Autonomy and robustness of translocation through the nuclear pore complex: a single-molecule study (original) (raw)
Abstract
All molecular traffic between nucleus and cytoplasm occurs via the nuclear pore complex (NPC) within the nuclear envelope. In this study we analyzed the interactions of the nuclear transport receptors kapα2, kapβ1, kapβ1ΔN44, and kapβ2, and the model transport substrate, BSA-NLS, with NPCs to determine binding sites and kinetics using single-molecule microscopy in living cells. Recombinant transport receptors and BSA-NLS were fluorescently labeled by AlexaFluor 488, and microinjected into the cytoplasm of living HeLa cells expressing POM121-GFP as a nuclear pore marker. After bleaching the dominant GFP fluorescence the interactions of the microinjected molecules could be studied using video microscopy with a time resolution of 5 ms, achieving a colocalization precision of 30 nm. These measurements allowed defining the interaction sites with the NPCs with an unprecedented precision, and the comparison of the interaction kinetics with previous in vitro measurements revealed new insights into the translocation mechanism.
Introduction
The exchange of ions, small and large molecules between cytoplasm and nucleus is mediated by the nuclear pore complex (NPC), a highly symmetrical supramolecular complex traversing the nuclear envelope (NE) (for review see Peters, 2005; Lim and Fahrenkrog, 2006; Tran and Wente, 2006; Stewart, 2007). A complex octagonal cylindrical central framework with an outer diameter of 125 nm and a length of 40 nm is covered by eight cytoplasmic filaments of ∼50 nm in length and eight nuclear filaments of ∼80 nm in length, forming a basket-like structure (Beck et al., 2004). The NPC is composed of ∼30 different polypeptides designated nucleoporins, which yield a total mass of ∼125 MDa in vertebrates (Rout et al., 2000; Cronshaw et al., 2002). A large number of nucleoporins contain repetitive sequences, the phenylalanine-glycine (FG) repeats, which are intrinsically unfolded (Denning et al., 2003).
Small molecules, which do not specifically interact with the FG repeats, permeate the NPC at rates inversely related to their Stokes radius (Keminer and Peters, 1999). The transport rates are consistent with restricted diffusion through a channel within the NPC center ∼10 nm in diameter and ∼45 nm in length. The translocation of molecules, which specifically interact with the FG repeats, e.g., the transport receptors importin β or karyopherin β1 (kapβ1) and transportin 1 or karyopherin β2 (kapβ2) is facilitated (Stewart, 2007). Larger substrates, containing a nuclear localization signal (NLS), do not interact directly with the NPC but bind in the cytoplasm to transport receptors of the karyopherin/importin β family. Import complexes are translocated through the NPC and dissociate in the nucleus upon binding of RanGTP. Conversely, substrates containing a nuclear export signal (NES) form ternary complexes with a transport receptor and RanGTP in the nucleus, which are translocated through the NPC, and hydrolysis of the GTP induces their dissociation within the cytoplasm. Surprisingly, passive and facilitated transport through NPCs do not interfere with each other, suggesting that the two modes of transport are most probably spatially segregated (Naim et al., 2007).
Restricted and facilitated diffusion as well as receptor-mediated transport of NLS-containing cargo molecules through the NPC are passive, bidirectional processes (Kopito and Elbaum, 2007). Receptor-mediated transport can proceed against concentration differences, and depends on a variety of parameters such as cytoplasmic and nucleoplasmic concentration of the cargoes, and the RanGDP and RanGTP levels in the respective cellular compartments. Probably, the mechanism by which molecules migrate through the NPC is simple thermal motion (Peters, 2007; Stewart, 2007). The FG repeats are thought to play a decisive role in the exclusion of larger molecules from the nuclear interior. Simultaneously the transport of large molecules, with which the FG repeats interact by means of transport receptors, is facilitated. However, both the spatial arrangement of binding sites within the NPC and the mechanism of exclusion of unrelated large substrate molecules are still a matter of dispute (Rout and Aitchison, 2001; Frey et al., 2006; Frey and Gorlich, 2007; Lim et al., 2007; Patel et al., 2007; Weis, 2007).
Single-molecule analysis provides unique information on spatial properties and kinetic processes that are lost by averaging over large populations of unsynchronized molecules (Zlatanova and van Holde, 2006; Ishii and Yanagida, 2007; Peters, 2007). Optical far-field single-molecule microscopy using fast and sensitive CCD camera systems is particularly suited for biological applications (Schmidt et al., 1999). Single molecules are imaged as diffraction-limited spots, and their positions can be determined with a precision in the size range of a typical protein diameter by image analysis. The localization precision that can be achieved depends predominantly on the signal-to-noise ratio (SNR), and may be as small as a few nanometers under optimal conditions (Thompson et al., 2002; Ober et al., 2004). If imaging is performed fast enough, single molecule detection allows observing the movement of single molecules even in aqueous solution (Grunwald et al., 2006a). The technique has most recently been applied to analyze the movement of single receptor and cargo molecules across the NPC predominantly in digitonin-permeabilized cells (Yang et al., 2004; Kubitscheck et al., 2005; Yang and Musser, 2006).
It might be anticipated that nuclear transport would show a quite different spatio-temporal behavior in living cells compared with digitonin-permeabilized cells. This was suggested by molecular crowding and viscosity differences in a live cell compared with digitonin-permeabilized cells, where most soluble cytoplasmic components are washed out (Banks and Fradin, 2005; Hu et al., 2007). Also, transcription is almost completely halted in digitonin-permeabilized cells, which can be deduced from the change in the intranuclear dynamics of splicing factors (Grunwald et al., 2006b), and certainly results in a significant reduction in nuclear export processes. Sustained export processes, however, should interfere with nuclear import by modifying the available intra-pore binding sites, and thus presumably, translocation times.
In this study we analyzed the translocation of single transport receptors and substrates in vivo. All experiments were based on introducing the fluorescently labeled probe molecules by microinjection into the cytoplasm of living cells. We defined the respective binding site distributions and the interaction times with the pore with an unprecedented precision. We found that all examined molecules bound in a central and spatially confined region of the NPC for a time span of roughly 5–7 ms. The interaction characteristics did not depend on the specific transport pathway. Hence, nucleocytoplasmic traffic in living cells is autonomous and robust with regard to persistent simultaneous import or export, molecular crowding, and cytoplasmic viscosity.
Results
Monitoring the association of single nuclear transport receptors and cargo molecules with GFP-labeled nuclear pore complexes
The interaction of AlexaFluor 488–labeled kapα2, kapβ1, kapβ1ΔN44, kapβ2, and BSA-NLS with the NPC was studied in living HeLa cells stably expressing GFP-POM121 (Bodoor et al., 1999; Kubitscheck et al., 2005). Cells were microinjected with low concentrations of the labeled protein at room temperature. Immediately after the delivery of the probe, the equatorial plane of the nucleus was brought into focus. While illuminating a cellular region centered at the NE with 488-nm laser light at irradiance between 5.5 and 7 kW/cm2, high-speed movies were acquired at a frame rate kacq of ∼200 Hz. Complete and irreversible bleaching of the POM121-GFP fluorescence occurred within an illumination period of 5 s, corresponding to 1,000 single frames. The first 100 images of such an image sequence were used to determine the position of the NE. For this purpose these images were averaged yielding a single image showing the GFP-labeled NPCs with an excellent signal-to-noise ratio (SNR). After bleaching the dominating GFP fluorescence single AlexaFluor 488–labeled probe molecules became visible while moving within cytoplasm and nucleoplasm, and also upon binding to the NE. Several video stills from video sequences illustrating such events are shown in Fig. 1. In this figure, the average image illustrating the position of the NE was plotted in green (designated as: GFP image), and selected images acquired later in the movie are overlaid in red. Single probe molecules bound to the GFP-marked NE are indicated by white arrows. Fig. 1 A shows an example sequence of an experiment, where the import receptor kapα-AlexaFluor 488 was interacting with the NE, and Fig. 1 B shows an image series of a relatively long-lasting binding period of BSA-NLS-AlexaFluor 488. In separate control experiments, unrelated molecules (BSA-AlexaFluor 488 without NLS) did not show any significant interaction with the NE. Because all proteins mentioned above are involved in nucleocytoplasmic transport, we conclude that the observed binding was directly to NPCs. Thus, such image series contain information on the spatial position of the single fluorescent probe molecules with regard to the NPCs, as well as temporal information on the interaction duration of the molecules with the NPCs.
Figure 1.

Imaging single molecules at the NE. Single frames from a movie showing the binding of AlexaFluor 488–labeled kapα (A) and BSA-NLS (B) to the NE. Kapα is the import receptor for the import substrate BSA-NLS. The NE was marked by a GFP-POM121 conjugate stably expressed in HeLa cells, which is part of the NPC. The GFP signal (green) was obtained by averaging the first 100 frames of each movie. Frame rate was ∼200 Hz. After bleaching of the dominant GFP fluorescence, single molecules of import receptors or import substrate became visible later in the same movies. Several consecutive images showing single AlexaFluor 488–kapα and AlexaFluor 488–BSA-NLS (red) are overlaid to the GFP-POM121 average. Single molecule signals at the NPC are indicated by white arrows. Images of single molecules were filtered by the Spot Enhancing filter (1.8) (Sage et al., 2005) in ImageJ (Abramoff et al., 2004) and converted to 8 bit; furthermore, brightness and contrast was adjusted for each individual frame. The image of the NE was converted to 8 bit and filtered using a rolling ball (10-pixel width). NE (green) and SMT (red) data were merged as RGB.
Association periods of transport receptors and transport substrates at the NPC
Individual molecules of kapα2, kapβ1, kapβ1ΔN44, kapβ2, and BSA-NLS, labeled with AlexaFluor 488 were observed for varying numbers of frames directly at the NPCs. These numbers, n, correlated with the length of the individual binding events and were translated into binding times, τ, with τ = n/kacq. These data were used to establish decay curves N(τ) quantifying the number of the probe molecules, which were still bound at a specific site after time τ. When plotted in this manner the ordinate value of the first data point corresponds to the total number of evaluated molecules. The decay curves allowed the quantification of the dissociation of molecules from the NE. For kapβ1, kapα2, BSA-NLS, and kapβ1 ΔN44 the dissociation curves exhibited binding times τkapβ1 = 6.6 ± 0.2 ms, τkapα = 7.5 ± 0.8 ms, τBSA-NLS = 6.2 ± 0.3 ms, and τkapβ1ΔN44 = 11.8 ± 0.6 ms (Fig. 2, A–D, full lines; see also Table I). Similar dwell times were recently determined in digitonized cells for the transport receptors p10/NTF2 and kapβ2, and also for an NLS substrate and kapβ1 (Yang and Musser, 2006). A small deviation from a mono-exponential decay behavior was seen in the semi-logarithmic plots of Fig. 2. Although this deviation was negligible for kapα2 and BSA-NLS, a second decay component comprising a fraction of ∼10% of the observed binding events was obtained for kapβ1 and kapβ1ΔN44 (Table I). This was in agreement with a previous study, where binding times of single transport receptors were examined in digitonin-permeabilized cells (Kubitscheck et al., 2005). Also in this system binding events were observed, which lasted significantly longer than expected from an exponentially distributed binding duration.
Figure 2.

Binding durations of single molecules at the NE. The dwell time of a molecule could be determined by counting the number of frames the molecule was observed at the NE. These data were transformed into a dissociation curve as described in the Dwell time measurements section of Materials and methods. A fit to a mono-exponential decay function satisfactorily described all datasets except kapβ2 (E), which required a bi-exponential fit. Data are shown as black dots, logarithmic scales were used to pronounce the mono-exponential decays. Black lines indicate fit results. Slight deviations from mono-exponential decays were detectable. The corresponding fits yielded decay times τkapβ1 = 6.6 ± 0.2 ms (A), τkapα = 7.5 ± 0.8 ms (B), τBSA-NLS = 6.2 ± 0.3 ms (C), and τkapβ1ΔN44 = 11.8 ± 0.6 ms (D), accordingly. The Kapβ2 (E) fit yielded τkapβ2,1 = 4.6 ± 0.1 ms and for the second component τkapβ2,2 = 103 ± 6 ms with amplitudes of Aτ1 = 90% and Aτ2 = 10%.
Table I. Binding site distribution and dwell times of import receptors and model cargo.
| τdwell [ms] | Fraction [%] | xc [nm] | FWHM [nm] | Nobservation | Ntraces | |
|---|---|---|---|---|---|---|
| BSA-NLS | 6.2 ± 0.3 | 100 | −13 ± 1 | 140 ± 5 | 293 | 75 |
| Kapα | 7.5 ± 0.8 | 100 | −6 ± 2 | 160 ± 5 | 387 | 136 |
| kapβ1 | 6.6 ± 0.2a | 100a | −10 ± 2 | 160 ± 5 | 402 | 110 |
| kapβ1ΔN44 | 11.8 ± 0.6b | 100b | −8 ± 1 | 90 ± 2 | 244 | 60 |
| kapβ2 | 4.6 ± 0.1 | 90 ± 1 | 5 ± 2 | 150 ± 5 | 305 | 97 |
| 103 ± 6 | 10 ± 3 |
The mutant kapβ1ΔN44 has a RanGTP binding deficiency (Gorlich et al., 1996). In the association analysis this mutant showed a nearly twofold prolonged dwell time at the NPC compared with wt kapβ1. A notably extended dwell time was expected, as Ran-GTP binding is required for the termination of the import event, i.e., the dissociation of kapα2 from kapβ1 and release of the import substrate from the nuclear pore into the nucleoplasm (Gorlich et al., 1996).
A monoexponential fit failed to describe the decay curve of kapβ2. A two-component fit was required for a good description of the data, and yielded a short dwell time, τkapβ2,1 = 4.6 ± 0.1 ms, and a longer time, τkapβ2,2 = 103 ± 6 ms (Fig. 2 E, Table I). The requirement of two decay components for kapβ2 was in accordance with our previous study in digitonin-permeabilized cells, where we found extended interaction times of more than 100 ms for 2.5% of the observed events (Kubitscheck et al., 2005). The slow kapβ2-dissociation was more frequent in live cells (10%) compared with digitonized cells (2.5%).
Distances between binding sites of single probe molecules and the NE can be measured with a precision in the nanometer range
Image sequences as those shown in Fig. 1 were used to determine both the position of the NE and the various used single fluorescent probe molecules as previously described (Kubitscheck et al., 2005). As an example, an overlay of the GFP image (background image), the corresponding deduced position of the NE (green dotted line), and the binding sites of several individual kapβ1ΔN44-AlexaFluor 488 molecules (red dots) deduced from one movie sequence is shown in Fig. 3 A. Approximately 30 distinct spots of GFP fluorescence were visible along the NE section of 13 μm length. Assuming a focal depth of 0.8 μm, this would correspond to a pore density of ∼3 nuclear pores per μm2, which is in agreement with our previously published data (Kubitscheck et al., 1996; Kues and Kubitscheck, 2002). During the 5-s observation time ∼10 out of the 30 discernable NPCs were visited by kapβ1ΔN44 molecules. These kapβ1ΔN44 molecules were almost exclusively observed at sites of strong POM121-GFP fluorescence corresponding to labeled NPCs, whereas at NE sections devoid of GFP fluorescence almost no kapβ1ΔN44 molecules were observed. This suggests that all NPCs are labeled by POM121-GFP. In Fig. 3 B the position of the NE (full line) and those of the observed individual kapβ1ΔN44-AlexaFluor 488 molecules (open circles) were plotted in a single graph. From such data the individual distances from the NPCs were determined as the shortest separation distances between the sites of receptor binding and the nearest point on the curved line indicating the NE. The distribution of these distances for kapβ1ΔN44 molecules binding to the NE is shown in Fig. 3 C. Negative values indicate a position at the cytoplasmic face of the NPC, positive values correspond to positions on the nucleoplasmic face. The distribution has a very symmetrical shape with a mean at x = −3 ± 1 nm. The full width at half maximum (FWHM) of the binding site distribution for kapβ1ΔN44 of this cell is very low with only 80 ± 2 nm.
Figure 3.

Single kapβ1 ΔN44 molecules at the NE. For a single cell, all fitted positions (open red circles) were overlaid with the image of the NE. The estimate of the NE position was indicated by a green line in A and B. kapβ1ΔN44 molecules were preferentially observed at the GFP-labeled NPCs, while molecules were only very rarely observed within unlabeled regions of the NE. The fitted positions of both the NE and the single molecules were plotted to scale in B. The individual distances of the kapβ1ΔN44 molecules from the NE were calculated as the shortest distances between the fitted position of the single molecule and the nearest point on the fit through the NPC signals marking the NE. The resulting distance distribution of kapβ1ΔN44 molecules to the NE is shown in C. The position of the NE was defined as zero. Then, negative values described a single molecule position at the cytoplasmic face of the NPC, positive values at the nucleoplasmic face. A fit to a Gaussian yielded a maximum position of the distribution at −3 ± 1 nm with a width (FWHM) of 80 ± 2 nm.
Transport receptors and the model transport substrate bind primarily to the cytoplasmic filaments and in the central domain of the NPC
For all used proteins we determined the distances between observation sites and the nearest point of the NE. These distances were accumulated from all acquired measurements, and plotted in binding site histograms (Fig. 4). As above, negative values indicate a cytoplasmic position, and positive values correspond to sites on the nucleoplasmic face of the NPC. These histograms represent a novel tool to determine NPC binding sites with an unprecedented localization precision. In our previous work using two-color fluorescence, the analysis was largely limited by a lower colocalization precision caused by the two detectors and by sample instabilities (Kubitscheck et al., 2005).
Figure 4.

Binding sites of single molecules at the NE. For all single molecule positions the shortest distance to the NE was calculated, and the results accumulated in distance distribution histograms. Fits of Gaussian functions to the histograms yielded the center position (indicated by an arrow) and width of the distributions. As above, negative distances corresponded to the cytoplasmic side of the NE, positive values to the nucleoplasmic side. (A) kapβ1; (B) kapα2; (C) BSA-NLS; (D) kapβ1ΔN44; and (E) kapβ2. Results of the data analysis are given in Table I.
Fig. 4 shows that all examined molecules bind preferably to the NPCs. The enhanced binding extends over a distance of 150 nm along the axial symmetry axis from about −80 to +70 nm with an approximate Gaussian shape. The peak positions were located at the very center of the NPC, slightly on the cytoplasmic side of the NPC (with the exception of kapβ2; see Table I). As shown previously, the tips of the cytoplasmic filaments extend ∼75 nm from POM121-GFP (Kubitscheck et al., 2005), whereas the closure of the nuclear cage is presumably located at a distance of 80 nm from POM121-GFP (Beck et al., 2004). Therefore, the observed binding sites corresponded exactly to the anticipated region of the NPC, with a preference for the cytoplasmic face. Noticeably, the binding site distribution of the mutant kapβ1 ΔN44 deficient in RanGTP binding was significantly narrower than all other distributions. This may be partly due to the longer dwell time resulting in a better mean SNR of this dataset.
Discussion
Nucleocytoplasmic transport is accomplished and regulated by receptor proteins, which bind to the cargo and interact with components of the nuclear pore complexes. In the last few years it was demonstrated that single-molecule microscopy is an excellent and precise tool to study details of cellular nanomachines like the NPC (Peters, 2007). Extensive single-molecule measurements of transport receptors and model substrates have been performed in digitonin-permeabilized cells because this approach avoided the additional complication of in vivo experiments. However, a number of important aspects were certainly absent from in vitro studies compared with the situation in living cells. First, digitonin permeabilization causes a loss of most soluble cytosolic and smaller nuclear components, largely reducing overall viscosity and what is known as “molecular crowding” (Ellis, 2001). Because molecular crowding is known to modify binding constants and dynamics (Banks and Fradin, 2005; Hu et al., 2007), it can be anticipated that a complex process such as nucleocytoplasmic transport should behave differently in vivo than in plasma membrane–permeabilized cells. It can also be assumed that such differences should be aggravated by the fact that in permeabilized cells most intranuclear processes are ceased, whereas in living cells processes such as transcription and RNA processing are active; e.g., in experiments analyzing the intranuclear dynamics of the splicing factor U1 snRNP we recently detected clear differences between in vitro and in vivo situations (Kues et al., 2001; Grunwald et al., 2006b). Sustained transcription and RNA processing will result in continuous nuclear export of mRNA. Export of the usually quite bulky ribonucleoprotein particles could possibly massively impede nuclear import (Naim et al., 2007). Despite the anticipated differences between permeabilized and living cells, the dwell times of all examined molecules were in the same range as those which were recently determined in digitonized cells for the transport receptors p10/NTF2 and kapβ2 in our laboratory (Kubitscheck et al., 2005), and also for kapβ1 and a NLS-2xGFP model import substrate by others (Yang et al., 2004; Yang and Musser, 2006). The examined transport receptors, however, belong to quite different transport receptor families. Here we demonstrate a significant robustness of the translocation process with regard to the specific interaction molecular pattern. Musser and coworkers showed that the duration of import substrate interaction with the NPC was a function of the concentration of kapβ1 in permeabilized cells, and was in the range between 1 and 8 ms for free kapβ1 concentrations between 15 and 0.1 μM (Yang and Musser, 2006). In living cells we determine dwell times for kapα2, kapβ1, and BSA-NLS of ∼7 ms (Table I). Our finding suggests low concentrations of free kapβ1 in living cells. Only an import-deficient kapβ1 mutant, kapβ1ΔN44, and kapβ2 show a clearly extended interaction time as discussed in the following two paragraphs. All observed dissociation kinetics are predominantly mono-exponential with a minor component showing a greater binding duration in some cases. The fraction of the latter is small (<15%). This is in accordance with previous in vitro measurements. Our results argue for a transport mechanism without major interference between import and export processes, which can be explained by pores being alternatively involved in export or import. It is possible that exporting pores are temporarily unavailable for import, or that indeed two different NPC species exist, one for import or one for export. An alternative scenario would be that NPCs have independent pathways along the NPC channel for import and export (Tran and Wente, 2006). Our measurements, which have been taken under conditions that were as undisturbed as possible, support the notion that molecular crowding caused by ongoing export or further intrinsic cellular components within NPCs in live cells has little effect on translocation times.
The mutant kapβ1ΔN44 has a RanGTP binding deficiency (Gorlich et al., 1996). In the association time analysis this mutant showed a 1.8-fold extended dwell time at the NPC compared with wild-type kapβ1 (Fig. 2, Table I). A significantly extended binding time might have been expected, as Ran-GTP binding was suggested to be required for the termination of the import event, i.e., the dissociation of kapα2 from kapβ1 and release of the import substrate from the NPC into the nucleoplasm (Gorlich et al., 1996; Kutay et al., 1997; Sun et al., 2008). However, the ternary complex was not “waiting” for dissociation at the nucleoplasmic exit of the NPC. Rather, our finding is compatible with the assumption that intra-pore movements of transport receptors are persistent random walks along the NPC axis, meaning that kapβ1 and kapβ1ΔN44 are free to move within the pore. Kapβ1ΔN44, however, could not exit the pore at its nucleoplasmic face due to its incapability to bind RanGTP. A Monte Carlo simulation (for details see the online supplemental material, available at http://www.jcb.org/cgi/content/full/jcb.200806173/DC1) of this situation revealed that the residence time within a pore with one “blocked” end, compared with a pore of identical length with both ends open, is indeed prolonged by a factor of 1.75. In this simulation we assumed that the trajectory length inside the pore alone defined the residence time and any further attractive interactions were neglected. Consideration of additional attractive interactions modified the numerical value of prolongation, but the trend of an approximate factor of 2 was maintained. Hence, our result is in good agreement with the notion that RanGTP is required to trigger the release of the ternary import complex on the nucleoplasmic face of the NPC. Certainly, the simple relation between dwell time within the pore and number of “open ends” is only valid under the simplifying assumptions mentioned above. A detailed and sophisticated simulation of binding and transport efficiency of NPCs was recently performed (Zilman et al., 2007).
In some cases (Fig. 2, B, D, and E) mono-exponential dissociation kinetics are not perfectly consistent with the decay data. This observation is in accordance with our previous in vitro results, where we detected small fractions (1–6%) of binding events that were significantly longer than 100 ms, but at that time no attempt was undertaken to analyze these events further. We would like to point out that even a mono-exponential decay does not necessarily imply that the dissociation mechanisms on the nucleoplasmic and cytoplasmic face of the NPC were identical. Generally, only the reciprocal value of the sum of the two—possibly differing—dissociation rates can be determined as a dissociation time constant. Hence, import and export of the transport receptors may well follow different mechanisms with different dwell times, although experimentally only a single dissociation time constant can be monitored. In the present analysis, only kapβ2 could not be analyzed at all assuming mono-exponential decay (Fig. 2). Here, 10% of the binding events showed a decay time of 100 ms. Previously, we assumed that translocation attempts through clogged NPCs may result in significantly prolonged interactions. Possibly, such events might be the origin of the small, second fraction in the binding duration of kapβ2. The fact that in the in vivo experiments we do observe only moderate increase in the overall number of prolonged binding events compared with experiments in digitonin-permeabilized cells suggests that a strong interference between import and export processes does not occur.
NPCs were marked by POM121-GFP, and the transport receptors and substrates were labeled with the green fluorescent AlexaFluor 488. The latter were monitored after complete bleaching of the GFP fluorescence in the same fluorescence channel. No additional chromatic corrections or alignments had to be performed, which optimized the colocalization precision. We estimate the error for a single distance measurement to be ≈ 30 nm (for discussion of errors, see Materials and methods). The distances between the observation sites of the various transport factors to the GFP reference signal were plotted in a histogram representation (Fig. 4). All histograms have an approximate Gaussian shape, and therefore their maximum positions and widths were quantified by fitting Gaussian functions to the data. It is noteworthy that all transport receptors, namely kapβ1, kapα2, and kapβ2, show almost identical widths corresponding to a Gaussian standard deviation of σexp = 68 nm (σexp = FWHM/2.35). A similar distribution was previously obtained for p10/NTF2 (Kubitscheck et al., 2005). These data argue for a related environment for each involved import pathway because NTF2/p10, kapβ1, and kapβ2 belong to independent receptor families and hence pathways. Obviously, the already-discussed robustness of nuclear transport also holds true for the biochemical translocation pathway. As discussed in Materials and methods, the observed width σexp represented the true spread of binding sites, σb, convoluted with the colocalization precision, σcoloc ≈ 30 nm. The true standard deviation of the binding site distribution can therefore roughly be estimated by
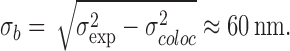
Furthermore, the center of the binding site distribution was in all cases in the range of ±10 nm from the reference site of POM121-GFP. Because the position of POM121 is known to be quite centrally within the NPC (Soderqvist and Hallberg, 1994), transport factors are observed only in the central region of the NPC. Binding occurred neither predominantly in the cytoplasmic nor in the nucleoplasmic filaments because in those cases a wider or even asymmetrical distribution would have been observed. For the same reason, no indication of a putative affinity gradient along the NPC was obtained (Ben-Efraim and Gerace, 2001). The distribution of the import cargo BSA-NLS-Alexa488 was slightly narrower than for the import receptors, corresponding to a binding site distribution width of σb,BSA ≈ 50 nm. Also, the shape of the distribution was more compact, and less blurred compared with those of kapβ1, kapα2, and kapβ2, indicating that the pure geometric distributions of binding sites for ternary import complexes is most realistically described by the histogram of Fig. 4 C.
The binding site distribution of kapβ1ΔN44 was clearly more confined to the center of the pore with σb,kapβ1ΔN44 ≈ 25 nm. The very small value of σb,kapβ1ΔN44 indicates a restricted intra-pore motion of kapβ1ΔN44. A free intra-NPC diffusion would extend over a larger distance (Ribbeck and Gorlich, 2002; Yang et al., 2004). Görlich and coworkers detected gold-labeled kapβ1ΔN44 arrested at the NPC, however, ∼40 nm from the central plane on the nucleoplasmic side of the NPC (Görlich et al., 1996). In contrast to these reports, Lyman et al. found a complex of GST-IBB-gold and kapβ1ΔN44 accumulating at a distance of 30 nm from the central plane on the cytoplasmic side of the NPC (Lyman et al., 2002). Presumably, details of the methodological approach and data analysis are responsible for these variations. Frey et al. (2006) performed their localization following microinjection into the cytoplasm of live Xenopus oocytes after 2 h incubation, whereas Lyman et al. examined the loci of the transport substrate 30 min after incubation in digitonin-permeabilized HeLa cells. Our in vivo experiments were performed only several minutes after microinjection into the cytoplasm of POM121-GFP expressing HeLa cells. Furthermore, we did not use bulky labels nor bulky substrates.
Numerous problems in biology and biophysics have been studied by single-molecule techniques in the past few years, but the application to living cell systems is still in its infancy (Zlatanova and van Holde, 2006; Ishii and Yanagida, 2007). Single-molecule microscopy and single particle tracking permit a completely new and fascinating insight into biological dynamics (Moerner, 2007). Here we demonstrate that the translocation of proteins across the NPC is autonomous with regard to the respective intracellular physical environment, and robust with regard to the specific biochemical pathway and simultaneously ongoing import–export processes. For the first time, fluorescence single-molecule microscopy provides a direct, real-time insight into molecular processes in living cells with near molecular resolution.
Materials and methods
Cell culture and microinjection
A HeLa cell line stably expressing the GFP-conjugate of POM121 was provided by Brian Burke (University of Florida, Gainesville, FL). HeLa cells were grown in DMEM supplemented with 10% FCS. For live cell analysis, cells were seeded in microscopy dishes (MatTek) up to a confluence of 50%, and examined at room temperature. Microinjection of the probe molecules was performed with an Eppendorf injection and micromanipulation setup using an injection time of 0.8 s at an injection pressure of 100 hPa, and a holding pressure 8 hPa. Before final use of protein probes in microinjection experiments, all probes were diluted and centrifuged at 4°C, 14,000 rpm for 20 min in transport buffer (50 mM Hepes/KOH, pH 7.3, 110 mM potassium acetate, 5 mM sodium acetate, 2 mM magnesium acetate, 1 mM EGTA, and 2 mM DTT). The concentration of labeled protein in the injection medium was 60 nM for all experiments. Experiments using different types of molecules were performed sequentially to avoid uncertainties about the observed protein species. Different import factors or substrates were never used on the same cell sample.
Preparation of import factors and substrate
His-tagged kapα2 and GST-kapβ1 were prepared according to Moroianu et al. (1995). His-tagged karyopherin β2 was purified according to Chook et al. (2002). His-tagged kapβ1ΔN44 was purified as described by Gorlich et al. (1996). BSA was purchased from Sigma-Aldrich, and conjugated to AlexaFluor 488 (Invitrogen) and SV40 large T antigen peptide (Bachem California, Inc.) as described previously (Newmeyer and Wilson, 1991).
GST pull-down assay
Functionality of kapα2, kapβ1, and kapβ1ΔN44 after covalent fluorescence labeling with AlexaFluor 488 was confirmed by GST pull-down assays. For this purpose a recombinant import substrate consisting of the SV40-NLS fused to GST was immobilized on Glutathione beads, and then used in a pull-down assay. Therefore, the GST-Sepharose beads were gently washed four times in transport buffer at 4°C, and 150 μl of beads per sample were centrifuged at 4°C for 3 min at 7,000 rpm. The loaded beads were incubated with either the labeled or unlabeled kapβ1 and unbound proteins were removed by gently washing with transport buffer several times. After adding kapα2 and kapα2-488, the beads were incubated for 30 min at 4°C on a rotator and then washed gently four times with transport buffer at 4°C. Beads with bound proteins were dissolved in sample buffer (200 mM Tris-HCl, pH 6.8, 6% (wt/vol) SDS, 15% glycerin, 10% β-mercaptoethanol, and 0.05% [wt/vol] Bromphenol blue) and resolved on a 10% polyacrylamide gel. As a control, the binding of unlabeled kapα2/kapβ1 and kapα2/kapβ1ΔN44 to GST-NLS was analyzed.
Single-molecule microscopy
Single-molecule tracking experiments were done with a custom-built single-molecule microscope based on an Axiovert 100TV (Carl Zeiss, Inc.) equipped with a 63X NA 1.4 oil immersion objective lens (for details, see Siebrasse et al., 2007). GFP and AlexaFluor 488 fluorescence was excited by an Ar+ laser emitting at 488 nm. Laser illumination was switched on only during image acquisition by means of an acousto-optical tunable filter. For single-molecule image acquisition an iXon DV 860 BI camera (Andor Technologies) in combination with a 4X magnifier was used. Single-molecule imaging was started 5 min after microinjection of the probes into the cytoplasm to allow cells to recover. Cell recovery after microinjection was monitored by observing the cellular morphology by digitally contrast-enhanced bright field imaging. Then, the green fluorescent signal of the nuclear envelope was used to set the focus at the equatorial plane. Data acquisition was started immediately. After bleaching of the dominant GFP fluorescence, single-molecule signals became perceptible in the cytoplasm, nucleoplasm, and at the NE. Generally, 2,000 frames were recorded in a single movie with an integration time of 5 ms, and a frame rate of kacq = 198 Hz.
Image processing of video images
Images were filtered by the Spot Enhancing filter (Sage et al., 2005) assuming a signal width of 1.8 pixels in ImageJ (Rasband, W.S., ImageJ, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/). Filtered and raw image movies were aligned site by site and used for visual identification of signal containing frames. Raw data sequences that contained single-molecule data were separated and later loaded for tracking in Diatrack 3.0 (Semasopht), a commercial image processing program for the identification and localization of single particle signals and trajectories (Vallotton et al., 2003). All tracks within a distance of 13 pixels from the image border were discarded. Tracks were stored as text files and analyzed using Origin 7.5 (Microcal).
Distance determination from NE
To determine the position of the NE, we fitted the POM-121 signal in Origin 7.5 (Microcal) using a user-defined one-dimensional Gauss fit with a linear ramp (Kubitscheck et al., 2005). The result, a list of NE positions with pixel resolution, was used to interpolate the shape of the NE using the corresponding spline fit in Origin. A total of 5,000 data points were generated to represent each NE. For each single molecule position we searched the nearest position of this punctuated NE representation and defined the distance to this point as the distance to the NE. For NE sections, where no GFP signal was observed the NE was linearly interpolated based on the nearest GFP signals available. In such sections a higher uncertainty with regard to the colocalization precision was given. However, single molecules were almost exclusively observed at labeled NPCs. Hence, we assume the precision of this method to be within the limits given in Kubitscheck et al. (2005). The distance values were binned with a 20-nm bin width. This value was chosen according to the localization precision of our data. Histogram data were smoothed using a nearest neighbor running average. Finally, fits to Gaussians were used to determine the center position and the width of the distributions.
Generally, the width of the distance distributions (Fig. 4) was determined by three factors. The first was the limited localization precision of a single molecule signal, which is the consequence of a finite SNR (Thompson et al., 2002), together with the localization precision for the NE signal. We estimate this colocalization precision to be in the range of 30 nm (Kubitscheck et al., 2005); therefore, the performed averaging of the histogram values for distances over ±20 nm did not smear the outcome. The second factor was the true topology of the different binding sites along the NPC axis. The third factor was the number of molecules, which were observed on their pathways toward and away from the NPC. Because we did only observe very few molecules diffusing to or away from the NPC as already stated in error discussion of the binding durations, the third possible contribution to a broadening of the binding site distribution was negligible. Qualitatively, the binding site distributions approximated Gaussians. Therefore, we used Gaussian functions to find the maxima of the distributions and to quantify their widths in terms of the full width at half maximum (FWHM) and standard deviation σ, with FWHM = 2.35 σ.
Dwell time measurements
To determine the binding durations at the NPC we counted the number of subsequent images of single molecules at a specific NE site within a distance of ±225 nm from the NE. This number, n, was the length of a translocation trajectory. Then, n was translated into a binding time, τ, by τ = n/kaq. The values of τ were used to calculate a decay curve N(τ) giving the number of particles, which were still bound after time τ. The resulting datasets were plotted on semi-log scales and fitted with exponential decay functions.
Systematic errors in the determination of the binding duration of the analyzed proteins might be introduced by the particle detection routine, blinking of the conjugated fluorophore, the finite resolution of our time measurement as given by the single frame integration time, and the fact that some traces were observed where molecules were diffusing to, or away from the NPC rather than being bound for the total time of observation. To estimate each of these possible contributions we carefully examined data and controls. Particle detection was done half-automated by screening the movies visually, and only using such frames for particle tracking that unambiguously contained a signal. All fitted positions were double-checked against raw data. By this procedure we excluded errors due to false positive particles found by the automated signal detection algorithm in Diatrack. A total of two traces showed blinking (see Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200806173/DC1), which was defined as a one-frame gap in a binding sequence. To confirm this low frequency of blinking events we imaged single proteins on a glass surface under comparable illumination conditions as used for in vivo imaging. Due to the lack of oxygen reducing systems and less scattering of light in case of the glass surface than inside a cell, resulting in a higher effective illumination intensity, we assumed this to be a worst-case condition. Molecules transiently attaching to the glass surface could be easily imaged for at least 10 frames without blinking events. Molecules attached to the surface could be observed for up to 50 frames showing only one blinking event. We concluded that we were limited neither by photostability nor blinking of the dyes.
Monte Carlo simulation of binding times
The impact of inhibited release for kapβ1ΔN44 on the nucleoplasmic face of the NPC in comparison to kapβ1 was tested with a Monte Carlo simulation. We generated a sequence of random numbers, ri, that were normally distributed around 0 with a standard deviation σ = 1. Such a distribution corresponds to the jump distance distribution of a particle in a one-dimensional diffusion process with 2Dt = σ2. The Gaussian distributed random numbers were used to construct one-dimensional trajectories {x(i)} that were defined by x(I + 1) = x(i) + ri. A “pore” was defined in the interval from −L ≤ x ≤ L, and the starting position of the particle was set to x(0) = −L − 2. The randomly moving particles could therefore enter the pore initially from left, and leave it on either side. The number of positions, which were occupied in a sequence within the pore were registered as “binding time”. We compared two different situations: (A) particles could enter and leave the pore on either side, or (B) particles could enter and leave only on the left side (x = −L) because they were reflected at x = L. We varied the ratio between σ and L from 1:2.5 to 1:20 to check for possible effects of the discretization. 2,500 binding events were used to construct binding time histograms, which were fitted by single exponential decay functions. Two exemplary histograms showing the binding times for situations A and B for a σ/L = 10 are shown in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200806173/DC1). The ratio of the binding times was 1.8 in this case. A significantly more detailed and sophisticated simulation of binding and transport efficiency of NPCs was recently performed by Zilman et al. (2007).
Online supplemental material
Photostability of labeled protein (Fig. S1) was tested by immobilization of single proteins on a glass surface, which were imaged under similar conditions as used in the live cell experiments. A Monte Carlo simulation on the effect of release inhibition of the import complex from the NPC (Fig. S2) was performed to estimate the effect on import complex dwell times at the NPC. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200806173/DC1.
Supplementary Material
[Supplemental Material Index]
Acknowledgments
We thank Marion Schmidt and Jan-Peter Siebrasse for critical reading of the manuscript, and Simone Scholl for help with live cell transport measurements of import factors.
Grants from the Deutsche Forschungsgemeinschaft to D. Grünwald, U. Kubitscheck, and R. Peters are gratefully acknowledged. This work was also supported by grant 1 R01 GM071329-01 of the National Institutes of Health (to R. Peters).
T. Dange and D. Grünwald contributed equally to this paper.
T. Dange's present address is Albert Einstein College of Medicine, Department of Biochemistry, 1300 Morris Park Ave., Bronx, NY, 10461.
D. Grünwald's present address is Albert Einstein College of Medicine, Anatomy and Structural Biology, 1300 Morris Park Ave., Bronx, NY, 10461.
A. Grünwald's present address is Albert Einstein College of Medicine, Department of Medicine, Nephrology, 1300 Morris Park Ave., Bronx, NY, 10461.
Abbreviations used in this paper: FG, phenylalanine-glycine; FWHM, full width at half maximum; NE, nuclear envelope; NPC, nuclear pore complex; SNR, signal-to-noise ratio.
References
- Abramoff, M.D., P.J. Magelhaes, and S.J. Ram. 2004. Image processing with ImageJ. Biophotonics International. 11:36–42. [Google Scholar]
- Banks, D.S., and C. Fradin. 2005. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 89:2960–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, M., F. Forster, M. Ecke, J.M. Plitzko, F. Melchior, G. Gerisch, W. Baumeister, and O. Medalia. 2004. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science. 306:1387–1390. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim, I., and L. Gerace. 2001. Gradient of increasing affinity of importin beta for nucleoporins along the pathway of nuclear import. J. Cell Biol. 152:411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodoor, K., S. Shaikh, D. Salina, W.H. Raharjo, R. Bastos, M. Lohka, and B. Burke. 1999. Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J. Cell Sci. 112:2253–2264. [DOI] [PubMed] [Google Scholar]
- Chook, Y.M., A. Jung, M.K. Rosen, and G. Blobel. 2002. Uncoupling Kapbeta2 substrate dissociation and ran binding. Biochemistry. 41:6955–6966. [DOI] [PubMed] [Google Scholar]
- Cronshaw, J.M., A.N. Krutchinsky, W. Zhang, B.T. Chait, and M.J. Matunis. 2002. Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158:915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning, D.P., S.S. Patel, V. Uversky, A.L. Fink, and M. Rexach. 2003. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA. 100:2450–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, R.J. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597–604. [DOI] [PubMed] [Google Scholar]
- Frey, S., and D. Gorlich. 2007. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell. 130:512–523. [DOI] [PubMed] [Google Scholar]
- Frey, S., R.P. Richter, and D. Gorlich. 2006. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 314:815–817. [DOI] [PubMed] [Google Scholar]
- Gorlich, D., N. Pante, U. Kutay, U. Aebi, and F.R. Bischoff. 1996. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 15:5584–5594. [PMC free article] [PubMed] [Google Scholar]
- Grunwald, D., A. Hoekstra, T. Dange, V. Buschmann, and U. Kubitscheck. 2006. a. Direct observation of single protein molecules in aqueous solution. Chemphyschem. 7:812–815. [DOI] [PubMed] [Google Scholar]
- Grunwald, D., B. Spottke, V. Buschmann, and U. Kubitscheck. 2006. b. Intranuclear binding kinetics and mobility of single native U1 snRNP particles in living cells. Mol. Biol. Cell. 17:5017–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Z., J. Jiang, and R. Rajagopalan. 2007. Effects of macromolecular crowding on biochemical reaction equilibria: a molecular thermodynamic perspective. Biophys. J. 93:1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, Y.Y., and T. Yanagida. 2007. How single molecule detection measures the dynamic actions of life. HFSP J. 1:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keminer, O., and R. Peters. 1999. Permeability of single nuclear pores. Biophys. J. 77:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito, R.B., and M. Elbaum. 2007. Reversibility in nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA. 104:12743–12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck, U., P. Wedekind, O. Zeidler, M. Grote, and R. Peters. 1996. Single nuclear pores visualized by confocal microscopy and image processing. Biophys. J. 70:2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck, U., D. Grunwald, A. Hoekstra, D. Rohleder, T. Kues, J.P. Siebrasse, and R. Peters. 2005. Nuclear transport of single molecules: dwell times at the nuclear pore complex. J. Cell Biol. 168:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kues, T., and U. Kubitscheck. 2002. Single molecule motion perpendicular to the focal plane of a microscope: Application to splicing factor mobilitydynamics within the cell nucleus. Single Molecules. 3:218–224. [Google Scholar]
- Kues, T., A. Dickmanns, R. Luhrmann, R. Peters, and U. Kubitscheck. 2001. High intranuclear mobility and dynamic clustering of the splicing factor U1 snRNP observed by single particle tracking. Proc. Natl. Acad. Sci. USA. 98:12021–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay, U., E. Izaurralde, F.R. Bischoff, I.W. Mattaj, and D. Gorlich. 1997. Dominant-negative mutants of importin-beta block multiple pathways of import and export through the nuclear pore complex. EMBO J. 16:1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, R.Y., and B. Fahrenkrog. 2006. The nuclear pore complex up close. Curr. Opin. Cell Biol. 18:342–347. [DOI] [PubMed] [Google Scholar]
- Lim, R.Y., B. Fahrenkrog, J. Koser, K. Schwarz-Herion, J. Deng, and U. Aebi. 2007. Nanomechanical basis of selective gating by the nuclear pore complex. Science. 318:640–643. [DOI] [PubMed] [Google Scholar]
- Lyman, S.K., T. Guan, J. Bednenko, H. Wodrich, and L. Gerace. 2002. Influence of cargo size on Ran and energy requirements for nuclear protein import. J. Cell Biol. 159:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner, W.E. 2007. New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. USA. 104:12596–12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu, J., G. Blobel, and A. Radu. 1995. Previously identified protein of uncertain function is karyopherin alpha and together with karyopherin beta docks import substrate at nuclear pore complexes. Proc. Natl. Acad. Sci. USA. 92:2008–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim, B., V. Brumfeld, R. Kapon, V. Kiss, R. Nevo, and Z. Reich. 2007. Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J. Biol. Chem. 282:3881–3888. [DOI] [PubMed] [Google Scholar]
- Newmeyer, D.D., and K.L. Wilson. 1991. Egg extracts for nuclear import and nuclear assembly reactions. Methods Cell Biol. 36:607–634. [DOI] [PubMed] [Google Scholar]
- Ober, R.J., S. Ram, and E.S. Ward. 2004. Localization accuracy in single-molecule microscopy. Biophys. J. 86:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, S.S., B.J. Belmont, J.M. Sante, and M.F. Rexach. 2007. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 129:83–96. [DOI] [PubMed] [Google Scholar]
- Peters, R. 2005. Translocation through the nuclear pore complex: selectivity and speed by reduction-of-dimensionality. Traffic. 6:421–427. [DOI] [PubMed] [Google Scholar]
- Peters, R. 2007. Single-molecule fluorescence analysis of cellular nanomachinery components. Annu. Rev. Biophys. Biomol. Struct. 36:371–394. [DOI] [PubMed] [Google Scholar]
- Ribbeck, K., and D. Gorlich. 2002. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 21:2664–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M.P., and J.D. Aitchison. 2001. The nuclear pore complex as a transport machine. J. Biol. Chem. 276:16593–16596. [DOI] [PubMed] [Google Scholar]
- Rout, M.P., J.D. Aitchison, A. Suprapto, K. Hjertaas, Y. Zhao, and B.T. Chait. 2000. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148:635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage, D., F. Neumann, F. Hediger, S. Gasser, and M. Unser. 2005. Automatic tracking of individual fluorescence particles: Application to the study of chromosome dynamics. IEEE Trans. Image Process. 14:1372–1383. [DOI] [PubMed] [Google Scholar]
- Schmidt, T., P. Hinterdorfer, and H. Schindler. 1999. Microscopy for recognition of individual biomolecules. Microsc. Res. Tech. 44:339–346. [DOI] [PubMed] [Google Scholar]
- Siebrasse, J.P., D. Grunwald, and U. Kubitscheck. 2007. Single-molecule tracking in eukaryotic cell nuclei. Anal. Bioanal. Chem. 387:41–44. [DOI] [PubMed] [Google Scholar]
- Soderqvist, H., and E. Hallberg. 1994. The large C-terminal region of the integral pore membrane protein, POM121, is facing the nuclear pore complex. Eur. J. Cell Biol. 64:186–191. [PubMed] [Google Scholar]
- Stewart, M. 2007. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 8:195–208. [DOI] [PubMed] [Google Scholar]
- Sun, C., W. Yang, L.C. Tu, and S.M. Musser. 2008. Single-molecule measurements of importin α/cargo complex dissociation at the nuclear pore. Proc. Natl. Acad. Sci. USA. 105:8613–8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, R.E., D.R. Larson, and W.W. Webb. 2002. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E.J., and S.R. Wente. 2006. Dynamic nuclear pore complexes: life on the edge. Cell. 125:1041–1053. [DOI] [PubMed] [Google Scholar]
- Vallotton, P., A. Ponti, C.M. Waterman-Storer, E.D. Salmon, and G. Danuser. 2003. Recovery, visualization, and analysis of actin and tubulin polymer flow in live cells: A fluorescent speckle microscopy study. Biophys. J. 85:1289–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis, K. 2007. The nuclear pore complex: oily spaghetti or gummy bear? Cell. 130:405–407. [DOI] [PubMed] [Google Scholar]
- Yang, W., and S.M. Musser. 2006. Nuclear import time and transport efficiency depend on importin beta concentration. J. Cell Biol. 174:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, W., J. Gelles, and S.M. Musser. 2004. Imaging of single-molecule translocation through nuclear pore complexes. Proc. Natl. Acad. Sci. USA. 101:12887–12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilman, A., S. Di Talia, B.T. Chait, M.P. Rout, and M.O. Magnasco. 2007. Efficiency, selectivity, and robustness of nucleocytoplasmic transport. PLoS Comput. Biol. 3:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova, J., and K. van Holde. 2006. Single-molecule biology: what is it and how does it work? Mol. Cell. 24:317–329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Material Index]