or damaged cargo is comprised of several major stages:
- Integration of signaling pathways to activate autophagy.
- Sequestration of cargo and formation of the autophagosome.
- Maturation (trafficking, fusion, acidification) to autolysosomes.
- Lysosomal degradation and release of macromolecular building blocks.
- Successful reutilization of liberated biomolecules.
Although the last stage involves processes not traditionally considered in the discussion of degradation pathways, successful transcriptional and biosynthetic utilization of autophagic products is an integral part of the process, which we propose determines beneficial versus detrimental outcomes of autophagy induction (Figure 2). There is also emerging evidence that a fine balance of autophagic recycling regulates neurite morphology and synaptic function. When considering disease mechanisms and potential autophagy-related therapies, it is important to recognize that maintaining or restoring balance between induction (Stages 1-2) and successful completion of autophagy (Stages 3-5) may be essential for promoting neuronal health and function.
Figure 2. Physiologic and Pathologic Alterations in Autophagy.

Neurons require a basal degree of autophagic degradation to mediate replacement of damaged or effete cellular components or to facilitate neuritic/synaptic remodeling. Physiologic induction of increased autophagic degradation (a) may be balanced by increased biosynthetic activity to regenerate the degraded components, or result in atrophy of soma and/or neuritic arbor. By definition, physiologic atrophy is reversible upon restoration of anabolic signals. Insufficient autophagy (b) results in accumulation of ubiquitinated proteins and neurodegeneration in experimental systems, suggesting that therapeutic interventions to increase autophagic degradation may be effective for neurodegenerative diseases.
However, diseased neurons often demonstrate increased autophagosomes or autolysosomes reflective of autophagic stress (see-saws). Autophagic stress is defined as an imbalance between rates of autophagosome formation and the capacity of the cell to complete the process of degradation and autophagic recycling [2](Box A). The imbalance may occur due to age- or disease-related decreases in the efficacy of autophagosome trafficking and lysosomal fusion/degradation (c), and/or excessive induction of autophagy due to disease-related injury and increased demand for autophagy (d). We hypothesize that the outcome of autophagic stress is determined by the balance between the rates of sequestration versus degradation and the transcriptional/biosynthetic capacities of the stressed neuron. New synthesis of essential components sequestered but not degraded in autophagosomes, upregulation of lysosomal and heat shock proteins, and/or regeneration of essential proteins and organelles that have been degraded all represent compensatory responses that would favor survival. However, age- and disease-related impairment of biosynthetic/regenerative capacity would exacerbate autophagic stress, leading to neurodegeneration and cell death. Thus, strategies that robustly induce autophagy may backfire in the disease context. Instead, modest levels of autophagy induction combined with therapies to promote successful completion of autophagic recycling may be necessary in aged or diseased subjects.
In the following sections, we will review data indicating that autophagy plays an important neuroprotective role and discuss mechanisms that contribute to a state of autophagic stress [2] (Box A). This will be followed by a discussion of human central nervous system diseases and models that illustrate three major patterns of autophagy dysregulation: insufficiency of autophagy induction, autophagic stress due to reduced lysosomal function and autophagic stress related to pathologic activation of autophagy, keeping in mind that these are not mutually exclusive mechanisms for any given disease. Discussion of promising therapeutic advances will be considered along with issues requiring future investigation.
Box A. Autophagic Stress: An imbalance between initiation and completion of autophagic recycling.
- Autophagic stress can be morphologically recognized by accumulations of autophagic vacuoles in injured cells [2], and operationally defined if either blunting autophagy induction or promoting lysosomal fusion/function can confer protection from the degenerative parameter being studied.
- There are three major mechanisms that contribute to autophagic stress, which are not mutually exclusive:
- In some disease states, robust induction of autophagy overwhelms the cell’s ability to successfully complete autophagic degradation. This may be due to aging- or injury-associated decline in fusion/degradation efficiency that is not limiting in the basal state.
- Despite reduced efficiency, protein and organelle degradation (autophagic flux) could increase with increased autophagy induction. If the rates of mitochondrial or synaptic structure degradation cannot be matched by reutilization of degraded components in regenerative biosynthesis, this would contribute to another form of autophagic stress.
- In other diseases, impairment in trafficking, fusion, or lysosomal degradation become severe enough to interfere with basal levels of autophagic turnover resulting in autophagy failure.
- The therapeutic relevance of the concept of autophagic stress lies in promoting research into combination therapies that modulate not only autophagy induction, but also lysosomal efficiency. A balanced induction of autophagy at levels compatible with the degradative reserves of the diseased neurons, combined with efforts to stimulate biosynthesis of healthy new mitochondria or axodendritic components would represent viable therapeutic goals for neurodegenerative diseases.
AUTOPHAGIC STRESS AND PATHOLOGIC ALTERATIONS IN AUTOPHAGY
There are two major lines of research related to acute and chronic brain disorders that converge to implicate autophagy modulation as a promising neuroprotective strategy for the treatment of pediatric and adult brain injuries and neurodegenerative diseases. The first involves the study of protein aggregation in which landmark in vivo studies demonstrated that enhancing autophagy confers protection in Huntington’s neurodegeneration [7], while genetic ablation of basal autophagy spontaneously causes neurodegeneration in mice [8, 9]. The second relates to a growing number of studies showing that autophagy serves a gatekeeping function upstream of multiple apoptotic and non-apoptotic neuronal cell death pathways [10-13], suggesting that therapies targeting autophagy could regulate neuron loss due to multiple death stimuli [14].
In subsequent sections, discussion of autophagy in neuroprotection and neurodegeneration will be organized according to the conceptual scaffold that we developed to integrate apparently disparate conclusions about protective and disease-promoting roles for autophagy in a rapidly accelerating field (Fig. 2). Different perturbations in the initiation and completion of autophagic recycling have been implicated in a wide spectrum of neurodegenerative conditions. These include deficits in induction and degradation (Fig. 2b) and relative imbalances between induction and clearance, which combine to create autophagic stress (Fig. 2c, 2d; Box A) [2]. Just as the outcome of physiologic increases in autophagic flux (See Glossary, Box B) is determined ultimately by whether or not biosynthetic activities go up or down (Fig. 2a), we hypothesize that the outcome of autophagic stress also relates to whether or not the aged- or diseased- neuron is capable of upregulating compensatory biosynthetic responses. This is supported by a recent study in nematodes suggesting that autophagy may not be sufficient for lifespan extension in the absence of transcriptional signals to regulate recycling of the raw material [15].
Box B. Glossary.
Atg5 - this protein is covalently conjugated to Atg12 in a ubiquitin-like conjugation reaction, and may play an E3-like role in promoting efficient LC3 lipidation [102].
Atg7 - an essential component of the core autophagy machinery that functions as an E1-like activating enzyme for both Atg12 and LC3.
Autophagic flux - a dynamic measure of how much cargo is successfully degraded/unit time.
Autophagic vacuole (AV) - a term used in mammalian autophagy systems to describe the range of autophagy-related structures, which are formed from maturation of the early autophagosome into autolysosomes through trafficking and fusion mechanisms. Frequently, intermediate compartments derived through fusion with endocytic vesicles (amphisome or multivesicular body) are also observed. Because some of these compartments are distinguished by pH and morphologic factors that may form a continuum, it is convenient to consider them as a group, or as early AVs versus degradative AVs.
AV content - a static description of the levels of autophagic vacuoles at various stages of maturation at a given time point.
Beclin 1 (Atg6) - the mammalian homolog to yeast Atg6, which is involved in class III phosphoinositide 3-kinase protein complexes that regulate autophagy and endocytosis.
LC3 (Atg8) - microtubule associated protein light chain 3, one of several mammalian Atg8 homologs. LC3 conjugation to phospholipids is essential for induction of autophagic sequestration. Membrane associated LC3 is also widely used as a specific marker of phagophores and autophagosomes. LC3 has also been reported to bind protein aggregates, and it has been proposed that this could represent a mechanism for autophagic cargo recognition.
mTOR - The mammalian target of rapamycin is a kinase that forms at least two complexes within cells to coordinate cell growth, including effects on protein synthesis, autophagy, and regulation of the cytoskeleton. Since mTPR suppresses autophagy, inhibition of mTOR by rapamycin activates autophagy in many cell types.
RNAi - RNA interference is a method to decrease expression of specific proteins by harnessing an endogenous cellular mechanism in which double-stranded RNAs trigger the destruction or silencing of specific RNAs.
INSUFFICIENT AUTOPHAGY IN NEURODEGENERATION
In contrast to other organ systems, the central nervous system displays only rare autophagic vacuoles (AVs, Box B), which can be dramatically increased in injured or degenerating neurons [16]. Low steady state AV levels could reflect either low basal activity or high activity with extremely efficient clearance. While, additional methods are needed to accurately measure the degree of autophagic flux (Box B) in the living brain, a pair of recent studies indicates that autophagy is critically important for neuronal health [8, 9]. When neuronal autophagy induction is consitutively blocked through nestin-targeted knockout of either Atg5 or Atg7, two proteins that mediate membrane rearrangement to generate autophagosomes, the mice develop ubiquitinated protein aggregates, neuronal cell loss, behavioral abnormalities and early death. These observations suggest that a basal level of autophagy is required for survival of neurons.
In Drosophila, there is age-related decline in the expression of Atg proteins (Box B), and enhancing the expression of Atg8 extends lifespan and confers resistance to oxidative stressors [17]. Drosophila lacking Atg7 are hypersensitive to starvation and oxidative stress, exhibiting decreased lifespan and increased ubiquitin-positive brain aggregates [18]. While nonfunctional mutations in Atg proteins have yet to be identified as a cause for human neurodegeneration, some studies indicate that mutant huntingtin protein sequesters beclin 1 [19], a Bcl-2 binding protein whose complexes regulate phosphoinositide 3-kinase-dependent autophagic and endocytic membrane dynamics. Moreover, aged human subjects and early Alzheimer’s disease patients show reduced expression of beclin 1 [19, 20]. As haploinsufficiency in beclin 1 decreases autophagic activity in vivo, these observations suggest that insufficient autophagy induction may contribute to certain forms of neurodegeneration.
Insufficient autophagy: Implications for neuroprotection
In line with these observations, it has been shown that the autophagy-inducing drug rapamycin confers protection in models of neurodegenerative diseases [7]. This concept arose from data showing that many neurodegenerative disease mutations result in aggregate-prone proteins, and that induction of autophagy can reduce the levels of protein aggregates in multiple systems [21-24]. Furthermore, the striking pathology exhibited by Atg gene knockout mice indicate that autophagy plays a major role in determining the burden of polyubiquitinated proteins in neurons [8, 9].
It is not clear that the effects of mTOR inhibitors are due exclusively to degradation of protein aggregates, as decreased protein synthesis and reduced aggregate formation have also been described in rapamycin treated cells [25]. Improvement in motor symptoms [7] could also be explained by reduction in levels of toxic soluble oligomers and proto-fibrils [26, 27]. Formation of larger aggregates and inclusions may actually represent a protective mechanism limiting exposure to oligomers as suggested in models of Huntington’s disease or diffuse Lewy body disease [28, 29], although inclusion formation in liver cells contributes to toxicity caused by autophagy deficiency [30]. As the relationship between decreased aggregate formation and increased clearance may be difficult to detangle, combination therapies that combine autophagy inducing and chaperone-like functions may prove promising [31].
One potential limitation of rapamycin-related therapies is that induction of autophagy depends upon the degree to which TOR activity is suppressing autophagy in a given system. Thus additional pharmacologic agents that can modulate autophagy in a TOR-independent manner would be beneficial [32]. Another is that a given drug may simultaneously affect the activity of two pathways with opposing effects on autophagy [33, 34]. These issues may be resolved by combination therapies, as illustrated by a recent publication showing that rapamycin synergizes with lithium in clearing protein aggregates [34]. As both lithium and rapamycin are used in patients for the treatment of other conditions, these represent attractive targets for autophagy-inducing therapies.
Autophagy induction appears to be a promising therapy for protein aggregation diseases, particularly autosomal dominant diseases such as Huntington’s disease in which patients at risk are readily identified at an early age. For other diseases, particularly those with a later age of onset such as Alzheimer’s or Parkinson’s disease, further study into mechanisms underlying dysfunction of autophagy (Figure 3) are necessary to determine areas requiring additional intervention, as discussed below.
Figure 3. Factors proposed to increase the susceptibility of the aging neuron to autophagic stress and neurodegeneration.
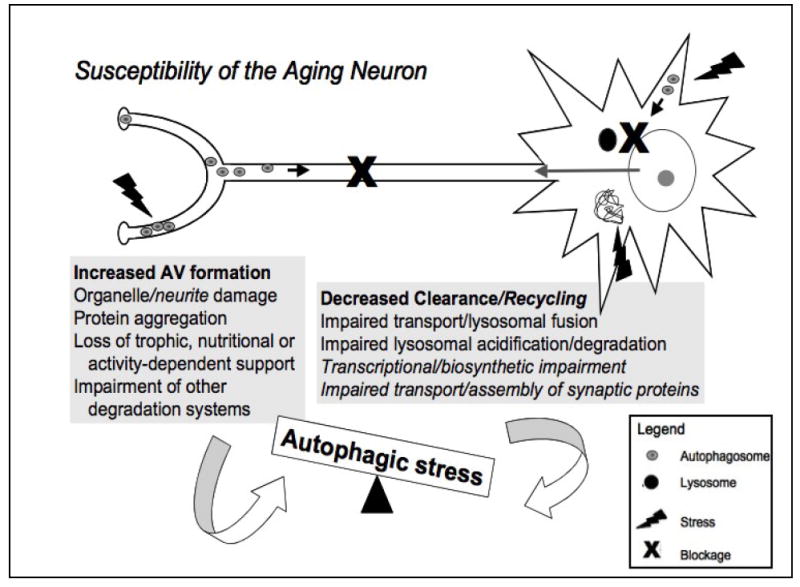
The neuron represents the most highly polarized cell type in the human body. It is exquisitely sensitive to disturbances in protein, organelle and membrane transport, and is crucially dependent upon activity-dependent signals derived from its contacts with other cells. The grey boxes summarize potential factors promoting increased autophagy induction (AV formation) and decreased autophagy completion (clearance/recycling), which can both contribute to autophagic imbalances. The high metabolic demand of maintaining functional synapses, which can be meters away from the cell body, and relatively low antioxidant defenses in the brain contribute to oxidative damage and protein aggregation, increasing demand for autophagy [10]. Additionally, aging- and disease-related deficits in the ubiquitin-proteasome system and other pathways of lysosomal degradation (chaperone-mediated autophagy) result in further induction of (macro)autophagy [58, 61, 104, 105]. At the same time, progressive accumulation of damage in non-mitotic cells due to aging/disease mechanisms results in impaired retrograde trafficking, lysosomal fusion and degradation of AVs [104]. Decreased transcriptional and biosynthetic efficiency observed with oxidative stress and aging and impaired anterograde delivery of synaptic components result in failure to compensate for autophagic stress, leading to neurite degeneration and neuronal cell death. Figure modified from Reference [2] and reproduced with permission from the Journal of Neuropathology and Experimental Neurology.
AUTOPHAGIC STRESS DUE TO IMPAIRED MATURATION/DEGRADATION
Since successful autophagy requires functional lysosomes to degrade the sequestered cytoplasmic components, we and others have proposed that lysosomal dysfunction can lead to autophagic stress characterized by accumulation of autophagic intermediates [1, 2, 35]. This mechanism of autophagy dysfunction has been advanced primarily through study of lysosomal storage diseases and Alzheimer’s disease. The different ages of onset of neurodegeneration could relate to the severity and kinetics of lysosomal impairment. While chemical ablation of lysosomal acidification can cause acute programmed cell death [36], the mechanisms operating in chronic human brain disorders require further elucidation. Nevertheless, gradual impairment in the efficiency of AV trafficking, AV-lysosome fusion or lysosomal acidification and degradation that are insufficient to trigger cell death, may predispose neurons to further neurodegenerative insults by more than one mechanism [37, 38].
Lysosomal storage diseases (LSDs) are genetic disorders arising from mutation in lysosomal enzymes or trafficking proteins that impair lysosomal processing of specific constituents [39, 40]. Degradative impairment in LSDs leads to an increase in early AVs and to a decrease in AV fusion/degradation [37, 41-43], although increased autophagy induction may also contribute to autophagic stress in LSDs [44-46].
Mechanisms of neurodegeneration in these diseases are just beginning to be understood. Alterations in phosphoinositide metabolism associated with compromised autophagic recycling in cathepsin D deficient mice may result in deficient pro-survival Akt signaling [43]. In addition, tonic deficiencies in autophagic quality control lead to accumulation of impaired mitochondria. For example, cells from patients with mucolipidosis type IV exhibit fragmented mitochondria with impaired calcium buffering capacity, which leads to increased sensitivity to cell death elicited by calcium mobilizing agents [38]. This may represent a common mechanism of neurodegeneration in LSDs as similar findings have been described in other LSDs including mucolipidosis types II and III and neuronal ceroid lipofuscinosis 2 [38, 42].
It has been shown that fibroblasts from these patients show normal levels of AV induction, as measured by appearance of LC3 and MDC stained puncta upon starvation, but the clearance of these structures upon re-feeding is signfiicantly delayed (Supplemental figure in Reference [38]). Interestingly, ultrastructural analysis of these cells does not reveal a backup of autophagocytosed mitochondria, suggesting that chronic lack of AV clearance may also decrease sequestration of impaired mitochondria [39]. These findings suggest that autophagic stress due to deficient completion of autophagy may in turn lead to insufficient sequestration of abnormal cellular constituents.
Alzheimer’s disease presents another situation for which stress from defective completion of autophagy has been implicated. Granuolovacuolar degeneration and lysosomal expansion have long been recognized as features of Alzheimer’s disease {Cataldo, 1996 #2035}. An autopsy study of Alzheimer’s disease reveals high levels of intermediate AVs accumulating in dystrophic neurites [47], which greatly exceeds levels observed in Parkinson’s/Lewy body disease [48, 49]. Trafficking along microtubules is essential for autophagic degradation in mammalian systems [50, 51], and is likely to be particularly important in neurons given the distances from neuronal processes, where endosomes and autophagosomes are formed, and the neuronal soma where the lysosomes are concentrated (Figure 3). In addition to disruption to efficient autophagic recycling, the expansion in AV intermediates itself can be harmful, serving as sites of microbial replication [52], enzyme leakage [53] or, in the case of Alzheimer’s disease, as sites of increased pathogenic Aβ peptide production [54]. While most of the Aβ generated is degraded in the lysosome, some is released extracellularly, potentially leading to the Aβ plaques found in AD brains [1]. Of course, as discussed above, the mechanisms of autophagic perturbation are not mutually exclusive, and there is some data suggesting that chronic deficiencies in lysosomal degradation could feedback to suppress autophagy induction.
Autophagic stress from impaired degradation: Implications for neuroprotection
The key factors here would involve enhancing lysosomal degradation of autophagic cargo, which requires additional investigation into the mechanism(s) by which trafficking, fusion, acidification or degradation are impaired. Unless these deficits are addressed, we would predict that therapies based solely on modulating initiation of autophagy would be unsuccessful, and perhaps contribute to further damage by exacerbating autophagic stress in diseased neurons.
Stimulation of autophagy can bypass microbe-induced deficits in macrophage phagolysosome maturation, resulting in microbial elimination [55], and decreased insulin receptor signaling has been shown to promote clearance of (Aβ) through promotion of AV maturation [56]. It unknown, however, whether deficits in AV maturation can be reversed. Administration of ascorbic acid to cultured glial cells promotes increased autophagic flux, enhancing the efficiency of lysosomal degradation by stabilizing acidification [57]. However, whether these techniques can operate under more rigorous in vivo buffering conditions is unknown. To alleviate the blockage, future strategies may involve promoting expression of trafficking proteins [58], proteins involved in promoting AV-lysosomal fusion, such as those regulating endosomal fusion and multivesicular bodies [59, 60], or normalizing age-related declines in lysosomal protein and membrane composition. If it is shown that sequestration of autophagy regulators or other essential factors contributes to harmful effects of AV accumulation, stimulating biosynthesis of these macromolecules may represent an alternative strategy to compensate for the maturation blockage.
Given that autophagic stress reflects an imbalance, rather than an absolute level of flux, it is conceivable that reducing input into the system may represent an alternative method to ameliorate harmful effects of autophagic stress. For example, aging- and disease- conditions create increased demand for macroautophagy (Figure 3). Thus, antioxidant or chaperone-mediated therapies that reduce oxidative damage to organelles and proteins, or approaches that promote proteasomal function and/or chaperone-mediated autophagy may lead to decreased demand for and induction of macroautophagy [61], restoring balance to a partially impaired system.
AUTOPHAGIC STRESS DUE TO DYSREGULATED AUTOPHAGY INDUCTION
The question of whether or not there is “excessive” autophagy mediating “autophagic cell death” is perhaps one of the most controversial areas in autophagy research. It is possible that these phenomena are observed only in impaired cells, and the effects of autophagy in damaged or diseased contexts cannot be compared with physiologic stresses in otherwise healthy cells. In medical research, it is well established that essential physiologic processes are tightly regulated -- deviation in either direction would result in pathologic consequences. We propose that balance, rather than absolute levels, is the determining factor for “excessive.” Normal levels of physiologic autophagy induction may serve to tip the balance in a cell that is marginally compensated for age- or disease-related deficits in clearing autophagosomes. Likewise, increased autophagic flux, which promotes survival in transformed tumor cells capable of surviving anaerobic conditions [62], may contribute to neurodegeneration if it exceeds the ability of damaged or aged neurons to resynthesize degraded mitochondria [63]. Many neurodegenerative diseases show evidence of impaired nuclear trafficking/retention of transcriptional regulators [64], including the antioxidant response protein Nrf2 in Alzheimer’s disease [65] and the neuroprotective transcription factor CREB in Parkinson’s disease [66].
Evidence indicating that autophagy can mediate harmful effects has been largely circumstantial or pharmacologic; however, in recent years, knockout or knockdown studies of essential Atg proteins indicate that autophagy can mediate harmful effects of neurodegeneration and cell death. Cell death in apoptosis-deficient cells were shown to be mediated by autophagic mechanisms [67], leading to early speculation that autophagic cell death might only be seen under limited experimental conditions. However, similar studies in apoptosis-competent neuronal cells in 2007 suggest otherwise. RNA interference (RNAi) knockdown of Atg 5, 7 or LC3 in neuronal cells conferred partial, but significant, protection from the Parkinsonian toxin MPP+ [10] and RNAi targeting Atg5 and beclin1 protected retinal photoreceptors from hydrogen peroxide injury [68]. Neuronal cell death caused by ion channel hyperactivity in nematodes is also exacerbated by TOR inhibition and partially reversed by inactivation of Atg1, Atg6 and Atg8 homologs [12]. Oxidative toxicity in neuronal cell lines and primary dopaminergic neurons was shown to elicit a form of beclin 1-independent autophagy that was not inhibited by wortmannin or other phosphoinositide 3-kinase inhibitors [10]. Given that interactions between Bcl-2 and beclin 1 may serve to restrict autophagy to normal physiologic levels [69], this study suggests that differences in the upstream regulation of autophagy induced by physiologic and pathologic stimuli may underlie development of harmful levels of autophagy or mitophagy [70].
A number of studies also indicate that autophagy serves an essential gatekeeping role upstream of apoptosis [71-73], and mice deficient in brain Atg7 show reduced neonatal hypoxic-ischemic injury [13]. Whether or not autophagy observed in other models of acute brain injury or neurodegenerative diseases results in beneficial or harmful outcomes remains to be determined [74-81].
Degradative and biosynthetic imbalance in synaptic dysfunction
A fine balance of degradative and biosynthetic processes, and of retrograde and anterograde trafficking of vesicles and organelles, plays a key role in activity-dependent synaptic remodeling and function [82, 83]. In a model of excitotoxicity, excessive induction of autophagy is associated with axonal dystrophy, even in the absence of an altered rate of degradation [84]. Autophagy mediates neurite retraction following nerve growth factor withdrawal in cervical ganglion neurons [85]. Additionally, autophagic degeneration occurs before cell death, suggesting that autophagy plays a role in neurite degeneration independent of cell death. More recently, it has been shown that autophagy actively mediates neurite retraction induced by pathologic mutation in leucine rich repeat kinase 2 [86], a gene implicated in both familial and sporadic Parkinson’s disease, under conditions that do not cause significant cell death. Autophagic stress has also been implicated in neurons exposed to methamphetamine [87], which causes neurite dystrophy in the absence of cell death, and elevated autophagic activity is associated with decreased dopaminergic neurotransmission (Daniela Hernandez, Zsolt Talloczy, and David Sulzer, personal communication). While excess autophagy is implicated in these models, other studies show that basal autophagy is necessary for maintenance of normal axonal and synaptic structures in Purkinje neurons [88, 89].
We propose a model in which neurons are able to sustain minor increases in autophagosome production necessitated by increased age-related demand for autophagy (Figure 3). However, blockage of intracellular trafficking that accompanies inefficient AV maturation or residual aggregates in neurites may secondarily prevent neurotropic factors from traversing the axon, resulting in impaired nerve terminal to nuclear signaling. Additionally, newly formed synaptic proteins and mitochondria may by inefficiently delivered to pre- and post-synaptic areas. Beyond considerations of cargo carried by vesicular transport, it is likely that membrane recycling itself is of prime importance in maintaining proper neuritic and synaptic morphologies [88, 89]. With this in mind, we hypothesize that the overall level of autophagic flux in the cell is less important than relative rates of autophagy initiation, degradation, and recycling for maintenance of neuronal viability and the elaborate neuritic arbor essential for normal brain function.
Autophagic stress from imbalanced autophagy induction: therapeutic implications
While 3-methyladenine and wortmannin are commonly used to inhibit autophagy in experimental systems [90], more selective mechanisms to downregulate autophagy while sparing other membrane trafficking systems requiring phosphoinositide phosphates would be desirable. Moreover, blunting or partial knockdown of the autophagic response without eliminating basal activity would be necessary, as chronic functional deficiency of beclin 1 interacting proteins leads to lethal neurodevelopmental abnormalities [91]. Downregulation of beclin 1 levels during cardiac overload can serve to prevent harmful overactivation of autophagy [92]. However, these strategies may not work on all forms of pathologic autophagy [70].
Much research remains to be conducted in autophagy regulation under both physiologic and pathologic conditions, and factors that promote or alleviate autophagic stress. Nevertheless, recent studies implicating different signaling mechanisms in mitochondrially targeted injuries [10] suggest the possibility of future therapies that decrease pathologic inducers of autophagy while sparing physiologic autophagic functions. Efforts to address potential limitations in synthesis of neuroprotective factors, chaperones, mitochondria and proteins that promote trafficking, vesicular fusion and/or stabilize lysosomal function may also prove necessary.
Future Perspectives
It is clear from the discussion above that modulating autophagolysosomal function represents a promising source of potential therapies for age-related diseases and both pediatric and adult neurodegenerative diseases. The age of onset of a neurodegenerative disease may reflect the severity of the insult and rates at which imbalances in autophagolyososomal function develop. In consideration of future therapeutic challenges, different therapeutic goals may be necessary depending upon the possible mechanisms for pathology induced by insufficient or excessive autophagic flux (Table I).
Table I.
Possible therapeutic goals and situations for application.
| Goal | Situations |
|---|---|
| Promote induction of autophagy | Lack of induction due to deficits in signaling or autophagic machinery Lack of induction due to defective recognition of damaged proteins/organelles Feedback inhibition of induction from chronic autophagic stress |
| Promote lysosomal function to prevent/alleviate autophagic stress | Age- and disease-associated decline in lysosomal function Expansion of AV intermediates responsible for toxic species generation Prolonged sequestration of essential components, making them unavailable for recycling |
| Blunt or slow autophagy induction | Autophagic stress due to high levels of induction Autophagic/degradative stress overwhelming the capacity of cell to recycle membranes or to synthesize/regenerate important cellular components Autophagy induced for cell death execution (will need to balance loss of irreplacable cell against benefit of ridding organism of defective cell) |
| Enhance degradation of specific cargo without increasing overall autophagic flux | Critical components degraded through bystander effect as result of autophagy induced for other reasons Aggregate-prone proteins or damaged mitochondria in aging subject with less efficient biosynthetic capacity |
With the discovery of proteins that specifically regulate autophagy [93], a framework has been constructed for more selective modulation of autophagy. The details required for rational identification of promising targets, however, require further elucidation. Future studies may focus on identifying specific molecules that modulate each step in the autophagy pathway. Small molecules and pharmacologic agents that can more selectively modulate certain aspects of autophagic stress may also help usher in the first wave of disease-specific therapies [94, 95].
Ideally small molecule regulators would affect only certain aspects or targets of the autophagy pathway, since global inhibition or enhancement of protein turnover could be problematic. In situations with substantial aggregation, however, a global induction of autophagy may be required provided this does not outstrip the degradative capacity of the aged or diseased cell. Promoting expression of biomolecules required for both induction and clearance of autophagosomes may serve to prevent potential autophagic stress. Alternatively, transient, periodic “housecleaning“ with intervening time for the cellular systems to return to homeostasis, could conceivably serve to promote healthy aging and reduce development of symptomatic disease. In order to create such therapeutic strategies, and to gain a better understanding of autophagy regulators, further investigation into the regulation of basal and induced autophagy, the potential cross-regulating signaling networks involved, and mechanisms by which imbalances in autohpagy affect synaptic function, regenerative remodeling and neuronal survival are needed.
Identifying differences between physiological and pathological stimuli for autophagy will be important for a new wave of therapies focused on balanced correction of dysfunctional autophagy, while preserving essential basal functions. Studies in this area may involve a more complete understanding of induction and degradation steps, and factors regulating expression level of Atg proteins, such as the role of mitochondrial stress [96]. Modulation of gene expression could be one potential mode of enhancing beneficial autophagy with reduced off-target effects on other pathways.
The final, but perhaps most important, area that requires additional elucidation is determining mechanisms that can function to modulate target specificity of autophagy. Promising work in this direction includes the elucidation of specific molecular bridges between protein aggregates and the autophagy machinery. The p62/sequestosome is an ubiquitin binding protein necessary for formation of larger ubiquitinated aggregates [30]. In addition, p62 directly interacts with LC3, and is proposed to target ubiquinated aggregates to the phagophore [28, 30, 97]. An autophagy-linked FYVE protein (Alfy) and histone deacetylase 6 (HDAC6) may also be involved in targeting aggregates for degradation [58, 59, 98]. Mitochondrial signals to trigger autophagy likely include oxidative alterations, kinase signals or depolarization [10, 99-101] {Scherz-Shouval, 2007 #1903}. Such research will allow us to target aggregate prone proteins, or damaged mitochondria, while sparing the cell the demands of high levels of nonselective autophagic flux. Studies are also need to address whether defective proteins can be actively targeted in a therapeutic manner to prevent aggregates from reaching the pathologic threshold. While these areas of focus are aimed at ameliorating brain disorders, the information gained from these endeavors would be broadly applicable to cancer, cardiovascular disease, infectious and autoinflammatory disorders.
Conclusions
Autophagy is a highly regulated catabolic pathway that must balance rates of induction and degradation in order to execute its cellular roles. Disruption of the balance can lead to neurodegeneration through convergent pathways that predispose the cell to further stresses. While the roles for autophagy during neurodegeneration, stroke and other causes of neuronal loss remain elusive, recent research has highlighted autophagy as a potential therapeutic target for multiple neurological diseases. Successful therapies will target the autophagy pathway in order to maintain or restore balance to the system, allowing neurons to remain functional in the face of rising levels of stress in aging cells. In particular, moderately increased levels of autophagy induction combined with therapies to promote successful completion of autophagic recycling may be necessary in aged or diseased subjects.
Acknowledgments
The authors’ research in autophagy, mitochondria and neurodegeneration is supported by the National Institutes of Health (AG026389, NS053777, DC009120).
Executive Summary
Autophagic recycling and pathologic alterations in autophagy
- Autophagy is involved in basal recycling of long-lived proteins and organelles, and can be induced by stresses that result in unneeded, effete or damaged cellular constituents.
- Pathologic changes may involve insufficient or excessive autophagy. The effects of autophagy are context dependent, which may explain dual roles of autophagy in both neuroprotection and neurodegeneration.
- We propose that (over)induction of autophagy in a cell with relatively compromised ability to complete autophagic recycling, results in a state of “autophagic stress.”
Insufficient autophagy in neurodegeneration
- Experimental animals that are constitutively defective in autophagy develop neurodegeneration accompanied by ubiquitinated protein aggregates, indicating that basal levels of autophagy are essential for neuronal health.
- Age- and disease-associated reductions in expression of the autophagy regulatory protein beclin 1 have been reported in patient brain samples.
- Drug treatments that promote autophagy have been shown to reduce levels of aggregated proteins in several in vivo and in vitro models of neurodegeneration.
- While long term effects of activating nonselective autophagy are unknown and may prove to be therapeutically limiting, rapamycin and lithium are among clinically used compounds that can enhance degradation and reduce synthesis of proteins which form toxic oligomers in neurodegenerative diseases.
Autophagic stress due to impaired maturation/degradation
- Chronic impairment in lysosomal degradation predisposes neurons to developing autophagic stress and promotes cell death accompanied by accumulation of AVs.
- Cells from lysosomal storage disease patients show delayed clearance of starvation-induced AVs, implying inefficient maturation and degradation.
- Build up of AVs at intermediate stages of maturation results in expansion of cellular compartments involved in production of pathogenic Aβ peptide and contribute to dystrophic axonal/synaptic swellings that may interfere with function.
- Strategies to promote lysosomal function and/or slow or blunt autophagic responses to alleviate “backing up” of AV intermediates remain to be developed.
Autophagic stress due to dysregulated autophagy induction
- In experimental systems, reducing the magnitude of autophagy induction in response to oxidative, neurotoxic, hypoxic-ischemic and ion channel excitotoxic injuries confers neuroprotection, implicating overactivation of autophagy.
- Autophagy plays a Janus-faced gatekeeping role upstream of multiple neuronal cell death pathways, reducing cell death in some systems and promoting cell death in other systems.
- Recent studies suggest that normal synaptic function depends upon finely regulated levels of autophagy. While chronic impairment in autophagy causes axonal/synaptic degeneration, increased levels of autophagy also lead to neurite retraction and reduced neurotransmission.
- Beclin 1-independent autophagy in parkinsonian models indicate that regulation of damage- or cell death-associated autophagy differs from commonly studied physiologic pathways. These differences remain to be confirmed, but may be exploited to develop more selective therapeutic strategies.
Future perspectives
- Modulation of autophagy holds significant promise for future neuroprotective therapies for a wide range of acute and chronic brain disorders, although much remains to be elucidated.
- Studies to define cross-regulatory mechanisms that maintain balance between induction and completion of autophagy, and allow successful biosynthetic reutilization of raw materials, will be important.
- Small molecule therapies that selectively enhance autophagic clearance of toxic or damaged organelles and protein aggregates without causing excessive induction of nonselective autophagy represent exciting areas for further investigation.
- We predict that success of autophagy-based neuroprotection will depend upon the capacity of the cell to effectively complete degradation and mount regenerative transcriptional pathways. Therapeutic efforts to increase levels of autophagy should include consideration of trafficking, degradative, and biosynthetic reserves of the neuron in specific aging- or disease-related contexts so that potential downstream deficits can be simultaneously addressed.
References
- 1.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–91. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 2.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–32. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 4.Bandhyopadhyay U, Cuervo AM. Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp Gerontol. 2007;42:120–8. doi: 10.1016/j.exger.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 5.Farre JC, Subramani S. Peroxisome turnover by micropexophagy: an autophagy-related process. Trends Cell Biol. 2004;14:515–23. doi: 10.1016/j.tcb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–41. doi: 10.1038/sj.cdd.4401728. [DOI] [PubMed] [Google Scholar]
- 7.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 9.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 10.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–12. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 12.Toth ML, Simon P, Kovacs AL, Vellai T. Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci. 2007;120:1134–41. doi: 10.1242/jcs.03401. [DOI] [PubMed] [Google Scholar]
- 13.Koike M, Shibata M, Tadakoshi M, et al. Inhibition of Autophagy Prevents Hippocampal Pyramidal Neuron Death after Hypoxic-Ischemic Injury. Am J Pathol. 2008 doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu CT. Eaten alive: autophagy and neuronal cell death after hypoxia-ischemia. Am J Pathol. 2008 doi: 10.2353/ajpath.2008.071064. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A Role for Autophagy in the Extension of Lifespan by Dietary Restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen A, Larsen KE, Behr GG, et al. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1243–54. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- 17.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2007;4 doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 18.Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–6. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata M, Lu T, Furuya T, et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281:14474–85. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 20.Pickford F, Masliah E, Britschgi M, et al. The autophagy protein beclin 1 is reduced in early Alzheimer’s disease and regulates Abeta accumulation in vivo. J Clin Invest. doi: 10.1172/JCI33585. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 22.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 23.Iwata A, Christianson JC, Bucci M, et al. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A. 2005;102:13135–40. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia K, Hart AC, Levine B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy. 2007;3:21–5. doi: 10.4161/auto.3528. [DOI] [PubMed] [Google Scholar]
- 25.King MA, Hands S, Hafiz F, Mizushima NM, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008 doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- 26.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 27.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 28.Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei J, Fujita M, Nakai M, et al. Enhanced lysosomal pathology caused by beta-synuclein mutants linked to dementia with Lewy bodies. J Biol Chem. 2007;282:28904–14. doi: 10.1074/jbc.M703711200. [DOI] [PubMed] [Google Scholar]
- 30.Komatsu M, Waguri S, Koike M, et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–31. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar S, Krishna G, Imarisio S, Saiki S, O’Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet. 2008;17:170–8. doi: 10.1093/hmg/ddm294. [DOI] [PubMed] [Google Scholar]
- 35.Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbagen.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Shacka JJ, Klocke BJ, Shibata M, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006;69:1125–36. doi: 10.1124/mol.105.018408. [DOI] [PubMed] [Google Scholar]
- 37.Shacka JJ, Klocke BJ, Young C, et al. Cathepsin D deficiency induces persistent neurodegeneration in the absence of Bax-dependent apoptosis. J Neurosci. 2007;27:2081–90. doi: 10.1523/JNEUROSCI.5577-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jennings JJ, Jr, Zhu JH, Rbaibi Y, Luo X, Chu CT, Kiselyov K. Mitochondrial aberrations in mucolipidosis Type IV. J Biol Chem. 2006;281:39041–50. doi: 10.1074/jbc.M607982200. [DOI] [PubMed] [Google Scholar]
- 39.Kiselyov K, Jennigs JJ, Jr, Rbaibi Y, Chu CT. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–62. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchison HM, Lim MJ, Cooper JD. Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol. 2004;14:86–96. doi: 10.1111/j.1750-3639.2004.tb00502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koike M, Shibata M, Waguri S, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Settembre C, Fraldi A, Jahreiss L, et al. A Block of Autophagy in Lysosomal Storage Disorders. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 43.Walls KC, Klocke BJ, Saftig P, et al. Altered regulation of phosphatidylinositol 3-kinase signaling in cathepsin D-deficient brain. Autophagy. 2007;3:222–9. doi: 10.4161/auto.3822. [DOI] [PubMed] [Google Scholar]
- 44.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 45.Liao G, Yao Y, Liu J, et al. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol. 2007;171:962–75. doi: 10.2353/ajpath.2007.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao Y, Espinola JA, Fossale E, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–93. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- 47.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 48.Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–81. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anglade P, Vyas S, Javoy-Agid F, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 50.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 51.Ravikumar B, Acevedo-Arozena A, Imarisio S, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–6. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 52.Jackson WT, Giddings TH, Jr, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, Dalkara T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:2189–94. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- 54.Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deretic V, Singh S, Master S, et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8:719–27. doi: 10.1111/j.1462-5822.2006.00705.x. [DOI] [PubMed] [Google Scholar]
- 56.Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy. 2007;3:569–80. doi: 10.4161/auto.4776. [DOI] [PubMed] [Google Scholar]
- 57.Martin A, Joseph JA, Cuervo AM. Stimulatory effect of vitamin C on autophagy in glial cells. J Neurochem. 2002;82:538–49. doi: 10.1046/j.1471-4159.2002.00978.x. [DOI] [PubMed] [Google Scholar]
- 58.Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 59.Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rusten TE, Vaccari T, Lindmo K, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–25. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 61.Martinez-Vicente M, Talloczy Z, Kaushik S, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008 doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding WX, Ni HM, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 63.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–40. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 64.Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol. 2007;66:873–83. doi: 10.1097/nen.0b013e318156a3d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramsey CP, Glass CA, Montgomery MB, et al. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–81. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 68.Kunchithapautham K, Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007;3:433–41. doi: 10.4161/auto.4294. [DOI] [PubMed] [Google Scholar]
- 69.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 70.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–6. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 72.Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 74.Lai Y, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- 75.Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morimoto N, Nagai M, Ohta Y, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–7. doi: 10.1016/j.brainres.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 77.Diskin T, Tal-Or P, Erlich S, et al. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22:750–62. doi: 10.1089/neu.2005.22.750. [DOI] [PubMed] [Google Scholar]
- 78.Dron M, Bailly Y, Beringue V, et al. Scrg1 is induced in TSE and brain injuries, and associated with autophagy. Eur J Neurosci. 2005;22:133–46. doi: 10.1111/j.1460-9568.2005.04172.x. [DOI] [PubMed] [Google Scholar]
- 79.Simon D, Seznec H, Gansmuller A, et al. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J Neurosci. 2004;24:1987–95. doi: 10.1523/JNEUROSCI.4549-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- 81.Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–33. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 82.Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17:1561–7. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 83.Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003;206:1985–92. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- 84.Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–68. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–88. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- 86.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–60. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–6. doi: 10.4161/auto.4964. [DOI] [PubMed] [Google Scholar]
- 90.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 92.Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 94.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang L, Yu J, Pan H, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alemi M, Prigione A, Wong A, et al. Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic Biol Med. 2007;42:32–43. doi: 10.1016/j.freeradbiomed.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 98.Simonsen A, Birkeland HC, Gillooly DJ, et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–51. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 99.Kim EH, Sohn S, Kwon HJ, et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–24. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- 100.Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein homolog phosphatase is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–24. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- 101.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 102.Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 103.Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094–104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen Q, Ding Q, Thorpe J, Dohmen RJ, Keller JN. RNA interference toward UMP1 induces proteasome inhibition in Saccharomyces cerevisiae: evidence for protein oxidation and autophagic cell death. Free Radic Biol Med. 2005;38:226–34. doi: 10.1016/j.freeradbiomed.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 105.Lang-Rollin I, Dermentzaki G, Vekrellis K, Xilouri M, Rideout HJ, Stefanis L. A novel cell death pathway that is partially caspase dependent, but morphologically non-apoptotic, elicited by proteasomal inhibition of rat sympathetic neurons. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2007.05165.x. [DOI] [PubMed] [Google Scholar]