Microglia Induce Neurotoxicity via Intraneuronal Zn2+ Release and a K+ Current Surge (original) (raw)
. Author manuscript; available in PMC: 2008 Oct 4.
Published in final edited form as: Glia. 2008 Jan 1;56(1):89–96. doi: 10.1002/glia.20592
Abstract
Microglial cells are critical components of the injurious cascade in a large number of neurodegenerative diseases. However, the precise molecular mechanisms by which microglia mediate neuronal cell death have not been fully delineated. We report here that reactive species released from activated microglia induce the liberation of Zn2+ from intracellular stores in cultured cortical neurons, with a subsequent enhancement in neuronal voltage-gated K+ currents, two events that have been intimately linked to apoptosis. Both the intraneuronal Zn2+ release and the K+ current surge could be prevented by the NADPH oxidase inhibitor apocynin, the free radical scavenging mixture of superoxide dismutase and catalase, as well as by 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron(III) chloride. The enhancement of K+ currents was prevented by neuronal overexpression of metallothionein III or by expression of a dominant negative (DN) vector for the upstream mitogen-activated protein kinase apoptosis signal regulating kinase-1 (ASK-1). Importantly, neurons overexpressing metallothionein-III or transfected with DN vectors for ASK-1 or Kv2.1-encoded K+ channels were resistant to microglial-induced toxicity. These results establish a direct link between microglial-generated oxygen and nitrogen reactive products and neuronal cell death mediated by intracellular Zn2+ release and a surge in K+ currents.
Keywords: zinc, oxidation, nitration, Kv2.1, potassium channel, apoptosis signaling kinase-1
INTRODUCTION
Microglia, rapidly activated in response to neuronal injury, are critically important in promoting neurodegeneration in many CNS disorders (Bessis et al., 2007; Block et al., 2007; Colton et al., 2000, 2005; Wojtera et al., 2005). In addition to cytokines, activated microglia can release neurotoxic levels of nitric oxide (·NO) and superoxide (O2·−), respectively, generated by inducible nitric oxide synthase (iNOS) and NADPH oxidase (Colton and Gilbert, 1987; Li et al., 2005; Mander and Brown, 2005). Peroxynitrite (ONOO−), rapidly and favorably formed by the cogeneration of · NO and O2·− (Espey et al., 2002), can trigger or exacerbate neuropathological injury (Bal Price et al., 2002; Beckman et al., 1990; Keynes and Garthwaite, 2004). At physiological pH, ONOO− becomes protonated and undergoeshomolytic cleavage to generate the protein tyrosine nitrating species nitrogen dioxide (·NO2) as well the highly oxidizing hydroxyl radical (·OH; Schopfer et al., 2003). In addition, peroxynitrite can react with CO2 to generate ONOOCO2, leading to ·NO2 and ·CO3 radicals, also via homolytic cleavage (Schopfer et al., 2003). However, it has been demonstrated that ONOO− can readily traverse membrane lipids at rates faster than its decomposition pathways (Marla et al., 1997). Thus, unlike O− 2· or ·OH, ONOO− can diffuse cellular dimension distances, providing the reactive species it generates widespread access to internal cellular structures (Lee et al., 1998; Marla et al., 1997).
In neurons, exogenous ONOO− applied under physiological conditions induces the liberation of Zn2+ from intracellular binding sites, which, in turn, initiates a cell death cascade mediated by the activation of the mitogen-activated protein kinase (MAPK) p38 (Zhang et al., 2004). In addition, ONOO− application also initiates a p38-dependent enhancement of voltage-dependent K+ currents (Bossy-Wetzel et al., 2004; Pal et al., 2004), a process that has been tightly linked to neuronal apoptosis (McLaughlin et al., 2001; Redman et al., 2007; Yu et al., 1997). Enhanced K+ currents enable cytosolic K+ efflux, necessary for the completion of the cell death cascade (Bortner and Cidlowski, 2004; Yu, 2003). Consistent with this, blocking K+ efflux following injury can halt neuronal apoptosis in a large number of experimental models (Yu, 2003; Yu et al., 1997). However, despite a pronounced K+ current surge in cortical neurons following exposure to ONOO donors (Pal et al., 2004), neurotoxicity could not be averted by preventing K+ channel function (Zhang et al., 2004). This observation raises the possibility that ONOO− donors cannot mimic potential physiological sources of the reactive species (Espey et al., 2002) and, consequently, overwhelm the affected neurons, such that K+ efflux is not necessary for the cell death process. Here, we used a co-culture model to investigate whether activated microglia, under conditions more akin to a pathophysiological insult in the brain, could initiate a cell death pathway in cortical neurons that was characterized by intraneuronal Zn2+ release and, especially, by a K+ current surge. Our results do indicate that reactive oxygen and nitrogen species originating from activated microglia induces neuronal death via a process that is fully dependent on Zn2+ release and an enhancement of K+ currents.
MATERIALS AND METHODS
Cell Culture
Cortical cultures were prepared from embryonic, day 16 Sprague-Dawley rats as previously described (Hartnett et al., 1997). Briefly, cortices were dissociated, and the resultant cell suspension was adjusted to 600,000 cells/well (six-well tissue culture plates containing five, 11-mm poly-L-ornithine-treated coverslips/well) in a growth medium composed of a volume-to-volume mixture of 80% Dulbecco’s modified minimal essential medium (MEM), 10% Ham’s F12-nutrients, 10% bovine calf serum (heat-inactivated, iron-supplemented; Hyclone, Logan, UT) with 25 mM HEPES, 24 U/mL penicillin, 24 μg/mL streptomycin, and 2 mM L-glutamine. Cultures were maintained at 37°C, 5% CO2, and the media was partially replaced on a monday–wednesday–friday schedule. Glial cell proliferation was inhibited after 2 weeks in culture with 1–2 μM cytosine arabinoside, after which the cultures were maintained in growth medium containing 2% serum and without F12-nutrients. Cultures were utilized at 18–22 days in vitro (DIV) and contained ~20% neurons (Rosenberg and Aizenman, 1989). In these cultures, neuronal somata and proximal processes are phase bright and protrude above a phase dark, flat astrocyte layer that covers distal processes (Harris and Rosenberg, 1993). Immortalized rat brain microglial cells (Cheepsunthorn et al., 2001) were generously supplied by J. Connor (Pennsylvania St. University, Hershey, PA). Microglia were maintained in Dulbecco’s modified MEM supplemented with 10% heat-inactivated fetal bovine serum, and plated in trans-well inserts (Corning, Corning, NY) at a density of 0.5 ×106 cells/well for 24 h prior to activation (Li et al., 2005). For the Zn2+ imaging and electrophysiological studies, trans-well inserts containing microglia were placed on top of neuron-containing coverslips in 24-well plates immediately prior to microglial activation. To ensure a robust and consistent activation of microglia, 10 U/mL interferon-γ (IFN-γ, Chemicon, Temecula, CA) and 1 μg/mL lipopolysaccharide were added directly into the well inserts (Duport and Garthwaite, 2005). Chemical activation of this microglial cell line leads to activation of both iNOS and NADPH oxidase in less than 30–60 min (Cheepsunthorn et al., 2001; Qian et al., 2007). The insert-containing co-cultures were then immediately transferred to the incubator and maintained in the dark at 37°C and 5% CO2 for the duration of the exposure (see later) to minimize the light-dependent consumption of microglial-generated NO by cell culture media components (Keynes et al., 2003).
Intracellular Zinc Measurements
After a 60-min exposure to activated microglia, trans-well inserts were removed and cortical cultures, in the absence of microglia, were thoroughly rinsed and loaded with the Zn2+-sensitive fluorescent reporter Fluozin-3 AM (5 μM, Molecular Probes, Carlsbad, CA) for 30 min in buffered solution (144 mM NaCl, 3 mM KCl, 10 mM HEPES, 5.5 mM glucose, with 5 mg/mL bovine serum albumin; pH 7.3). Coverslips were then transferred to a recording chamber (Warner, Hamden, CT) mounted on an inverted epifluorescence microscope and superfused with MEM, supplemented with 25 mM HEPES and 0.01% BSA. Images were acquired with 490 nm excitation light using a computer-controlled monochromator (Polychrome II, TILL Photonics, Martinsried, Germany) and a CCD camera (IMAGO, TILL Photonics). After baseline images were acquired, free intracellular Zn2+ was chelated with 20 μM tetrakis-(2-pyridylmethyl)ethylenediamine (TPEN). The relative Zn2+ fluorescence for all neuronal cell bodies in a single field (n = 5–30) was determined by subtracting the fluorescence signal after TPEN application from the baseline signal (Δ_F_TPEN). Zn2+ signals emanating from supporting astrocytes are minimal under these conditions, as these cells appear to buffer intracellular Zn2+ more effectively than neurons (Dineley et al., 2000).
Electrophysiology
After a 60-min exposure to activated microglia, trans-well inserts were removed and cortical cultures were returned to the incubator for an additional 3 h, a time-point by which we have observed pronounced K+ current surges in neurons undergoing apoptosis (McLaughlin et al., 2001). Following this time, cultures were rinsed thoroughly and transferred to a recording chamber. All recordings were performed from neurons at room temperature using whole cell recordings, with 2–3 MΩ patch electrodes as previously described (McLaughlin et al., 2001). The extracellular recording solution consisted of 115 mM NaCl, 2.5 mM KCl, 2.0 mM MgCl2, 10 mM HEPES, 10 mM D-glucose, pH 7.2; 0.25 μM TTX was added. The electrode solution consisted of (in mM) 120 K-gluconate, 11 EGTA, 10 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 0.22 ATP; pH 7.2. Currents were amplified with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA), filtered at 2 kHz, and digitized at 10 kHz. Potassium currents were evoked with 80-ms voltage steps from −70 mV in 10-mV increments. For analysis, steady-state current amplitudes were measured at + 10 mV and normalized to cell capacitance. Series resistance was compensated in all cases (~80%).
Neuronal Transfection
Neurons were transfected with either apoptosis signal-regulating kinase-1 dominant negative (DN) vector (ASK1 DN; gift from H. Ichijo, Tokyo Medical University, Japan), metallothionein III (MT-III), or a DN truncated mutation of Kv2.1 (gift from J. Nerbonne, Washington University, St. Louis, MO) in 18–22 DIV neurons using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as described by Pal et al. (2003). Transfection rates for neurons in this type of culture are normally around 1% (Santos and Aizenman, 2002), but can be as high as 5% (unpublished). For electrophysiological recordings, neurons were also transfected with a plasmid encoding enhanced green fluorescent protein (eGFP). For toxicity assays, neurons were co-transfected with a luciferase-expressing vector (Boeckman and Aizenman, 1996; Pal et al., 2003). The MT-III plasmid was constructed by amplifying full-length cDNA from rat brain. The primers used were as follows: sense, agaagcttgccaccatggaccctgagacctgccc; antisense, gaggatcctggcagcagctgcatttct. The MT-III cDNA obtained was inserted into an eGFP-expressing vector. Transfections were performed 24 h prior to activated microglial exposure.
Toxicity Assay
Cell death was measured using a luciferase activity assay (Boeckman and Aizenman, 1996; Pal et al., 2003). Twenty-four hours after transfecting neurons with a luciferase expressing vector plus or minus a vector of interest, microglia (50,000 cells/mL) were plated directly on top on neurons and activated for 60 min as described previously. Trans-well inserts were not utilized under these conditions to minimize expenses, since individual neurons did not have to be identified for recordings (i.e., Zn2+ measurements or electrophysiology). Cell viability was assayed 48 h later on transfected neurons as previously described (Pal et al., 2003). An overnight exposure to staurosporine (0.5 μM) or NMDA (200 μM) in the absence of microglia was used as a positive control for maximal neuronal cell death. We also took advantage of the fact that neuroprotective manipulations of neurons could be performed without affecting microglial function. As such, the overexpression of DN vectors for either the p38 upstream MAPK apoptosis signal regulating kinase-1 (ASK-1), or Kv2.1, could be directed to neurons prior to co-culture and activation of microglia. These conditions were important to control since some of the signaling pathways triggered in neurons during cell death are also involved within microglia to promote their activation, including both p38 phosphorylation and changes in K+ channel function (Kaushal et al., 2007; Koistinaho and Koistinaho, 2002).
Immunocytochemistry
Cortical cultures were examined for the presence of 3-nitrotyrosine under control conditions or after exposure to activated microglia (Mander and Brown, 2005). Ninety minutes after microglia exposure as described earlier, neurons were fixed for 10 min in 4% paraformaldehyde. Following a 5-min wash in phosphate buffered saline (PBS), neurons were treated with 0.3% triton in PBS, washed again, and blocked in 1% bovine serum albumin. Cultures were then incubated overnight with 3.3 μg/mL of antinitrotyrosine monoclonal antibody (Upstate, Lacke Placid, NY) at 4°C. A FITC-conjugated secondary antibody (Sigma-Aldrich, St. Louis, MO) was used to detect cells labeled with the primary antibody.
RESULTS
Activated Microglia Induce an Increase in Intraneuronal Zn2+
Neurons previously exposed to microglia activated with LPS/IFN-γ (see Materials and Methods) exhibited a significant increase in TPEN-sensitive Zn2+ fluorescence (Δ_F_TPEN) when compared with untreated control cells (Figs. 1 and 2). This increase in Δ_F_TPEN was not evident in neurons treated for 60 min with LPS/IFN-γ in the absence of microglia, or in neurons exposed for 60 min to resting microglia (Fig. 2A). To determine the origin of increased cytosolic Zn2+ concentration ([Zn2+]i), we added a cell impermeable Zn2+ chelator Ca2+-EDTA while neurons were exposed to activated microglia. Under these conditions, we still observed a significant increase in Δ_F_TPEN, suggesting that influx of extracellular Zn2+ is not necessary for the increase in [Zn2+]i (Fig. 2B). We did note, however, that even in control neurons the overall Zn2+ fluorescence in Ca2+-EDTA exposed cells was generally lower in intensity compared with neurons that remained in a normal buffer. This may have resulted from the removal of exchangeable Zn2+ pools normally available to neurons (Frederickson et al., 2002). Nonetheless, as [Zn2+]i was still substantially elevated in microglia-exposed cells in the presence of Ca2+-EDTA, we conclude that measurable changes in Zn2+ fluorescence are primarily due to Zn2+ release from intracellular sources (Aizenman et al., 2000).
Fig. 1.
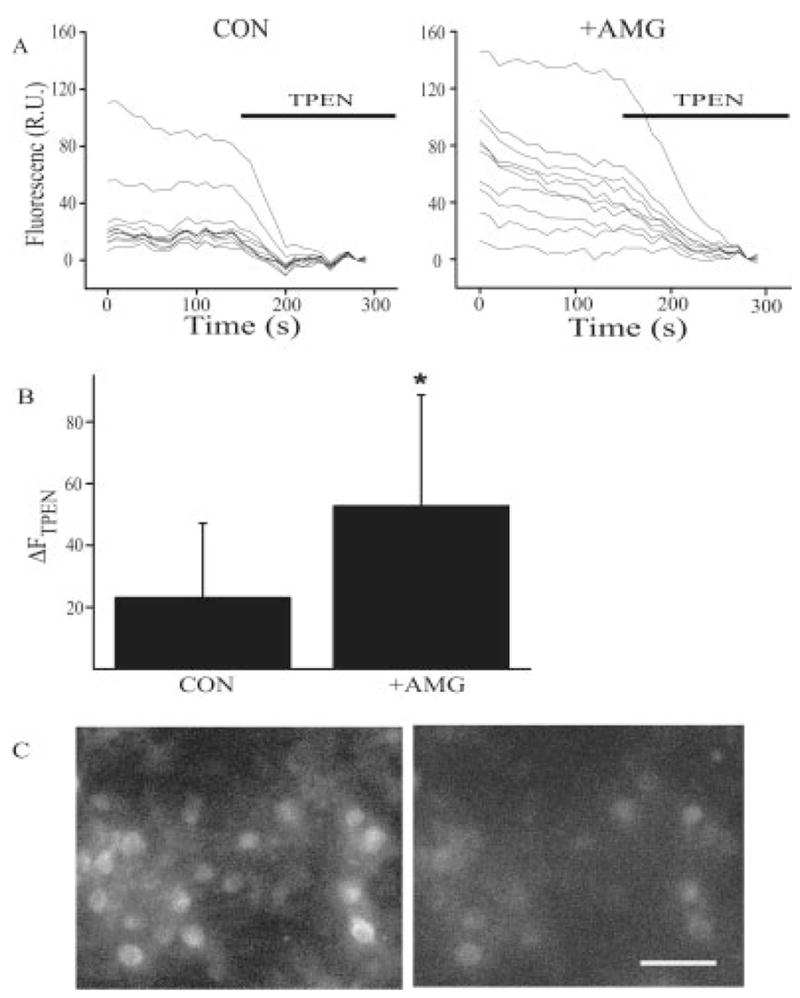
Intraneuronal Zn2+ accumulation as a result of activated microglia. (A) Fluorescence traces from individual neurons before and after addition of the Zn2+ chelator TPEN (20 μM). Compared with control cells, neurons previously exposed to activated microglia (AMG) have higher baseline Zn2+ fluorescence and greater quenching by TPEN. (B) Average (mean ± SD) TPEN-sensitive Zn2+ fluorescence (Δ_F_TPEN) from the control neuron-containing coverslip shown earlier (n = 9 cells) and the neuronal culture exposed to activated microglia (n = 9 cells; P < 0.05; _t_-test). (C) Examples of fluorescence images of rat cortical neurons loaded with 5 μM fluozin-3 AM following a 60-min exposure to activated microglia (10 U/mL IFN-γ + 1 μg/mL LPS), before (left) and after (right) treatment with 20 μM TPEN. The picture shown is representative of the range in responses observed in a typical coverslip. Bar: 60 μm.
Fig. 2.

Properties of microglial-induced intraneuronal Zn2+ release. (A) Pooled (mean ± SEM) TPEN-sensitive Zn2+ fluorescence (Δ_F_ TPEN) from control neurons, neurons exposed to microglia (MG) activated with LPS/IFN-γ, neurons exposed to LPS/IFN-γ alone or to quiescent microglia (n = 13–18 coverslips per group, P < 0.05; ANOVA, Dunnett). (B) Δ_F_TPEN in control and activated microglia-exposed cortical neurons in the presence of 1 mM Ca-EDTA to chelate extracellular Zn2+ (n = 8–11 coverslips per group, P < 0.01; t_-test). (C) Δ_F TPEN in neurons exposed to activated microglia in the absence (n = 10) or presence of the NADPH oxidase inhibitor apocynin (APO, 1 μM) (n = 10), SOD/CAT (SOD 500 U/mL, catalase 100 U/mL) (n = 9) or FeTPPS (5 μM) (n = 10) (P < 0.05; ANOVA, Dunnett). (D) Immunostaining of tyrosine nitration in rat cortical neurons. Top panels, phase contrast images of control neurons (a) and neurons exposed to activated microglia (b). Bottom panels, fluorescent images of the same fields shown earlier demonstrating that 3-nitrotyrosine immunoreactivity is virtually nonexistent in control neurons (c) whereas extensive labeling is observed in neurons previously exposed to activated microglia (d). Bar: 20 μm.
To characterize the origin of the reactive species responsible for intraneuronal Zn2+ release, selective scavengers and inhibitors of microglial-generated free radicals were used. When the O− 2 scavenging mixture of superoxide dismutase/catalase was added to the neuronal culture medium during activated microglia exposure, Δ_F_TPEN was attenuated compared with neurons exposed to activated microglia alone (Fig. 2C). Similarly, Δ_F_ TPEN measured after exposure to apocyanin-treated activated microglia was also significantly decreased, suggesting that NADPH oxidase was the primary source of O− 2 production (Fig. 2C). Because NADPH-derived microglial O− 2 contributes to ONOO− formation (Li et al., 2005), neurons were also treated with 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron(III) chloride (FeTPPS) during activated microglial exposure. FeTPPS is widely believed to be an ONOO− decomposition catalyst with minimal superoxide dismutase activity, and does not appear to complex with NO (Lee et al., 1998; Misko et al., 1998). Following this treatment, we observed a significant reduction of the liberation of intraneuronal Zn2+ caused by activated microglia (Fig. 2C). On the basis of these results, we suggest that activated microglia produce oxygen and nitrogen reactive species, which causes an increase in [Zn2+]i in co-cultured neurons. Importantly, evidence that the nitrating species ·NO2 was generated in our system was confirmed by the presence of 3-nitrotyrosine immunoreactivity in neurons exposed to activated microglia (Fig. 2D).
Activated Microglia Trigger a Surge in Voltage-Gated Potassium Currents in Neurons
Oxygen and nitrogen-derived reactive species induce intracellular Zn2+ release and enhance voltage-gated K+ current during apoptosis (Aizenman et al., 2000; Bossy-Wetzel et al., 2004; McLaughlin et al., 2001; Pal et al., 2004, 2006). Therefore, we investigated whether microglial-generated reactive products would also induce a surge in neuronal K+ currents. K+ current densities in neurons exposed to activated microglia were significantly larger than the current densities recorded from control neurons (Figs. 3 and 4). Furthermore, this current enhancement could be prevented by apocyanin or FeTPPS (Fig. 4A). To determine whether the microglia-mediated increase in [Zn2+]i caused K+ current enhancement via the p38 upstream MAPK ASK-1 (Aras and Aizenman, 2005), neurons were transfected with metallothionein-III (MT-III), to bind excess intracellular Zn2+, or a DN vector for ASK-1. We observed that microglia-exposed neurons expressing either MT-III or ASK-1 DN exhibited K+ current densities similar to control cells (Fig. 4B). Likewise, both empty vector-expressing neurons and untransfected cells behaved similarly when exposed to activated microglia. These results demonstrate that an increase in [Zn2+]i triggered by activated microglia is capable of inducing a key step in the neuronal cell death process, namely an ASK-1-mediated K+ current surge.
Fig. 3.
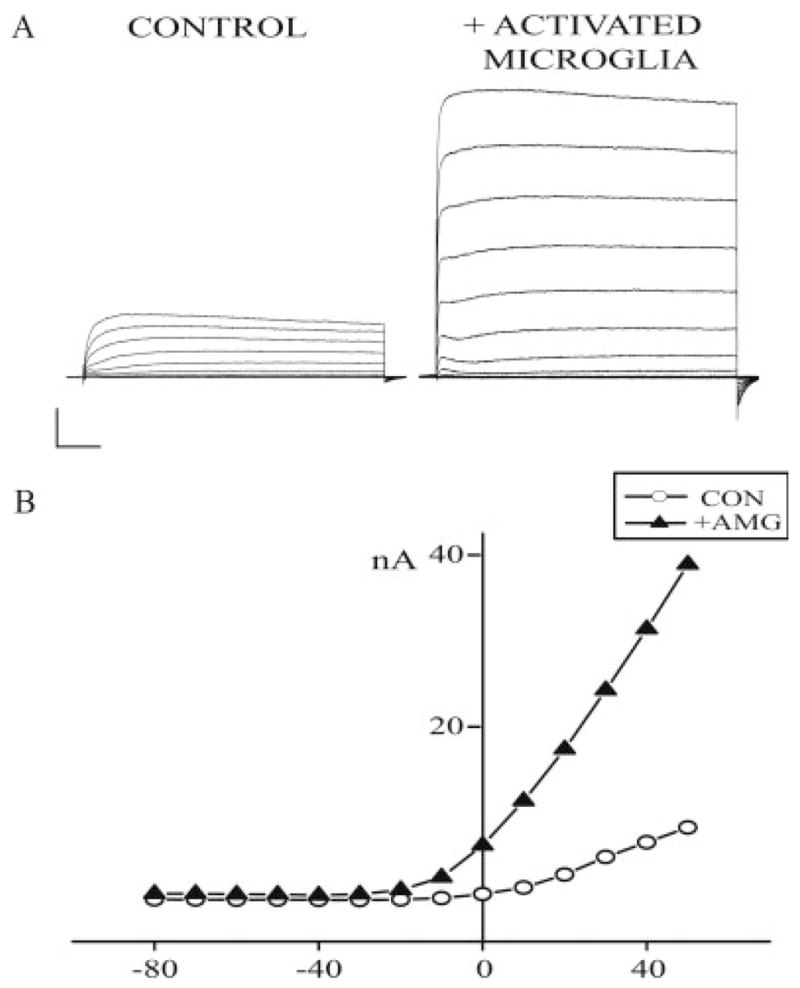
Exposure to activated microglia induces a voltage-gated K+ current surge in neurons. (A) Representative K+ currents evoked by a series of 10-mV incremental voltage steps to + 70 mV, from a holding potential of −70 mV in a control neuron (left) and a neuron previously exposed to activated microglia (right). Calibration: 25 ms, 5 nA. (B) Steady-state current–voltage relationship from traces shown in A.
Fig. 4.
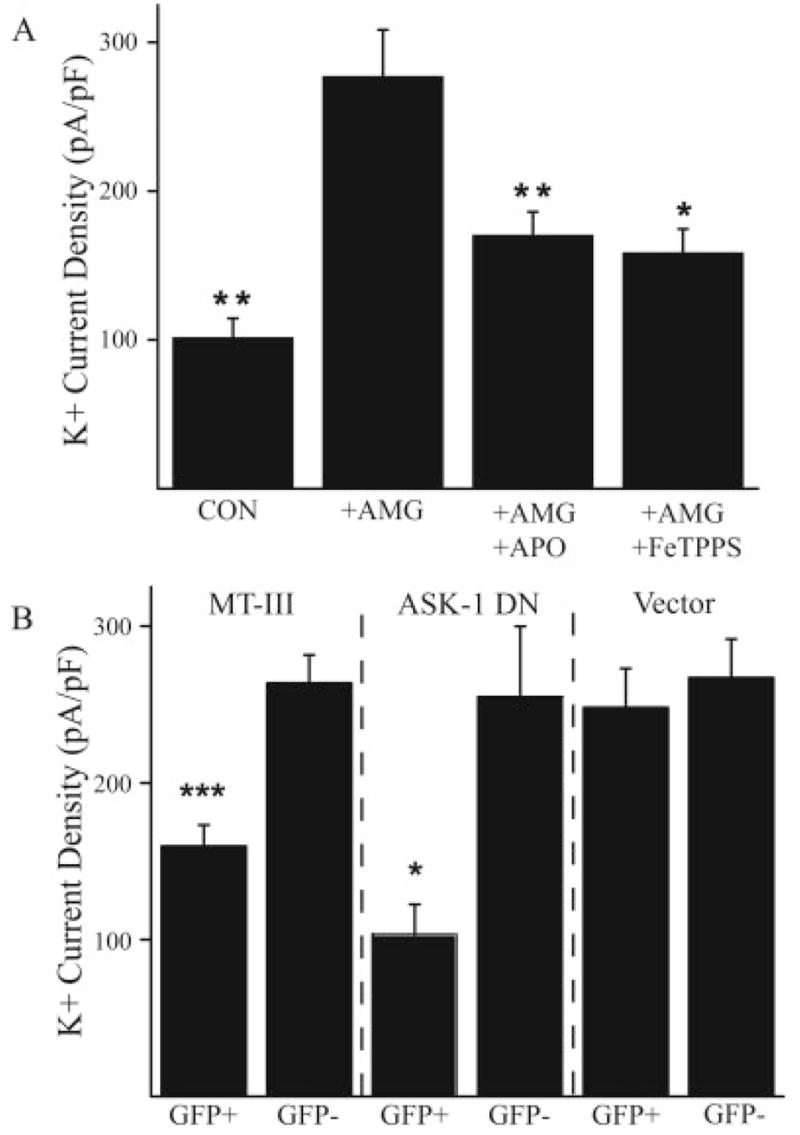
Molecular components of the K+ current surge mediated by microglia. (A) Mean (±SEM) current densities (single voltage step to 10 mV) in control neurons (n = 29, P < 0.01), and in neurons exposed to activated microglia in the absence (n = 37) or presence of apocyanin (APO, 1 μM) (n = 22, P < 0.01) or FeTPPS (50 μM, n = 13, P < 0.05); **P < 0.01, ANOVA, Dunnett. (B) Mean (±SEM) current densities (single voltage step to 10 mV) comparing neurons overexpressing either MT-III (n = 13, P < 0.001), ASK-1 DN(n = 9, P < 0.05) or an empty eGFP-expressing vector (n = 10) to untransfected neurons on the same coverslip (n = 9 for each group); ***P < 0.001; _t_-test.
Microglial-Induced Neuronal Cell Death
We hypothesized that reactive species produced by activated microglia, capable of increasing neuronal [Zn2+]i and producing an ASK1-dependent K+ current surge, would cause neuronal cell death via this pathway (Bossy-Wetzel et al., 2004; McLaughlin et al., 2001). A cell viability assay was thus employed to investigate the mechanism of microglial-mediated neurotoxicity. We first observed that activated microglial-induced neuronal death could be prevented by apocyanin (Fig. 5A), confirming the role for NADPH oxidase in this process (Block et al., 2007). FeTPPS appeared to interfere with the luciferase viability assay. However, treating co-cultures with the iNOS inhibitor 1,400 W effectively attenuated microglia-mediated neurotoxicity (Fig. 5A). The fact that inhibition of either NADPH oxidase or iNOS was sufficient to halt neuronal death strongly suggests that microglial-derived reactive oxygen and nitrogen species are responsible for mediating neuronal damage.
Fig. 5.

Molecular components of microglial neurotoxicity. (A) Mean (±SEM) viability (expressed as % control) in neurons exposed to activated microglia in the absence (n = 9) or presence of apocyanin (1 μM, n = 3) or 1,400 W (25 μM, n = 6) (P < 0.01; ANOVA, Dunnett). (B) Mean (±SEM) viability (expressed as % control) in control neurons (n = 10) and in neurons overexpressing metallothionein-III (MT-III) (n = 3, P < 0.01), ASK-1 DN (n = 4, P < 0.05) or Kv2.1 DN (n = 3, P < 0.05); *,**P < 0.05, 0.01, ANOVA, Dunnett.
Finally, we investigated the molecular progression of microglial-mediated neuronal death by neuronal overexpression of either MT III, ASK-1 DN, or Kv2.1 DN (Fig. 5B). A nonpharmacological approach was necessary to investigate the progression of this pathway, as inflammatory microglia also exhibit activation of p38 MAP kinase and voltage-gated K+ currents, two of the key signaling components proposed in our neuronal death pathway (Fordyce et al., 2005). Overexpressing metallothionein-III effectively increased neuron viability, indicating that the increased availability of a high affinity Zn2+ buffer was sufficient to rescue neurons from the microglial-triggered cell death process. We also observed that interfering with ASK-1 function, required for Kv2.1-mediated K+ current surge (Aras and Aizenman, 2005), attenuated cell death. Finally, overexpression of a DN vector for Kv2.1, the K+ channel responsible for mediating neuronal apoptosis in cortical, midbrain dopaminergic, and cerebellar granule neurons (Jiao et al., 2007; Pal et al., 2003; Redman et al., 2006), was also highly neuroprotective. Taken together, these results demonstrate that microglial-derived reactive species induce neuronal cell death via a process that involves the release of Zn2+ from intracellular sources, ASK-1 activation, and a surge in Kv2.1-mediated voltage-gated K+ currents.
DISCUSSION
We provide evidence that two important components of a previously characterized cell death cascade in neurons are intimately related to microglial-mediated neuronal injury, namely, intracellular Zn2+ release and a surge in voltage-dependent K+ currents (Aizenman et al., 2000; McLaughlin et al., 2001). Interfering with these processes proved to be highly effective in blocking neuronal cell death as a result of microglial activation. There are increasing numbers of studies suggesting a close association between microglia activation and neuronal injury in central nervous system disorders (Bessis et al., 2007; Colton et al., 2000, 2005; Duport and Garthwaite, 2005; Wojtera et al., 2005). In acute injury, such as that occurring during stroke, microglial-derived oxygen and nitrogen reactive species may represent critical components for initiating or compounding neuronal damage as it normally precedes but can amplify the production of proinflammatory cytokines (Block et al., 2007; Qin et al., 2004). Naturally, the production of microglial cytokines is a well-established mechanism of neuronal injury and has been implicated with many chronic neurodegenerative disorders (Block et al., 2007). Additionally, the production of reactive species by microglia is likely to impair other cellular components, such as membrane lipids, that may or may not necessarily involve the liberation of intracellular Zn2+. Of note however, aldehydes, which are lipid peroxidation products, can react with Zn2+-thiolate clusters in proteins and readily liberate the metal (Hao and Maret, 2006). We therefore suggest that targeting intraneuronal Zn2+ dysregulation, or the cell death signaling transduction cascades activated by Zn2+, could provide effective therapeutic strategies for minimizing neuronal damage in stroke and related disorders.
Since Zn2+ can be readily liberated from intracellular metal-binding proteins by oxygen and nitrogen-derived stressors (Maret, 2006; Sidorkina et al., 2003), and can subsequently trigger neuronal cell death (Aizenman et al., 2000; Bossy-Wetzel et al., 2004; McLaughlin et al., 2001), it is also entirely possible that the processes described here will be shown to be widely expressed in many forms of neurodegeneration. In fact, release of intracellular Zn2+ during neuronal injury has now been reported to be involved in a number of neurodegenerative conditions, including epileptic seizures (Lavoie et al., 2007; Lee et al., 2000), cerebral ischemia (Calderone et al., 2004), and target deprivation (Land and Aizenman, 2005). Importantly, microglial activation has been shown to be a potential crucial component of the neuropathological changes observed in all of these conditions (Boer et al., 2006; De Simoni et al., 2000; Ekdahl et al., 2003; Milligan et al., 1991b; Rizzi et al., 2003; Wang et al., 2007). Microglia have also been shown to be activated during normal brain development (Milligan et al., 1991a), where they play a critical role in the programmed death of neurons (Marin-Teva et al., 2004). Interestingly, it has been reported that both intraneuronal Zn2+ accumulation and enhanced K+ currents are cellular markers of developmental neuronal cell death (Hribar et al., 2004; Lee et al., 2006). It must be mentioned, however, that microglia activated in a manner similar to that described here were unable to generate sufficient NO to injure neurons in hippocampal slice cultures (Duport and Garthwaite, 2005). This preparation, however, is markedly insensitive to NO-mediated damage (Keynes et al., 2004), and thus other regulatory processes may be at play, including effective scavenging of reactive oxygen species by endogenous substances, thereby possibly limiting the production of ONOO− and its reactive products. It is also important to consider the possibility that the responses observed here in neurons exposed to activated microglia may also be expressed during nonpathological conditions. As such, an upregulation of delayed rectifier neuronal currents following microglial signaling could lead to pronounced changes in intrinsic excitability (Misonou et al., 2005).
It has yet to be established if Zn2+ liberation under all injurious conditions precedes the characteristic K+ current surge that is widely present in neuronal apoptosis. It is also not known whether neuronal cell death can always be averted by preventing cellular K+ efflux. In fact, our own work has shown that following exposure to ONOO− donors these two processes, Zn2+ liberation and K+ current surge, are both not required together for cell death to ensue. Specifically, we were unable to rescue cortical neurons from exposure to the ONOO− generator 3-morpholino-sydnonimine (SIN-1) by preventing cellular K+ efflux (Zhang et al., 2004), even though a pronounced K+ current surge could be easily detected under similar circumstances (Pal et al., 2004). This suggests that an exogenous ONOO− neurotoxicity circumvents the K+ efflux requirement for completion of a cell death program. Other studies have shown that the toxic actions of ONOO− generators such as SIN-1 can be complex and that they can sometimes involve an excitotoxic component (Trackey et al., 2001). This additional injurious component may not necessarily require either Zn2+ release or K+ extrusion to be fully expressed. More significantly, however, addition of an exogenous ONOO− generator or ONOO− itself may prove to be a stimulus not analogous to the release of reactive species by activated microglia under pathological situations, as suggested by others (Espey et al., 2002).
The results presented in this study demonstrate that under conditions previously shown to be highly toxic to both neurons (Xie et al., 2002) and oligodendrocytes (Li et al., 2005), reactive oxygen and nitrogen species derived from activated microglia induce a molecular cell death cascade that requires both the release of intracellular Zn2+ and a MAPK-dependent enhancement of Kv2.1-mediated K+ currents. These two critical steps in neuronal apoptosis thus provide discrete molecular targets for combating neurodegenerative conditions, where activation of microglia could be deleterious to neurons.
Acknowledgments
NIH; Grant numbers: NS043277, DC004199; Grant sponsor: American Heart Association, Pennsylvania-Delaware Affiliate.
We thank P.A. Rosenberg, B.A. Freeman and K. He for helpful comments and suggestions, H. Ichijo, and J. Nerbonne for the gifts of plasmids, and J. Connor for the microglial cell line.
References
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Aras MA, Aizenman E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett. 2005;387:136–140. doi: 10.1016/j.neulet.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Matthias A, Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem. 2002;80:73–80. doi: 10.1046/j.0022-3042.2001.00675.x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boeckman FA, Aizenman E. Pharmacological properties of acquired excitotoxicity in Chinese hamster ovary cells transfected with N-methyl-D-aspartate receptor subunits. J Pharmacol Exp Ther. 1996;279:515–523. [PubMed] [Google Scholar]
- Boer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E. Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol. 2006;173:188–195. doi: 10.1016/j.jneuroim.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch. 2004;448:313–318. doi: 10.1007/s00424-004-1266-5. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheepsunthorn P, Radov L, Menzies S, Reid J, Connor JR. Characterization of a novel brain-derived microglial cell line isolated from neonatal rat brain. Glia. 2001;35:53–62. doi: 10.1002/glia.1070. [DOI] [PubMed] [Google Scholar]
- Colton CA, Brown CM, Vitek MP. Sex steroids, APOE genotype and the innate immune system. Neurobiol Aging. 2005;26:363–372. doi: 10.1016/j.neurobiolaging.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Colton CA, Chernyshev ON, Gilbert DL, Vitek MP. Microglial contribution to oxidative stress in Alzheimer’s disease. Ann NY Acad Sci. 2000;899:292–307. doi: 10.1111/j.1749-6632.2000.tb06195.x. [DOI] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, De Luigi A, Garattini S, Vezzani A. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci. 2000;12:2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Scanlon JM, Kress GJ, Stout AK, Reynolds IJ. Astrocytes are more resistant than neurons to the cytotoxic effects of increased [Zn(2+)](i) Neurobiol Dis. 2000;7:310–320. doi: 10.1006/nbdi.2000.0303. [DOI] [PubMed] [Google Scholar]
- Duport S, Garthwaite J. Pathological consequences of inducible nitric oxide synthase expression in hippocampal slice cultures. Neuroscience. 2005;135:1155–1166. doi: 10.1016/j.neuroscience.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Zhu C, Bonde S, Bahr BA, Blomgren K, Lindvall O. Death mechanisms in status epilepticus-generated neurons and effects of additional seizures on their survival. Neurobiol Dis. 2003;14:513–523. doi: 10.1016/j.nbd.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Espey MG, Xavier S, Thomas DD, Miranda KM, Wink DA. Direct real-time evaluation of nitration with green fluorescent protein in solution and within human cells reveals the impact of nitrogen dioxide vs. peroxynitrite mechanisms. Proc Natl Acad Sci USA. 2002;99:3481–3486. doi: 10.1073/pnas.062604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Suh SW, Koh JY, Cha YK, Thompson RB, LaBuda CJ, Balaji RV, Cuajungco MP. Depletion of intracellular zinc from neurons by use of an extracellular chelator in vivo and in vitro. J Histochem Cytochem. 2002;50:1659–1662. doi: 10.1177/002215540205001210. [DOI] [PubMed] [Google Scholar]
- Hao Q, Maret W. Aldehydes release zinc from proteins. A pathway from oxidative stress/lipid peroxidation to cellular functions of zinc. FEBS J. 2006;273:4300–4310. doi: 10.1111/j.1742-4658.2006.05428.x. [DOI] [PubMed] [Google Scholar]
- Harris KM, Rosenberg PA. Localization of synapses in rat cortical cultures. Neuroscience. 1993;53:495–508. doi: 10.1016/0306-4522(93)90214-z. [DOI] [PubMed] [Google Scholar]
- Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: A paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- Hribar M, Bloc A, Medilanski J, Nusch L, Eder-Colli L. Voltage-gated K+ current: A marker for apoptosis in differentiating neuronal progenitor cells? Eur J Neurosci. 2004;20:635–648. doi: 10.1111/j.1460-9568.2004.03520.x. [DOI] [PubMed] [Google Scholar]
- Jiao S, Liu Z, Ren WH, Ding Y, Zhang YQ, Zhang ZH, Mei YA. cAMP/protein kinase A signaling pathway protects against neuronal apoptosis and is associated with modulation of Kv2.1 in cerebellar granule cells. J Neurochem. 2007;100:979–991. doi: 10.1111/j.1471-4159.2006.04261.x. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keynes RG, Garthwaite J. Nitric oxide and its role in ischaemic brain injury. Curr Mol Med. 2004;4:179–191. doi: 10.2174/1566524043479176. [DOI] [PubMed] [Google Scholar]
- Keynes RG, Griffiths C, Garthwaite J. Superoxide-dependent consumption of nitric oxide in biological media may confound in vitro experiments. Biochem J. 2003;15:399–406. doi: 10.1042/BJ20020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Land PW, Aizenman E. Zinc accumulation after target loss: An early event in retrograde degeneration of thalamic neurons. Eur J Neurosci. 2005;21:647–657. doi: 10.1111/j.1460-9568.2005.03903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie N, Peralta MR, III, Chiasson M, Lafortune K, Pellegrini L, Seress L, Toth K. Extracellular chelation of zinc does not affect hippocampal excitability and seizure-induced cell death in rats. J Physiol. 2007;578:275–289. doi: 10.1113/jphysiol.2006.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hunt JA, Groves JT. Mechanisms of iron porphyrin reactions with peroxynitrite. J Am Chem Soc. 1998;120:7493–7501. [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: Evidence against synaptic vesicle origin. J Neurosci. 2000;20:RC79. doi: 10.1523/JNEUROSCI.20-11-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Hwang JJ, Park MH, Koh JY. Cytosolic labile zinc: A marker for apoptosis in the developing rat brain. Eur J Neurosci. 2006;23:435–442. doi: 10.1111/j.1460-9568.2005.04553.x. [DOI] [PubMed] [Google Scholar]
- Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci USA. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation. 2005;2:20. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal. 2006;8:1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Marla SS, Lee J, Groves JT. Peroxynitrite rapidly permeates phospholipid membranes. Proc Natl Acad Sci USA. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CE, Cunningham TJ, Levitt P. Differential immunochemical markers reveal the normal distribution of brain macrophages and microglia in the developing rat brain. J Comp Neurol. 1991a;314:125–135. doi: 10.1002/cne.903140112. [DOI] [PubMed] [Google Scholar]
- Milligan CE, Levitt P, Cunningham TJ. Brain macrophages and microglia respond differently to lesions of the developing and adult visual system. J Comp Neurol. 1991b;314:136–146. doi: 10.1002/cne.903140113. [DOI] [PubMed] [Google Scholar]
- Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem. 1998;273:15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Trimmer JS. Kv2.1: A voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicol. 2005;26:743–752. doi: 10.1016/j.neuro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, He K, Aizenman E. Nitrosative stress and potassium channel-mediated neuronal apoptosis: Is zinc the link? Pflugers Arch. 2004;448:296–303. doi: 10.1007/s00424-004-1256-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Takimoto K, Aizenman E, Levitan ES. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2006;13:661–667. doi: 10.1038/sj.cdd.4401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Tan KS, Wei SJ, Wu HM, Xu Z, Wilson B, Lu RB, Hong JS, Flood PM. Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol. 2007;179:1198–1209. doi: 10.4049/jimmunol.179.2.1198. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci USA. 2007;104:3568–3573. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, Jefferson BS, Ziegler CB, Mortensen OV, Torres GE, Levitan ES, Aizenman E. A vital role for voltage-dependent potassium channels in dopamine transporter-mediated 6-hydroxydopamine neurotoxicity. Neuroscience. 2006;143:1–6. doi: 10.1016/j.neuroscience.2006.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi M, Perego C, Aliprandi M, Richichi C, Ravizza T, Colella D, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol Dis. 2003;14:494–503. doi: 10.1016/j.nbd.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Santos S, Aizenman E. Functional expression of muscle-type nicotinic acetylcholine receptors in rat forebrain neurons in vitro. Methods Find Exp Clin Pharmacol. 2002;24:63–66. doi: 10.1358/mf.2002.24.2.677127. [DOI] [PubMed] [Google Scholar]
- Schopfer FJ, Baker PRS, Freeman BA. NO-dependent protein nitration: Acell signaling event or an oxidative inflammatory response? Trends Biochem Sci. 2003;28:646–654. doi: 10.1016/j.tibs.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of poly(ADP-RIBOSE) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic Biol Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: Peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtera M, Sikorska B, Sobow T, Liberski PP. Microglial cells in neurodegenerative disorders. Folia Neuropathol. 2005;43:311–321. [PubMed] [Google Scholar]
- Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD. Peroxynitrite mediates neurotoxicity of amyloid β-peptide1–42-and lipopolysaccharide-activated microglia. J Neurosci. 2002;22:3484–3492. doi: 10.1523/JNEUROSCI.22-09-03484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–386. doi: 10.1016/s0301-0082(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]