Polo-Like Kinase 1 Is Essential for Early Embryonic Development and Tumor Suppression (original) (raw)
Abstract
Polo-like kinases (Plks) are serine/threonine kinases that are highly conserved in organisms from yeasts to humans. Previous reports have shown that Plk1 is critical for all stages of mitosis and may play a role in DNA replication during S phase. While much work has focused on Plk1, little is known about the physiological function of Plk1 in vivo. To address this question, we generated Plk1 knockout mice. Plk1 homozygous null mice were embryonic lethal, and early Plk1−/− embryos failed to survive after the eight-cell stage. Immunocytochemistry studies revealed that Plk1-null embryos were arrested outside the mitotic phase, suggesting that Plk1 is important for proper cell cycle progression. It has been postulated that Plk1 is a potential oncogene, due to its overexpression in a variety of tumors and tumor cell lines. While the Plk1 heterozygotes were healthy at birth, the incidence of tumors in these animals was threefold greater than that in their wild-type counterparts, demonstrating that the loss of one Plk1 allele accelerates tumor formation. Collectively, our data support that Plk1 is important for early embryonic development and may function as a haploinsufficient tumor suppressor.
Polo-like kinases (Plks) are conserved multifunctional kinases critical for cell cycle progression. First identified in Drosophila melanogaster (9), Plks have since been found in a variety of organisms, including yeasts, worms, and all vertebrates, including mammals. Although only one member of the Plk family has been identified in yeast (CDC5 in budding yeast and Plo1 in fission yeast), there are four members of the Plk family in mammals (Plk1, Plk2/Snk, Plk3/Fnk/Prk, and Plk4/Sak) (28, 42).
All members of the Plks regulate cell cycle progression and function at overlapping, yet different, stages of various cell cycle phases. For example, Plk2 is believed to be required for centriole duplication during the G1/S transition (44), Plk3 is important for S-phase entry (49) and mitosis (6), and Plk4 functions in centriole duplication and mitotic exit (4, 11, 13). Plk1 is the best-studied member of the four mammalian Plks. Besides its role in DNA replication and DNA damage repair, Plk1 functions during all stages of mitosis, including centrosome maturation, bipolar spindle formation, chromosome segregation, and cytokinesis (reviewed in references 3, 30, and 42).
One of the functions of Plk1 is to promote the G2/M transition. Plk1 facilitates mitotic entry by activating the Cdk1/cyclin B complex, the master controller of M-phase progression. Plk1 phosphorylates CDC25 phosphatase and helps its nuclear localization, which subsequently leads to the removal of two inhibitory phosphorylation sites (Thr 14 and Thr 15 of human Cdk1) on the ATP binding domain of Cdk1 (20, 39). The two kinases Wee1 and Myt1, which are responsible for these inhibitory phosphorylation events on Cdk1, are also inhibited through phosphorylation by Plk1 at the same time (27, 45). There is also evidence that Plk1 directly phosphorylates cyclin B (14, 40), although whether this leads to nuclear localization of cyclin B and activation of the Cdk1/cyclin B complex remains to be elucidated. Collectively, these studies support a critical role for Plk1 at the G2/M transition.
Plk1 also operates at other stages of mitosis. Plk1 localizes to centrosomes and is required for microtubule nucleation during centrosome maturation (21). Plk1 is found associated with kinetochores, indicating that it may have additional roles in kinetochore assembly and the potential regulation of the spindle assembly checkpoint (2). Plk1 also phosphorylates cohesins for their dissociation from chromosomes (35) and activates the anaphase-promoting complex to facilitate the separation of sister chromatids (1, 19). Furthermore, Plk1 is indispensable for the successful completion of cytokinesis (5, 26). Therefore, it is clear that Plk1 plays important regulatory functions all the way through mitosis.
Mammalian Plks have also been implicated in cancer development. Plk1 has been shown to be upregulated in several human malignancies, including breast, esophageal, lung, and colorectal cancers (25, 38, 46, 47). Plk1 expression has been reported to correlate with the prognosis of, and metastatic potential in, cancer patients (17, 37, 48). Overexpression of Plk1 has been observed in tumors, which suggests that Plk1 may act as an oncogene. Because of this finding, Plk1 inhibitors have recently been developed and are being tested as potential anticancer agents (10, 22, 23, 33). Preclinical and clinical studies testing the efficacy of these drugs are ongoing. Paradoxically, mutations of the Plk1 gene have also been identified in several cancer cell lines (32). These Plk1 mutations are missense mutations in the C-terminal polo-box domain (32) which lead to the destabilization of Plk1 and indicate that downregulation of Plk1 may also induce tumor formation. The exact role of Plk1 in tumorigenesis remains to be determined.
To understand the physiological functions of the four Plks in mammals, mouse models of these Plks have recently been generated and studied. Deletion of Plk4 in mice is embryonic lethal (13), with Plk4−/− embryos arresting at approximately embryonic day 8.0 (E8.0) and possessing a marked increase in apoptosis and mitotic defects. Heterozygous Plk4 mice develop spontaneous tumors, primarily hepatocellular carcinomas and liver carcinomas, at an advanced age (18). Because loss of heterozygosity on human chromosome 4q28, which contains the PLK4 gene, is common in human hepatomas, it has been proposed that Plk4 is a haploinsufficient tumor suppressor that predisposes for tumor development because of mitotic errors accumulated in these cells that promote aneuploidy. In contrast, Plk2-deficient mice are born healthy, albeit at a smaller size than their wild-type (WT) littermates (14). Plk2−/− mice do not have a decreased survival rate compared to Plk2+/+ mice, and no known tumor phenotype has been reported. Cells derived from Plk2−/− mice do have a decreased S-phase population, potentially implicating Plk2 in G1/S phase progression (14).
Although Plk1 has been studied for many years, there are no knockout (KO) mice available for Plk1, which could be very useful for studying the physiological function of Plk1 in vivo. In this study, we generated Plk1 KO mice and identified Plk1 as essential for early embryonic development because of its regulation of proper cell cycle progression. Moreover, Plk1 heterozygotes developed tumors at threefold greater frequency than their WT counterparts, suggesting that Plk1 functions as a haploinsufficient tumor suppressor.
MATERIALS AND METHODS
Generation of Plk1 heterozygous mutant mice.
The gene trap embryonic stem (ES) cell line RRR358 was purchased from BayGenomics. The insertion site of the gene trap vector was mapped, using genomic PCR and sequencing. Chimeras were generated through injection of ES cells into blastocysts, and germ lined chimeras were backcrossed to C57BL/6 mice to obtain mice heterozygous for the Plk1 mutation. We established a colony of Plk1+/+ and Plk1+/− mice and maintained them on a normal diet. We euthanized mice ranging from 50 to 70 weeks of age by CO2 asphyxiation and subjected them to necroscopy. Tissue samples were collected and fixed in formalin and embedded in paraffin blocks prior to being cut and stained with hematoxylin and eosin (H&E). The statistical significance of the incidence of tumors was analyzed by chi-square test.
In vitro culture and genotyping of preimplantation embryos.
Preimplantation embryos were obtained from an intercrossing of mice heterozygous for Plk1. E3.5 embryos were flushed out of the uterus by using M2 medium (Sigma) and were cultured in Dulbecco's modified Eagle's medium supplemented with 15% fetal bovine serum, 0.1 mM beta-mercaptoethanol, 4 mM glutamine, and 103 units/ml recombinant leukemia inhibitory factor. Nested PCR was used to genotype the embryos, using methods described previously (24), with the following primers: first-round sense primer for both WT and KO mice, 5′-TATTTCCGCAATTACATGAGTGAGC-3′; first-round antisense primer for WT mice, 5′-TAGCAGAGTGAAGGGGCACCAGTCC-3′; first-round antisense primer for KO mice, 5′-CTCCTACATAGTTGGCAGTGTTTGG-3′; second-round sense primer for both WT and KO mice, 5′-CACGCAGCGCCATCATCCTGCACCT-3′; second-round antisense primer for WT mice, 5′-CCAGGAGGCTCAGGCGGTACGTTTG-3′; and second-round antisense primer for KO mice, 5′-GGCCATCCGGGCTACCGGCTAAAAC-3′. RNA was extracted from embryos, using Trizol reagents, and cDNA was generated using SuperScript III (Invitrogen). Nested PCR was used to amplify the cDNA of WT and KO alleles with the following primers: first-round sense primer for both WT and KO mice, 5′-TATTTCCGCAATTACATGAGTGAGC-3′; second-round sense primer for both WT and KO mice, 5′-CACGCAGCGCCATCATCCTGCACCT-3′; antisense primer for WT mice for both rounds, 5′-CCAGGAGGCTCAGGCGGTACGTTTG-3′; and antisense primer for KO mice for both rounds, 5′-ATCCGCCACATATCCTGATCTTCCA-3′.
Immunofluorescence staining, antibodies, and Western blotting.
Standard immunostaining procedures were used, except that fixed embryos were treated with 2 M HCl for 30 min to denature double-stranded DNA during BrdU staining. For BrdU incorporation assays, embryos were cultured in medium containing 40 nM BrdU (Sigma) for 16 h and immunostained with anti-BrdU antibody (BioSource). Anti-phosphorylated histone H3 Ser 10 antibodies were obtained from Millipore and anti-α-tubulin antibodies from Sigma. Liver samples were homogenized in NETN (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) on ice. Crude cell lysates were then centrifuged at 14,000 rpm for 10 min, and cleared lysates were collected. Samples were boiled in 2× Laemmli buffer, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed. Membranes were blocked in 5% milk-Tris-buffered saline-Tween 20 and then probed with antibodies as indicated. A Plk1 antibody was raised in rabbit against mouse Plk1 amino acids 6 to 24. Monoclonal β-actin was purchased from Sigma and β-galactosidase antibodies from Abcam.
Metaphase spread of splenocytes.
Spleens were mashed between two slides, and splenocytes were incubated with Dulbecco's modified Eagle's medium with 10 μg/ml lipopolysaccharide (Sigma) and 50 ng/ml colcemid (Gibco) for 12 h. Cells were then washed with phosphate-buffered saline and treated with 75 mM KCl for 15 min. They were fixed in Carnoy's solution (75% methanol and 25% acidic acid), and a 15-μl aliquot was dropped onto a precleaned slide, which was then stained with 5% Giemsa solution (Gibco). Chromosome numbers were determined under a light microscope.
RESULTS AND DISCUSSION
In order to characterize the function of Plk1 in vivo, mice heterozygous for Plk1 were generated by using gene trap ES cell line RRR358. In this ES cell line, one allele of Plk1 was disrupted by a gene trap vector inserted at exon 9, which abolished proper transcription of Plk1 (Fig. 1A). Exon 9 encodes part of the polo-box domain of Plk1, which is essential for its substrate binding (7, 8) and is required for targeting the kinase activity of Plk1 to various subcellular localizations (15, 31). Since the gene trap is on exon 9, a truncated mutant of Plk1 fused with β-geo should be translated. However, we could detect neither the fusion protein, using a β-galactosidase antibody, nor the truncated Plk1 mutant, using a Plk1 N-terminal antibody, by Western blotting using tissues from heterozygous mice (data not shown), suggesting that the fusion protein or truncated Plk1 could not be correctly translated or was extremely unstable. Thus, this Plk1 gene trap virtually abolishes Plk1's expression.
FIG. 1.
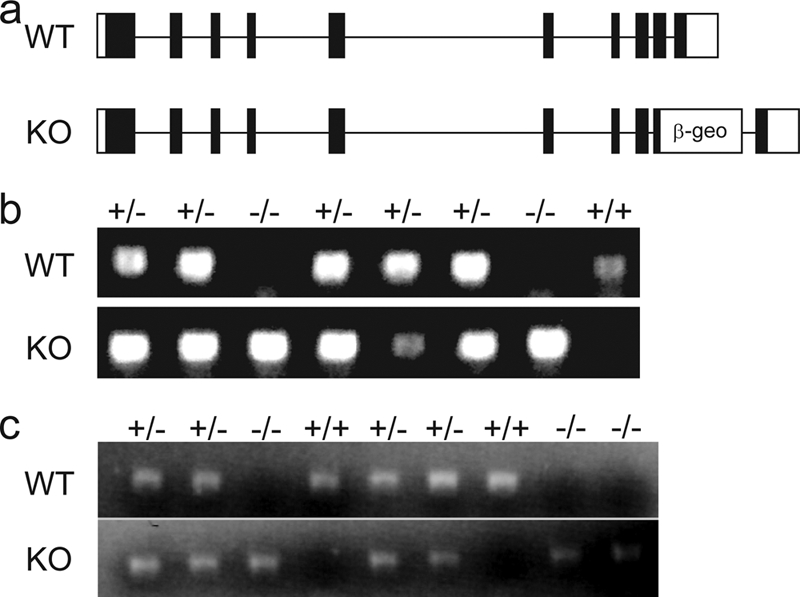
Presence of Plk1 homozygous mutant embryos at E3.5. (a) Gene structure of WT and mutant (KO) allele of Plk1 locus. β-geo represents the gene trap vector. (b) Typical genotyping results for E3.5 embryos from Plk1 heterozygous mutant intercrossed mice. (c) Typical reverse transcription-PCR results for E3.5 embryos from Plk1 heterozygous mutant intercross mice.
Mice heterozygous for Plk1 were obtained by injecting ES cells into blastocysts and breeding chimeras with C57BL/6 mice. After several intercrosses of heterozygous mice, no homozygous null Plk1 mice were obtained (Table 1), indicating that the complete deletion of Plk1 was embryonic lethal. To investigate at what stage Plk1-null embryos underwent cell death, we isolated embryos from matings of Plk1 heterozygous mice. The embryonic lethality occurred in the early embryonic stages, as no surviving Plk1 homozygous null embryos were obtained at E10.5 (Table 1). We then harvested E3.5 embryos and discovered a population of abnormal embryos with four or eight cells that were consistently identified together with normal blastocysts. PCR genotyping suggested that all of these developmentally delayed embryos, but none of the normally developed blastocysts, were homozygous null for Plk1 (Fig. 1B). Analysis by reverse transcription-PCR also confirmed that these abnormal embryos did not contain any WT Plk1 transcripts (Fig. 1C). Embryos were then cultured in vitro for 4 days. None of the abnormal embryos showed further growth, and all underwent apoptosis (Fig. 2), while normal blastocysts displayed proper hatching and outgrowth. These data suggest that Plk1 is essential for early embryonic development.
TABLE 1.
Summary of genotypes of offspring of Plk1 heterozygous mutant intercrossed mice
| Age of embryo/infant mice | No. of embryos | No. of embryos or infant mice with indicated actual genotype: | No. of embryos or infant mice with indicated expected genotype: | ||||
|---|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | +/+ | +/− | −/− | ||
| E3.5 | 23 | 6 | 13 | 4 | 6 | 11 | 6 |
| E10.5 | 18 | 5 | 13 | 0 | 4 | 10 | 4 |
| After birth | 185 | 70 | 115 | 0 | 46 | 93 | 46 |
FIG. 2.

Plk1 homozygous mutant embryos fail to survive after the eight-cell stage. Embryos harvested at E3.5 from heterozygous mutant intercrossed mice were cultured and imaged for 4 consecutive days. Pictures of typical abnormal and normal embryos are shown. The morphology and genotype of all embryos cultured are summarized in the table below.
Plk1 has multiple functions during the cell cycle and is required particularly for centrosome maturation, spindle assembly, and mitotic entry. To test if the embryonic lethality of these Plk1-null embryos was due to the inability to maintain a normal cell cycle, we performed immunostaining with an antibody against phosphorylated histone H3 serine 10, which is a marker for mitotic cells. All normal embryos displayed strong, concentrated staining in a portion of their cells, which were in mitosis. However, none of the abnormal embryos showed any positive staining (Fig. 3A). Although the number of total cells in the abnormal embryos was less than that in normal embryos, consistent observations of all the embryos suggested that this lower number of cells could not be the cause for the absence of phospho-H3S10. Further analysis of spindle assembly using an antibody against α-tubulin revealed that none of these abnormal embryos contained any assembled spindles, while normal spindle assembly was observed in normal embryos (Fig. 3B). These observations together indicate that Plk1 is indispensable for normal cell cycle progression in four-cell- or eight-cell-stage embryos.
FIG. 3.

Plk1 homozygous mutant embryos failed to enter mitosis. Pictures of stained, typical abnormal and normal embryos are shown. (a) Embryos harvested at E3.5 from heterozygous mutant intercrossed mice were stained with an antibody against phosphorylated histone H3 serine 10 (pH3). (b) Embryos harvested at E3.5 from heterozygous mutant intercrossed mice were stained with an antibody against α-tubulin. (c) Embryos harvested at E3.5 from heterozygous mutant intercrossed mice were cultured in medium containing 40 nM BrdU for 16 hours and immunostained with an anti-BrdU antibody. The morphology and staining patterns of all embryos are summarized in the table below. DAPI, 4′6-diamidino-2-phenylindole.
Since Plk1 was also reported to play a role in DNA replication (34, 41), we tested whether Plk1 is involved in DNA synthesis by using BrdU incorporation as a readout. Both normal and abnormal embryos incorporated BrdU (Fig. 3C), indicating that the initiation of DNA replication can occur in the absence of Plk1, although we were unable to conclude whether replication occurs normally or completely in Plk1-null cells.
From our in vitro studies using Plk1 embryos, we concluded that Plk1 deficiency leads to early embryonic lethality. However, Plk1+/− mice are born healthy and fertile, with no obvious effects except a slight decrease in Plk1 levels compared to those for Plk1 WT mice (Fig. 4A). Given the requirement of Plk1 for early embryogenesis and normal cell cycle progression, we hypothesized that loss of one Plk1 allele might cause problems with cell cycle control. Such defects may lead to chromosomal instability and promote tumorigenesis in these Plk1+/− mice. To test whether this is the case, we established a cohort of Plk1+/+ and Plk1+/− mice. We euthanized animals ranging from 50 to 70 weeks of age and performed necroscopies to search for tumorigenesis in these mice. The average age at euthanasia for both Plk1+/+ and Plk1+/− mice was 57 weeks (see Fig. S1 in the supplemental material). Interestingly and surprisingly, Plk1+/− mice developed tumors in various organs at a frequency of 27.5% (11 out of 40), compared with only 9% (3 out of 34) for Plk1+/+ mice. This increased incidence of tumors is highly significant by chi-square analysis (P < 0.001) (Fig. 4C). A significant portion of these tumors appeared to be lymphomas that invaded the lung and liver. Shown in Fig. 4B is a lymphoma that invaded the liver, with a corresponding view of an H&E-stained section of the tumor. The rest of the tumors were lung carcinomas, except for one squamous cell carcinoma and one ovarian sarcoma (Fig. 4C). The increased incidence of tumors could potentially be caused by chromosomal instability, since Plk1 is important for mitotic transitions. To test this possibility, we harvested spleens from 6-month-old Plk1 WT and heterozygous mice and prepared chromosome spreads to determine whether Plk1 heterozygosity leads to aneuploidy, which may account for the subsequent tumorigenesis. We found that the heterozygous splenocytes contained a higher percentage of aneuploidies, suggesting that chromosomal instability is indeed present in somatic cells (Fig. 4D and E), which may eventually result in tumor formation in these Plk1+/− mice.
FIG. 4.

Plk1 heterozygotes develop spontaneous tumors. (a) Plk1 levels are decreased in the livers of Plk1 heterozygous mice compared to those in the WT. IB, immunoblot. (b) Plk1+/+ and Plk1+/− mice were euthanized between the ages of 50 and 70 weeks and underwent necroscopy. Shown is a lymphoma invading the liver of a Plk1+/− mouse. The left panel is the tumor (indicated by arrow), and the right panel is an H&E-stained section of the tumor. (c) Summary of the types and numbers of tumors found in both Plk1 WT and Plk1 heterozygous mice. Chi-square analysis was applied; the left panel is a summary of the tumors found in Plk1+/+ and Plk1+/− mice (P < 0.001), and the right panel is a summary of the tumors found in Plk1+/+ p53−/− and Plk1+/− p53−/− mice (P < 0.05). (d) Representative metaphase spreads of normal karyotype in Plk1+/+ and aneuploidy in Plk1+/− splenocytes. (e) Increased aneuploidy in Plk1+/− splenocytes. Chromosomes from 100 metaphase spreads were counted for each sample. Spleens from three WT mice and three heterozygotes were used in this study.
We also crossed Plk1 heterozygous mice onto a p53−/− background and determined whether the loss of p53 would rescue the embryonic lethality observed in Plk1−/− mice. Loss of p53 did not rescue the embryonic lethality of the Plk1 deletion, as only Plk1 heterozygotes were obtained from p53−/− Plk1+/− crossings (data not shown). The Plk1+/− p53−/− mice developed tumors at a higher frequency than p53−/− mice, although the tumor spectrum between the mice remained similar. All eight Plk1+/−p53−/− mice developed tumors, mostly lymphomas and sarcomas (Fig. 4C). In comparison, four out of eight Plk1+/+p53−/− mice developed tumors, mainly lymphomas and sarcomas. While the number of animals used in these experiments was limited, this finding is significant according to chi-square analysis (P < 0.05).
In conclusion, our results suggest that Plk1 is critical for maintaining the normal cell cycle. The absence of Plk1 leads to early embryonic lethality, and Plk1 heterozygous mice develop spontaneous tumors, suggesting that a normal level of Plk1 is critical for maintaining chromosomal stability. Future studies using conditional KO or hypomorphic Plk1 mice will allow for further analysis of the role of Plk1 as a putative tumor suppressor.
Our observations are consistent with previous reports of KO models of Plk1 homologs in other organisms, all of which support a critical role for Plk1 in cell cycle regulation. In Drosophila, the polo 2 mutant was lethal at the larval stage, probably due to a defect at the onset of mitosis. Although the polo 1 mutant was viable, embryos from homozygous females showed a defect in spindle formation (36). In budding yeast, the cdc5 mutant was lethal and displayed a dumbbell-shaped morphology and the consistent presence of mitotic spindles, indicating a defect in mitotic exit (16). In fission yeast, the plo1 mutant was also lethal and displayed two distinct phenotypes, one with monopolar spindle formation and another with failed septation (29), suggesting multiple roles for Plo1 during mitosis. The differences in phenotypes among various KO models in these organisms could reflect multiple roles of Plk1 in mitosis as well as the degree that each organism could tolerate the absence of Plks.
There are four members of the Plk family in mouse and human, all of which function in controlling cell cycle progression. All four members of the Plk family contain a serine/threonine kinase domain and a polo-box domain. Among them, Plk1, Plk2, and Plk3 have a tandem polo-box repeat, while Plk4 has a single polo box. The similar domain architecture could result in functional redundancy among Plks, which may explain why Plk2 KO mice are viable, albeit 20% smaller at birth. However, Plk4-null mice are embryonic lethal and die around E7.5, with increased mitotic cells in mutant embryos, suggesting a delay in progression through anaphase and the blockage of cell division. In this study, we showed that Plk1-null embryos had a perturbed progression of the cell cycle and were arrested at the eight-cell stage. These embryos might be arrested in the G2 phase and fail to enter mitosis, which would be consistent with previous reports of a role for Plk1 at mitotic entry (14, 20, 27, 39, 40, 45). It was also documented that Plk1 is required for recovery from G2 DNA damage-induced arrest (43). However, recent studies suggest that Plk1 may not be absolutely required for mitotic entry; instead, cells without Plk1 activity showed long delays in late prophase before entering mitosis (12, 23). Moreover, Plk1 clearly has a critical role in cytokinesis (5, 26). Therefore, it is possible that these Plk1-null embryos might have a cytokinesis defect, which could eventually allow them to exit mitosis but be arrested at the tetraploidy G1 phase. Nonetheless, our Plk1 KO data clearly suggest that different Plks have overlapping yet distinct functions in mammalian cells.
Similar to Plk1 heterozygotes, Plk4 heterozygotes display an augmented frequency of tumors at advanced age (18 to 24 months). In comparison, Plk1 heterozygotes develop tumors at 13 to 14 months of age on average. This could be due to the fact that Plk1 is essential for mitosis; null embryos die at E3.5 without entering the blastocyst stage. Loss of one allele of Plk1 can perhaps cause a delay in mitosis or failed chromosome segregation, eventually leading to aneuploidy and tumorigenesis, which is supported by our observation that premalignant splenocytes from Plk1+/− mice harbor increased levels of aneuploidy. On the other hand, the phenotypes that occur in the absence of Plk4 are less severe. Plk4-null embryos are able to undergo mitosis but die at a later stage due to an elevated number of mitotic errors and the delayed progression of anaphase. This difference could be the reason why Plk4 heterozygous mice develop tumors at an advanced age, since the accumulation of mitotic errors may be less rapid in these mice.
It is intriguing that we observed increased tumor susceptibility in mice lacking one allele of Plk1, since Plk1 is normally considered to be an oncogene, due to its enhanced expression in a variety of human cancers. In fact, Plk1 inhibitors have been developed for potential use as anticancer agents (10, 22, 23, 33). Our study with Plk1 KO mice leads us to speculate that the levels of Plk1s must be tightly regulated in the cell; too much can tip the balance toward the promotion of tumorigenesis and, even when reduced by half, can also license tumor progression. Therefore, using Plk1 as a therapeutic target may not be as straightforward as previously thought. The potential negative impact of reduced Plk1 activity should be carefully considered and assessed before Plk1 inhibitors are used for the treatment of human cancers.
Supplementary Material
[Supplemental material]
Acknowledgments
We thank members of the labs of Eric Fearon and Benjamin Margolis for technical support.
This work was supported in part by grants from the American Cancer Society (RSG-08-125-01-CCG to X.Y.) and the National Institutes of Health (CA113381 to J.C.). J.L.W. is the recipient of National Research Service award F31 GM770802. X.Y. is a recipient of an AACR-Susan G. Komen for the Cure Cancer Development award and is supported by the developmental fund of the University of Michigan Cancer Center. J.C. is a recipient of an Era of Hope Scholar award from the Department of Defense and a member of the Mayo Clinic Breast SPORE program.
Footnotes
▿
Published ahead of print on 15 September 2008.
REFERENCES
- 1.Alexandru, G., F. Uhlmann, K. Mechtler, M. A. Poupart, and K. Nasmyth. 2001. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell 105459-472. [DOI] [PubMed] [Google Scholar]
- 2.Arnaud, L., J. Pines, and E. A. Nigg. 1998. GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma 107424-429. [DOI] [PubMed] [Google Scholar]
- 3.Barr, F. A., H. H. Sillje, and E. A. Nigg. 2004. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5429-440. [DOI] [PubMed] [Google Scholar]
- 4.Bettencourt-Dias, M., A. Rodrigues-Martins, L. Carpenter, M. Riparbelli, L. Lehmann, M. K. Gatt, N. Carmo, F. Balloux, G. Callaini, and D. M. Glover. 2005. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 152199-2207. [DOI] [PubMed] [Google Scholar]
- 5.Carmena, M., M. G. Riparbelli, G. Minestrini, A. M. Tavares, R. Adams, G. Callaini, and D. M. Glover. 1998. Drosophila polo kinase is required for cytokinesis. J. Cell Biol. 143659-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conn, C. W., R. F. Hennigan, W. Dai, Y. Sanchez, and P. J. Stambrook. 2000. Incomplete cytokinesis and induction of apoptosis by overexpression of the mammalian polo-like kinase, Plk3. Cancer Res. 606826-6831. [PubMed] [Google Scholar]
- 7.Elia, A. E., L. C. Cantley, and M. B. Yaffe. 2003. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2991228-1231. [DOI] [PubMed] [Google Scholar]
- 8.Elia, A. E., P. Rellos, L. F. Haire, J. W. Chao, F. J. Ivins, K. Hoepker, D. Mohammad, L. C. Cantley, S. J. Smerdon, and M. B. Yaffe. 2003. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 11583-95. [DOI] [PubMed] [Google Scholar]
- 9.Fenton, B., and D. M. Glover. 1993. A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos. Nature 363637-640. [DOI] [PubMed] [Google Scholar]
- 10.Gumireddy, K., M. V. Reddy, S. C. Cosenza, R. Boominathan, S. J. Baker, N. Papathi, J. Jiang, J. Holland, and E. P. Reddy. 2005. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 7275-286. [DOI] [PubMed] [Google Scholar]
- 11.Habedanck, R., Y. D. Stierhof, C. J. Wilkinson, and E. A. Nigg. 2005. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 71140-1146. [DOI] [PubMed] [Google Scholar]
- 12.Hanisch, A., A. Wehner, E. A. Nigg, and H. H. Sillje. 2006. Different Plk1 functions show distinct dependencies on polo-box domain-mediated targeting. Mol. Biol. Cell 17448-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudson, J. W., A. Kozarova, P. Cheung, J. C. Macmillan, C. J. Swallow, J. C. Cross, and J. W. Dennis. 2001. Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr. Biol. 11441-446. [DOI] [PubMed] [Google Scholar]
- 14.Jackman, M., C. Lindon, E. A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5143-148. [DOI] [PubMed] [Google Scholar]
- 15.Kang, Y. H., J. E. Park, L. R. Yu, N. K. Soung, S. M. Yun, J. K. Bang, Y. S. Seong, H. Yu, S. Garfield, T. D. Veenstra, and K. S. Lee. 2006. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell 24409-422. [DOI] [PubMed] [Google Scholar]
- 16.Kitada, K., A. L. Johnson, L. H. Johnston, and A. Sugino. 1993. A multicopy suppressor gene of the Saccharomyces cerevisiae G1 cell cycle mutant gene dbf4 encodes a protein kinase and is identified as CDC5. Mol. Cell. Biol. 134445-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kneisel, L., K. Strebhardt, A. Bernd, M. Wolter, A. Binder, and R. Kaufmann. 2002. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J. Cutan. Pathol. 29354-358. [DOI] [PubMed] [Google Scholar]
- 18.Ko, M. A., C. O. Rosario, J. W. Hudson, S. Kulkarni, A. Pollett, J. W. Dennis, and C. J. Swallow. 2005. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat. Genet. 37883-888. [DOI] [PubMed] [Google Scholar]
- 19.Kotani, S., S. Tugendreich, M. Fujii, P. M. Jorgensen, N. Watanabe, C. Hoog, P. Hieter, and K. Todokoro. 1998. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1371-380. [DOI] [PubMed] [Google Scholar]
- 20.Kumagai, A., and W. G. Dunphy. 1996. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science 2731377-1380. [DOI] [PubMed] [Google Scholar]
- 21.Lane, H. A., and E. A. Nigg. 1996. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol. 1351701-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lansing, T. J., R. T. McConnell, D. R. Duckett, G. M. Spehar, V. B. Knick, D. F. Hassler, N. Noro, M. Furuta, K. A. Emmitte, T. M. Gilmer, R. A. Mook, Jr., and M. Cheung. 2007. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol. Cancer Ther. 6:450-459. [DOI] [PubMed] [Google Scholar]
- 23.Lénárt, P., M. Petronczki, M. Steegmaier, B. Di Fiore, J. J. Lipp, M. Hoffmann, W. J. Rettig, N. Kraut, and J. M. Peters. 2007. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17304-315. [DOI] [PubMed] [Google Scholar]
- 24.Li, T., A. Inoue, J. M. Lahti, and V. J. Kidd. 2004. Failure to proliferate and mitotic arrest of CDK11p110/p58-null mutant mice at the blastocyst stage of embryonic cell development. Mol. Cell. Biol. 243188-3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macmillan, J. C., J. W. Hudson, S. Bull, J. W. Dennis, and C. J. Swallow. 2001. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann. Surg. Oncol. 8729-740. [DOI] [PubMed] [Google Scholar]
- 26.Mundt, K. E., R. M. Golsteyn, H. A. Lane, and E. A. Nigg. 1997. On the regulation and function of human polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun. 239377-385. [DOI] [PubMed] [Google Scholar]
- 27.Nakajima, H., F. Toyoshima-Morimoto, E. Taniguchi, and E. Nishida. 2003. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 27825277-25280. [DOI] [PubMed] [Google Scholar]
- 28.Nigg, E. A. 1998. Polo-like kinases: positive regulators of cell division from start to finish. Curr. Opin. Cell Biol. 10776-783. [DOI] [PubMed] [Google Scholar]
- 29.Ohkura, H., I. M. Hagan, and D. M. Glover. 1995. The conserved Schizosaccharomyces pombe kinase plo1, required to form a bipolar spindle, the actin ring, and septum, can drive septum formation in G1 and G2 cells. Genes Dev. 91059-1073. [DOI] [PubMed] [Google Scholar]
- 30.Petronczki, M., P. Lenart, and J. M. Peters. 2008. Polo on the rise—from mitotic entry to cytokinesis with Plk1. Dev. Cell 14646-659. [DOI] [PubMed] [Google Scholar]
- 31.Seong, Y. S., K. Kamijo, J. S. Lee, E. Fernandez, R. Kuriyama, T. Miki, and K. S. Lee. 2002. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem. 27732282-32293. [DOI] [PubMed] [Google Scholar]
- 32.Simizu, S., and H. Osada. 2000. Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat. Cell Biol. 2852-854. [DOI] [PubMed] [Google Scholar]
- 33.Steegmaier, M., M. Hoffmann, A. Baum, P. Lenart, M. Petronczki, M. Krssak, U. Gurtler, P. Garin-Chesa, S. Lieb, J. Quant, M. Grauert, G. R. Adolf, N. Kraut, J. M. Peters, and W. J. Rettig. 2007. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 17316-322. [DOI] [PubMed] [Google Scholar]
- 34.Stuermer, A., K. Hoehn, T. Faul, T. Auth, N. Brand, M. Kneissl, V. Putter, and F. Grummt. 2007. Mouse pre-replicative complex proteins colocalise and interact with the centrosome. Eur. J. Cell Biol. 8637-50. [DOI] [PubMed] [Google Scholar]
- 35.Sumara, I., E. Vorlaufer, P. T. Stukenberg, O. Kelm, N. Redemann, E. A. Nigg, and J. M. Peters. 2002. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 9515-525. [DOI] [PubMed] [Google Scholar]
- 36.Sunkel, C. E., and D. M. Glover. 1988. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 8925-38. [DOI] [PubMed] [Google Scholar]
- 37.Takai, N., T. Miyazaki, K. Fujisawa, K. Nasu, R. Hamanaka, and I. Miyakawa. 2001. Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett. 16441-49. [DOI] [PubMed] [Google Scholar]
- 38.Tokumitsu, Y., M. Mori, S. Tanaka, K. Akazawa, S. Nakano, and Y. Niho. 1999. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 15687-692. [DOI] [PubMed] [Google Scholar]
- 39.Toyoshima-Morimoto, F., E. Taniguchi, and E. Nishida. 2002. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 3341-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toyoshima-Morimoto, F., E. Taniguchi, N. Shinya, A. Iwamatsu, and E. Nishida. 2001. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 410215-220. [DOI] [PubMed] [Google Scholar]
- 41.Tsvetkov, L., and D. F. Stern. 2005. Interaction of chromatin-associated Plk1 and Mcm7. J. Biol. Chem. 28011943-11947. [DOI] [PubMed] [Google Scholar]
- 42.van de Weerdt, B. C., and R. H. Medema. 2006. Polo-like kinases: a team in control of the division. Cell Cycle 5853-864. [DOI] [PubMed] [Google Scholar]
- 43.van Vugt, M. A., A. Bras, and R. H. Medema. 2004. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 15799-811. [DOI] [PubMed] [Google Scholar]
- 44.Warnke, S., S. Kemmler, R. S. Hames, H. L. Tsai, U. Hoffmann-Rohrer, A. M. Fry, and I. Hoffmann. 2004. Polo-like kinase-2 is required for centriole duplication in mammalian cells. Curr. Biol. 141200-1207. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe, N., H. Arai, Y. Nishihara, M. Taniguchi, N. Watanabe, T. Hunter, and H. Osada. 2004. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 1014419-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolf, G., R. Elez, A. Doermer, U. Holtrich, H. Ackermann, H. J. Stutte, H. M. Altmannsberger, H. Rubsamen-Waigmann, and K. Strebhardt. 1997. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 14543-549 [DOI] [PubMed] [Google Scholar]
- 47.Wolf, G., R. Hildenbrand, C. Schwar, R. Grobholz, M. Kaufmann, H. J. Stutte, K. Strebhardt, and U. Bleyl. 2000. Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol. Res. Pract. 196753-759. [DOI] [PubMed] [Google Scholar]
- 48.Yuan, J., A. Horlin, B. Hock, H. J. Stutte, H. Rubsamen-Waigmann, and K. Strebhardt. 1997. Polo-like kinase, a novel marker for cellular proliferation. Am. J. Pathol. 1501165-1172. [PMC free article] [PubMed] [Google Scholar]
- 49.Zimmerman, W. C., and R. L. Erikson. 2007. Polo-like kinase 3 is required for entry into S phase. Proc. Natl. Acad. Sci. USA 1041847-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]