Viral and host factors induce macrophage activation and loss of Toll Like Receptor tolerance in chronic HCV infection (original) (raw)
. Author manuscript; available in PMC: 2008 Dec 3.
Published in final edited form as: Gastroenterology. 2007 Aug 2;133(5):1627–1636. doi: 10.1053/j.gastro.2007.08.003
Abstract
Background&Aims
Persistent inflammation contributes to progression of liver damage in chronic HCV (cHCV) infection. Repeated exposure to Toll like receptor (TLR) ligands results in tolerance, a protective mechanism aimed at limiting inflammation.
Methods
Monocytes/macrophages were repeatedly stimulated via pro-inflammatory cytokine-inducing TLRs and evaluated for activation markers.
Results
Unlike monocytes (Mo) of controls or patients with non-alcoholic steatohepatitis, the Mo of cHCV patients were hyper-responsive and failed to show homo- or hetero-tolerance to TLR ligands, manifested by elevated TNFα production. Serum levels of IFNγ, endotoxin (TLR4 ligand) and HCV core protein (TLR2 ligand) were elevated in cHCV patients suggesting potential mechanisms for in vivo monocyte pre-activation. Treatment of normal monocytes with IFNγ resulted in loss of tolerance to LPS or HCV core protein. Further, we found increased levels of MyD88-IRAK1 complexes and NFκB activity both in monocytes of cHCV patients and in normal monocytes that lost TLR tolerance after IFNγ+LPS pretreatment. In vitro differentiation of TLR tolerant cHCV monocytes into macrophages restored their capacity to exhibit TLR tolerance to LPS and HCV core protein and this could be reversed by administration of IFNγ. cHCV patients exhibited increased TNFα in the circulation and in the liver. In cHCV livers we found Kupffer cell/macrophage activation indicated by increased CD163 and CD33 expression.
Conclusions
We identified that host-derived factors (IFNγ and endotoxin) and viral factors (HCV core protein) act in tandem to induce and maintain monocyte/macrophage activation, thus favoring persistent inflammation in patients with cHCV infection.
Keywords: MyD88, IFNγ, HCV core protein, LPS, monocytes, Kupffer cells, NFκB, TNFα, inflammation
Introduction
Hepatitis C virus (HCV) infection leads to chronic disease in ~70% of infected patients (1). Many of the HCV-associated symptoms as well as progression to liver fibrosis are caused by persistent inflammation. Over-production of pro-inflammatory cytokines, in particular TNFα, was reported in patients with chronic HCV infection (2,3), however, the mechanisms leading to increased TNFα production and persistent inflammation during chronic HCV infection are yet to be fully understood.
Innate immunity plays a key role in the outcome of infections. Immune cells employ toll-like receptors (TLRs) to recognize specific pathogen-associated molecular patterns (4–7), such as dsRNA (TLR3), ssRNA (TLR7/8) that are present in the HCV life cycle (5), and lipopolysaccharide (LPS) (TLR4). TLR2 is activated by multiple ligands including HCV core protein (4,6,7). Upon engagement with specific ligands most TLRs, except for TLR3, recruit MyD88, IL-1 receptor-associated kinases (IRAK) and TNF receptor-associated factor 6 (TRAF6), and trigger NFκB and MAPK activation pathways to results in production of pro-inflammatory cytokines, including TNFα (4,6). Recently cross-tolerance between TLRs has been documented (8–10). Decreased production of TNFα in response to challenge with a TLR ligand with which the cells were pre-treated has been defined as “homotolerance”, while decreased activation to a TLR ligand that does not share homology with the pre-treatment ligand is called “heterotolerance”(8). TLR tolerance plays an important role in protection from hyperactivation of the immune system and can be overcome by conditioning of innate immune cells with cytokines, including interferons and growth factors (8–10).
Kupffer cells (KCs) are liver resident macrophages and a part of the body-wide distributed monocyte/macrophage system (11). KCs express TLRs and respond to pathogens with inflammatory cytokines, including TNFα (12). Activated KCs play a pivotal role in triggering and maintaining inflammation during alcoholic-and non-alcoholic liver diseases (13,14). During cHCV infection Kupffer cells exhibit an activated phenotype (15,16); however they do not support HCV replication in vitro (17). The role of Kupffer cells in inflammation during chronic infection with HCV is yet to be fully understood.
Here we hypothesized that activation of macrophages, including Kupffer cells, may favor the persistent inflammation in patients with chronic HCV infection. We identified that host-derived factors (IFNγ and endotoxin) and viral factors (HCV core protein) act in tandem to induce and maintain monocyte/macrophage activation, thus favoring persistent inflammation in patients with chronic HCV infection.
Materials and Methods
Reagents
LPS was from List Biological Laboratories (Campbell, CA), peptidoglycan (PGN) from Fluka (Milwaukee, WI), Pam2CSK4 and Pam3CSK4 from EMC Microcollections (Germany), Gardiquimod from Invivogen (San Diego, CA), fetal calf serum (FCS) from HyClone (Logan, UT), RPMI1640 from Gibco (Grand Island, NY), recombinant HCV core protein (genotype 1A aa 2–192) from Biodesign (Saco, MN).
Patients and Cells
The study was approved by the Committee for the Protection of Human Subjects in Research at UMass. Healthy controls (n=24), treatment-naïve patients with chronic HCV infection (cHCV patients, n=30) and patients with non-alcoholic steatohepatitis (NASH, n=6) were enrolled in our study after informed consent was obtained; patient’s characteristics are detailed in table 1.
Table 1.
The characteristics of patients included in the study.
| Parameter | HCV patients (Value) | NASH patients (Value) |
|---|---|---|
| Age (years) | 46±9 | 45±8 |
| Gender: Male Female | 22 | 2 |
| 8 | 4 | |
| AST (U/l) | 72±35 | 62±28 |
| ALT (U/l) | 84±41 | 76±31 |
| Viral load | 1.92×106 ± 0.3×106 | 0 |
| Viral genotype 1a 1b 2b 3aundetermined | 21 | Not detected |
| 3 | Not detected | |
| 2 | Not detected | |
| 1 | Not detected | |
| 3 | Not detected | |
| Liver biopsy performed | 26 | 6 |
| Liver fibrosisIshak Stage 1–2Ishak Stage 3–4 | 6 | 0 |
| 5 | 0 | |
| 1 | 0 | |
| Liver inflammationMild (Ishak Score 0–6)Moderate (Ishak Score 7–12) | 26 | 6 |
| 11 | 5 | |
| 14 | 1 |
Monocytes were separated from blood by centrifugation in Ficoll gradient and adherence to plastic as we previously described (6). Macrophages were differentiated in vitro from monocytes for 8 days in RPMI1640 with 1% non-essential aminoacids and 18% pooled normal human platelet-free serum.
For Kupffer cells isolation, C57Bl6 mice received anesthesia with Ketamine (100mg/kg) and Xylazine (10mg/kg) and the livers were perfused with saline for 5 minutes followed by digestion with liberase (20mg/L) for 5 minutes. The livers were excised, minced and further digested in vitro for 30min at 37°C. The hepatocytes were separated by centrifugation for 5min at 300xg, while the non-hepatocyte content was loaded on the top of the 50%-25% Percoll gradient and centrifuged 60 minutes at 800xg. The inter-cushions fraction was washed and adhered to plastic in DMEM+5%FBS; the non-adherent fraction was washed and the adherent population was adjusted to 2×106/ml in DMEM+10%FBS.
Quantification of cytokines, endotoxin, and HCV core protein
The cytokines were quantified using specific ELISA (BD Bioscience, San Jose, California), HCV core protein with Ortho HCV core antigen ELISA (Wako Chemicals USA, Richmond, VA) and endotoxin by Lymulus Amebocyte Lysate assay (Lonza Group Ltd, Switzerland).
Western blot analysis and Immunoprecipitation
Total cellular proteins (25_μ_g) were separated in SDS-PAGE, transferred to nitrocellulose membrane and immuno-blotted with indicated primary followed by horseradish peroxidase-conjugated secondary antibodies. The densitometric analyzed of the specific bands was performed with UVP system and LabWork 4.0 program (UVP BioImaging Systems). For immunoprecipitation, equal amounts of cellular proteins were incubated with anti-MyD88 antibody (Santa Cruz) for 16hrs at +4°C. The immunoprecipitated complexes were purified using Catch&Release system (Upstate, Charlottesville, VA) and subjected to western blot analysis as described above.
Electrophoretic mobility shift assay (EMSA)
Subcellular extracts and EMSA were performed as previously described (6). The densitometric analysis was performed using GDS-800 system and Labwork 4.0 program.
RNA Isolation and PCR analysis
RNA was isolated with RNeasy Kit (Qiagen, Valencia, CA). One μg of RNA was transcribed to cDNA in 20μl reaction volume. For real-time PCR, the SYBR Green Mix (Eurogentec, San Diego, CA) was combined with primers (sequences are shown in Table 2) and 1μl of cDNA and amplified using iCycler (Bio-Rad, Hercules, CA). Gene amplification was quantified based on SYBR Green fluorescence with iCycler software and analyzed using comparative ΔΔCt method using 18S as housekeeping control. For regular PCR, the products were separated in 1% agarose gels and the intensities of the bands were analyzed by densitometric analysis as for EMSA.
Table 2.
PCR primers.
| Target | Primer | Sequence 5′- 3′ |
|---|---|---|
| CD163 | forward | AGCTCTCTGGGACTGCACAT |
| reverse | ACACACAGTCCCCCACTCTC | |
| CD33 | forward | TGTTCCACAGAACCCAACAA |
| reverse | TTCCTCCTGTGGGTCTTCAC | |
| CXCL-9 | forward | AAAAGTGGGAGAAACAGGTCAGC |
| reverse | TTGTGAGTGGGATGTGGTTGG | |
| CXCL-10 | forward | AGTGGCATTCAAGGAGTACCT |
| reverse | ATCCTTGGAAGCACTGCATCG | |
| CXCL-11 | forward | AAGTAGCAGCAACAGCACCAGC |
| reverse | GGTCCTTTCACCCACCTTTCATC | |
| RIG-I | forward | CCAGATGCCAGACAAAGATGAAGAG |
| reverse | GCTCGGACATTGCTGAAGAAGTC | |
| 18S | forward+reverse | Ambion (Austin, TX) |
| TNFα | forward+reverse | MaximBio (San Francisco, CA) |
Statistical analysis
The Students t-test and Wilcoxon non-parametric analysis in StatView (SAS Inst, Cary, NC) were employed. A value of p<0.05 was considered significant.
Results
Monocytes of cHCV patients lack tolerance to TNF α-inducing TLR ligands
We have previously reported that monocytes of cHCV patients produced significantly higher levels of TNFα both at baseline and upon stimulation with TLR4 and TLR2 ligands compared to controls (6). Production of TNFα by immune cells contributes to the efficient control of invading pathogens, while excessive and uncontrolled production may lead to systemic chronic inflammation (2,3). Tolerance to TLR ligands is one of the protective mechanisms against chronic inflammation (10). Considering the presence of chronic inflammation in cHCV patients, we hypothesized that excessive production of TNFα in monocytes may represent loss of tolerance to TLR ligands. Monocytes of controls and HCV patients were primed in vitro with LPS, a TLR4 ligand, and re-stimulated with LPS to assess “homotolerance”. In agreement with previous reports (8,9), we found that LPS priming lead to low TNFα production in response to a re-challenge with LPS in normal monocytes (Fig 1A). However, homo-tolerance to TLR4 ligand was not found in monocytes from HCV patients (Fig 1B). We further identified that pretreatment with LPS induced hetero-tolerance to subsequent stimulation with TLR2 (PGN), TLR2/TLR6 (PAM3CSK4), TLR2/TLR1 (PAM2CSK), TLR3 (poly I:C) and TLR7/8 (Gardiquimod) ligands in controls (Fig 2A), and in patients with liver inflammation due to non-viral non-alcoholic steatohepatitis (NASH) (Fig 2C) but not in HCV-infected patients (Fig 2B). This data indicated that monocytes of HCV-infected patients have lost TLR tolerance.
Figure 1. Monocytes of cHCV patients fail to develop homo-tolerance to TLR4.

Monocytes of controls (n=16) (A) and HCV patients (n=15) (B) were kept in medium or stimulated with TLR4 ligand LPS (100ng/ml) for 24 hrs (1st stimulation) and re-challenged with LPS (100ng/ml) for an additional 24 hrs (2nd stimulation). The production of TNFα in culture supernatants was analyzed in ELISA. Average±SD is shown (*p<0.05).
Figure 2. Monocytes of cHCV patients, unlike controls and NASH patients, fail to develop hetero-tolerance to TNFα-inducing TLR ligands.

Monocytes from controls (n=16) (A), HCV patients (n=15) (B) and NASH patients (n=6) (C) were cultured in medium or stimulated with LPS (100ng/ml) for 24 hrs (1st stimulation) and re-challenged with LPS (100ng/ml), PGN (5μg/ml), Pam2CSK4 (100ng/ml), Pam3CSK4 (100ng/ml), poly I:C (100ng/ml) or Gardiquimod (5μg/ml) for an additional 24 hrs (2nd stimulation). The production of TNFα in culture supernatants was analyzed in ELISA. Average±SD is shown (* p<0.05).
HCV-infected patients exhibit elevated serum levels of IFNγ, endotoxin and HCV core protein
To dissect the mechanisms of loss of tolerance in monocytes of cHCV patients, we analyzed their serum in search of immunomodulatory molecules. We identified moderate but significantly higher levels of TNFα (data not shown) and IFNγ in the plasma of cHCV patients compared to controls (Fig 3A). IFNγ is among the most potent agents that may lead to abrogation of TLR-mediated tolerance (8,9). The plasma levels of IFNγ were biologically active in vivo, as indicated by increased expression of IFNγ-dependent genes (CXCL9, CXCL10, CXCL11, and RIG-I) in monocytes of patients compared to controls (Fig 3B). We also found increased levels of circulating endotoxin, a TLR4 ligand, in the plasma of HCV-infected patients compared to controls (Fig 3C). Finally, cHCV patients but not controls had detectable plasma levels of HCV core protein (Fig 3D), a potent immunomodulator (6,18). No correlation between HCV core protein and IFNγ in plasma was identified (data not shown).
Figure 3. cHCV patients exhibit elevated levels of IFNγ, endotoxin, and HCV core protein in the peripheral circulation.
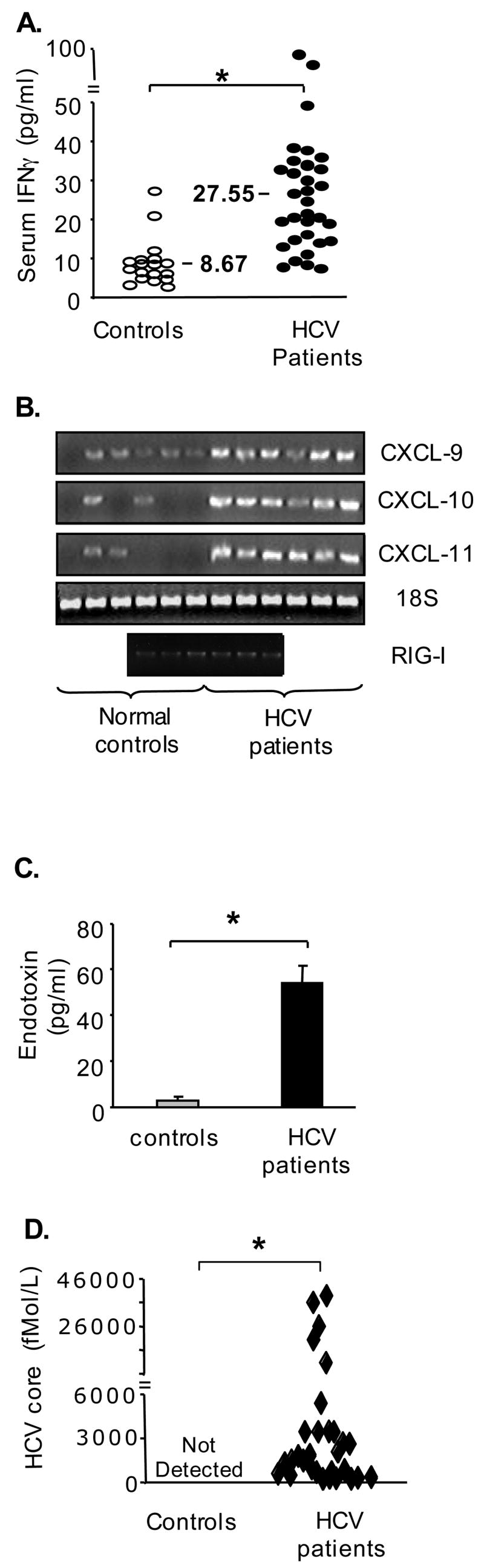
(A) The levels of IFNγ in plasma of HCV-infected patients (n=30) and controls (n=18) were quantified using a specific ELISA. The asterisk (*) represents p<0.05.
(B) The levels of RNA coding for CXCL9, CXCL10, CXCL11 and RIG-I in freshly isolated monocytes were quantified using PCR. Each band represents a pooled sample from 2 controls or 3 cHCV patients.
(C) The levels of endotoxin in plasma of cHCV patients (n=30) and controls (n=18) were quantified using LAL assay; average±SE is shown, the asterisk (*) indicates p<0.05.
(D) The levels of HCV core protein in the plasma of HCV-infected patients (n=30) and control (n=18) were quantified using HCV core ELISA; the asterisk (*) represents p<0.05.
IFNγ, endotoxin and HCV core protein modulate monocyte activation towards loss of TLR tolerance
Previous studies demonstrated that HCV core protein activates monocytes via TLR2 (6,7). Here we show that repeated stimulations with HCV core protein, similar to other TLR2 ligands (Fig 2), induced tolerance in normal monocytes (Fig 4A). The monocytes of HCV patients produced more TNFα compared to normal cells in response to HCV core protein (Fig 4A). However, in contrast to normals and to NASH patients, the monocytes from HCV-infected patients failed to develop tolerance upon repeated stimulations with HCV core protein (Fig 4A). Based on the finding of increased IFNγ levels in the peripheral circulation of cHCV patients (Fig 3A) and previous reports suggesting the role of IFNγ in loss of tolerance to TLR ligands (8–10), we further hypothesized that IFNγ may play a role in increased TNFα production and loss of tolerance to TLR ligands in cHCV infection. Both IFNγ and LPS alone, in amounts comparable to levels detected in the plasma of cHCV patients, induced low levels of TNFα in normal monocytes (data not shown). Further, combined IFNγ+LPS stimulation induced higher levels of TNFα compared to LPS alone (data not shown). Interestingly, when the _in vivo_-detected levels of endotoxin were used to pre-treat normal monocytes followed by stimulation with a higher dose of LPS, TNFα induction was inhibited, suggestive of tolerance (Fig 4B). Further, _in vivo_-detected levels of IFNγ could overcome the LPS tolerance (Fig 4B), and reverse both HCV core protein-induced homo-tolerance (Fig 4C) and LPS-rendered hetero-tolerance to HCV core protein (Fig 4D) in normal monocytes. These data suggest that HCV core, LPS and IFNγ favor loss of TLR tolerance and contribute to excessive production of TNFα in monocytes.
Figure 4. Tolerance to LPS and to HCV core protein is disrupted by IFNγ treatment.

(A) Monocytes from controls (n=16) and HCV patients (n=15) were kept in medium or stimulated for 24 hrs (1st stimulation) and re-challenged with HCV core protein for additional 24 hrs (2nd stimulation). The production of TNFα was analyzed in culture supernatants using ELISA and the * indicates p<0.05 (panels A–D).
(B) Monocytes of controls (n=3) were stimulated with LPS (100pg/ml) for 24 hrs (1ststimulation) and re-challenged with LPS for 24 hrs (2nd stimulation). IFNγ (100pg/ml) was added 4 hrs before and during the 1st stimulation.
(C) Monocytes of controls (n=3) were stimulated with HCV core (10000fMol/L) for24 hrs (1st stimulation) and re-challenged with HCV core (25000fMol/L), for 24 hrs (2nd stimulation). IFNγ (100pg/ml) was added for 4 hrs before and during the 1st stimulation.
(D) Monocytes of controls (n=3) were stimulated with LPS (100pg/ml) for 24 hrs (1st stimulation) and re-challenged with HCV core (25000 fMol/L) for 24 hrs (2nd stimulation). IFNγ (100pg/ml) was added for 4 hrs before and during the 1st stimulation.
TLR plus IFNγ condition elevated NFκB activation and upregulation of MyD88/IRAK1 complexes
Based on the findings of increased levels of both endotoxin and IFNγ in plasma of the cHCV patients (Fig 3) we hypothesized that TLR signaling pathways may also be amplified in cHCV patients. Activation of the NFκB complex is instrumental in TLR-induced TNFα production (19,20). We found that pre-treatment with IFNγ enhanced LPS-induced NFκB activity in normal monocytes (Fig 5A) consistent with increased amounts of TNFα produced in response to LPS+IFNγ-stimulation (Fig 4B). Similar to LPS+IFNγ-treated controls, the freshly isolated monocytes of cHCV patients exhibited increased levels of NFκB activity (Fig 5B). Upon ligand engagement most TLRs recruit MyD88 and IRAK1 to initiate activation of the NFκB pathway (4). Persistent NFκB activation is favored by sustained presence of the MyD88/IRAK complexes (9). Immunoprecipitation approach identified that LPS triggered formation of MyD88/IRAK1 complexes (Fig 5C), while LPS+IFNγ-treated cells showed elevated level of MyD88/IRAK1 complexes compared to IFNγ- or LPS-treated monocytes (Fig 5C). Similar to in vitro LPS+IFNγ-stimulated controls, we identified significantly higher levels of MyD88/IRAK complexes in freshly isolated monocytes of cHCV patients compared to controls (Fig 5D). Together, these data suggested that the monocytes of cHCV patients exhibit molecular signatures of impaired TLR tolerance indicated by the presence of in vivo pre-formed MyD88/IRAK1 complexes, persistent activation of NFκB, and increased production of TNFα. Further, the loss of TLR tolerance seen in cHCV monocytes could be reproduced in vitro by conditioning of normal monocytes with IFNγ and endotoxin.
Figure 5. HCV-infected patients’ monocytes show elevated NFκB activity and increased frequency of MyD88/IRAK complexes, similar to IFNγ+LPS-stimulated normal monocytes.

(A) Normal monocytes were pre-treated in vitro with IFNγ followed by stimulation with LPS for 1 hour, as indicated. Five μg of nuclear protein were analyzed for NFκB binding capacity in EMSA using a radioactive labeled NFκB-specific oligonucleotide. A cold NFκB oligonucleotide incubated with nuclear protein from LPS-stimulated sample was used to determine the specificity of NFκB band (Comp). A representative EMSA gel is shown on the top and densitometric analysis of n=4 is shown on the bottom.
(B) The NFκB binding capacity of normal and cHCV monocytes was analyzed in EMSA, as described above. Each band represents 5μf of nuclear proteins from a pool of 2 controls or 3 HCV patients. Normal monocytes (corresponding to the pool #2) were stimulated with LPS (100ng/ml for 1hr in vitro) and used as positive control for this assay (Normal+LPS). A cold NFκB oligonucleotide, incubated with nuclear protein from LPS-stimulated sample was used to determine the specificity of NFκB band (Comp). The densitometric analysis of the EMSA gel is shown on the top as average±SD and a representative gel is shown at the bottom.
(C) Normal monocytes were pre-treated in vitro with IFNγ followed by stimulation with LPS for 1 hour. Five hundred μg of protein were incubated with anti-MyD88 antibody at +4°C overnight; the loading of equal protein amounts included in the immunoprecipitation assay was conformed by immunoblotting against β-actin (input). The amount of IRAK1 immunopreciptated with anti-MyD88 antibodies was detected by western blot (bottom band) and quantified by densitometric analysis; shown is average±SD from n=3.
(D) Equal amounts (500 μg) of total protein from freshly isolated monocytes of controls (n=10) and cHCV patients (n=15) were incubated with anti-MyD88 antibody at +4°C overnight; each band represents a pool of 2 controls or 3 HCV patients. The amount of IRAK1 immunopreciptated with anti-MyD88 antibodies was detected and quantified as above; shown is average±SD.
The resistance to TLR tolerance in cHCV patients is reversible
Since monocytes are precursors for tissue macrophages, such as Kupffer cells (11), we differentiated monocytes in vitro with or without endotoxin, IFNγ or HCV core protein in order to identify if the loss of tolerance to TLR ligands seen in monocytes of cHCV patients is also a feature of macrophages. Similar to monocytes, normal macrophages responded with production of TNFα and showed both homo- and hetero-tolerance to endotoxin (Fig 6A) and HCV core protein (Fig 6B, C). However, IFNγ prevented the development of tolerance when added during macrophage differentiation and the first stimulation (Fig 6A). Surprisingly, in vitro differentiation of cHCV monocytes to macrophages restored their TLR tolerance: unlike freshly isolated cHCV monocytes (Fig 1B and Fig 2B), in vitro differentiated cHCV macrophages produced normal TNFα levels and showed tolerance to LPS (Fig 6A) and HCV core protein (Fig 6B, C). To approximate the in vivo conditions of the cHCV-infected host, we differentiated macrophages in vitro in the presence of IFNγ. Similar to control macrophages, IFNγ was capable of breaking both homo- and hetero-tolerance to LPS (Fig 6A) and to HCV core protein (Fig 6B, C) in cHCV macrophages, thus closely reproducing the lack of tolerance to TLR ligands seen in monocytes of cHCV patients (Fig 2B). These data suggested that the loss of tolerance to TLR ligands seen in cHCV monocytes is caused by in vivo environment and it is reversible; however it can be re-induced by modulatory agents, such as IFNγ, endotoxin and HCV core protein.
Figure 6. IFNγ is capable of breaking TLR tolerance in macrophages.

(A) Macrophages of controls (open bars) and cHCV patients (black bars) were differentiated in vitro, then stimulated with LPS (10pg/ml) for 24 hrs (1st stimulation) and re-challenged with LPS (100pg/ml), for additional 24 hrs (2nd stimulation). Where indicated IFNγ (100pg/ml) was added during the differentiation and 1st stimulation. The production of TNFα in the culture supernatants was analyzed in ELISA from n=6 (A–C).
(B) Macrophages were differentiated as above, then stimulated in vitro with LPS (10pg/ml) for 24 hrs (1st stimulation) and re-challenged with HCV core (25000 fMol/L) for additional 24 hrs (2nd stimulation).
(C) Macrophages were differentiated as above, then stimulated with HCV core (10000 fMol/L) for 24 hrs (1st stimulation) and re-challenged with HCV core (25000 fMol/L), for additional 24 hrs (2nd stimulation).
Patients with cHCV infection show evidence of macrophage activation in the liver
Finally, based on the activated phenotype of circulating cHCV monocytes and the stimulatory effect of IFNγ on macrophages, we hypothesized that the liver macrophages, Kupffer cells, may be activated in vivo during cHCV infection. Here we show that TNFα mRNA levels were significantly increased in livers of cHCV patients (Fig 7A). Kupffer cells produce TNFα and express CD163 and CD33, which are increased upon activation (16,17,21). We identified that CD163 (Fig 7B) and CD33 (Fig 7C) RNA levels were elevated in livers of cHCV patients compared to controls, suggesting Kupffer cell activation. We further employed mouse KCs to evaluate the relevance of the TLR tolerance. As shown in figure 7D, one-time in vivo encounter with LPS renders KCs tolerant to either LPS or HCV core protein, similar to normal monocytes (Fig 1A and 2A). However, Kupffer cells exposed to LPS in vivo at multiple occasions during prolonged periods of time failed to develop tolerance to either LPS or to HCV core protein, similar to monocytes of HCV-infected patients (Fig 1B and 2B). These data suggested that first, KCs develop tolerance to TLR ligands and second, in vivo endotoxinemia leads to loss of tolerance to subsequent TLR stimuli in KCs.
Figure 7. The patients with cHCV infection exhibit elevated levels of TNFα, CD163 and CD33 in livers.

The levels of RNA coding for TNFα(A), CD163 (B) and CD33 (C) were analyzed in livers of controls (n=3) and HCV patients (n=12) using real-time quantitative PCR. The asterisk (*) represents p<0.05.
(D) The mice were injected i/p with saline, LPS (5mg/kg) one time (the “1 dose of LPS” group) or 5 times (the “5 dose of LPS” group received 1mg/kg LPS every 3 days, total of 5 doses) and the Kupffer cells were isolated. In vitro the cells were stimulated with LPS (100ng/ml) or HCV core protein (25000 fMol/L) and the TNFα in culture supernatants was quantified using specific ELISA. The asterisk (*) represents p<0.05 using t-test from n=4 in saline and n=3 in LPS-treated groups.
Discussion
This study has provided three key observations related to the inflammatory cell activation during cHCV infection. First, we identified that monocytes, the precursors of tissue macrophages, are pre-activated in patients with chronic HCV infection. Second, we demonstrated that in vivo occurring concentrations of IFNγ, endotoxin and HCV core protein modulate monocyte functions and favor the increased frequency of MyD88/IRAK complexes, elevated NFκB activation, and excessive TNFα production seen in cHCV patient’s monocytes. Third, our data suggest that the mechanisms by which LPS, HCV core protein and IFNγ amplify the inflammatory monocytes/macrophages activation involve loss of TLR tolerance. Together these data provide evidence for both host and virus-derived factors modulate macrophages for perpetual inflammatory activation during cHCV infection.
Inflammation is a major component of the pathogenesis of cHCV infection (19,22); however its fine mechanisms are yet to be fully understood. TNFα, a potent inducer of inflammation and a key regulator of innate immunity, is beneficial for generation of the initial immune responses against viruses, including HCV (22). However TNFα has multiple harmful effects including induction of hepatocyte apoptosis, aggravation of liver fibrosis, and increase in the frequency of autoimmune diseases associated with HCV (23,24). Increased production of TNFα in T cells, NK, NKT cells and dendritic cells of cHCV-infected hosts was previously reported and may play a detrimental role in HCV clearance (6,22,25). Here we focused our attention on monocytes and tissue macrophages as potential sources for TNFα during cHCV infection to show that cells of patients with chronic HCV infection produce higher amounts of TNFα compared to controls upon stimulation with TNFα-inducing TLR ligands. Under normal regulation TLR tolerance limits TNFα production and it is protective against excessive immune activation and its harmful effects (10). Our data revealed that these protective mechanisms are de-regulated in HCV patients. We provide evidence that HCV patient’s monocytes fail to establish homo- and hetero-tolerance to pro-inflammatory cytokine-inducing TLR ligands, including TLR2/TLR1, TLR2/TLR6, TLR4, TLR3 and TLR7/8. Here we showed that HCV core protein, a TLR2 ligand (6,7), activated and induced tolerance in normal but not in cHCV monocytes. We also identified increased levels of endotoxin, a TLR4 ligand, in plasma of patients with cHCV infection, which is in agreement with previous reports (26). It is noteworthy that cHCV patients included in our study did not exhibit liver cirrhosis, yet low levels of endotoxemia occurred in patients with cHCV without advanced liver disease.
IFNγ is pivotal for efficient adaptive immune responses. We identified that IFNγ levels are higher in patients compared to controls, which is in agreement with other reports (27). Further, moderately elevated levels of IFNγ associated with endotoxinemia and HCV core protein present in the circulation during cHCV infection may play a detrimental role in the initiation and/or maintenance of the pro-inflammatory activation. In vitro, plasma levels of endotoxin comparable to those identified in cHCV patients induced LPS homo-tolerance, while IFNγ could reverse tolerance to TLR ligands. Our data on cHCV monocytes are in agreement with other researchers who observed that in vivo IFNγ restored the systemic TNFα response to endotoxin in LPS-desensitized mice and identified that monocytes played a critical role in this process (28). Both beneficial and detrimental effects of IFNγ during bacterial infection have been recognized (29). Our results suggest that moderately elevated levels of IFNγ seen in the circulation of cHCV patients may favor pro-inflammatory activation in monocytes and, possibly, tissue macrophages. Most importantly, IFNγ is key for establishment of anti-viral immunity and although the levels of IFNγ found in our patients may not be sufficient to support a robust adaptive immune response (22), we show that they are sufficient for pro-inflammatory activation of monocytes/macrophages.
The mechanism of TLR tolerance is complex (30) and it is yet to be fully determined how IFNγ breaks TLR tolerance. Upon stimulation with a specific ligand, most TLRs recruit the common TLR adaptor MyD88, IRAK and TRAF6 proteins and trigger MAPK and NFκB signaling to produce pro-inflammatory cytokines (4,8). TLR-mediated activation is regulated by expression of TLRs and by inhibitory molecules ST2, SIGIRR, SHIP, TWIST, Pi3K, and TTP. We failed to detect differences in protein expression of TLR2, TLR4 or any of the above-mentioned negative regulators in cHCV monocytes compared to controls (data not shown). In TLR ligand-induced tolerance the functions of IRAK1, MAPK, IKK and NFκB components are impaired (8,9). In contrast, cytokine-induced break in TLR tolerance is characterized by lack of changes in TLR expression, increased formation of MyD88/IRAK complex, increased NFκB activation and elevated levels of pro-inflammatory cytokine production (8,9,30). We identified that cHCV monocytes exhibited multiple prerequisites of excessive response to TLR ligands: high levels of MyD88/IRAK complexes, pre-activation of NFκB, and no changes at the levels of TLR proteins or at the level of negative regulators. We observed a similar pattern of changes in TLR-tolerant cells induced in vitro by IFNγ treatment, in agreement with other investigators (8,9). These results suggested that the in vivo cytokine milieu likely affects the ability of monocytes and tissue macrophages to respond to repeated encounter with TLR ligands during cHCV infection. Our data using _in vitro_-differentiated macrophages support this hypothesis: in contrast to freshly isolated cHCV monocytes, cHCV macrophages differentiated in vitro without the cHCV environment developed tolerance to TLR ligands. We also show that the cHCV _in vivo_-conditioning of the monocytes that renders them resistant to TLR-mediated tolerance is reversible upon elimination of the modulatory agents, and the presence of modulatory agents (IFNγ, LPS and HCV core protein) during macrophage differentiation can reproduce the lack of TLR tolerance seen in monocytes.
Based on our results with monocytes/macrophages we, thus, speculate that during cHCV infection HCV core protein may act together with host-derived factors, such as IFNγ and endotoxin, to modulate the function of tissue macrophages, including Kupffer cells. In agreement with other investigators (21), we show that cHCV patients present markers of macrophage activation, such as CD163 and CD33, in the livers, suggestve of in vivo KC activation. Using mouse KCs we identified that they are capable of establishing tolerance to TLR ligands in a manner similar to monocytes and macrophages. Further, we show that in vivo contact with LPS, a TLR4 ligand, modulates Kupffer cells functions: where short term endotoxinemia renders KCs resistant, while prolonged endotoxemia makes KCs sensitive to subsequent TLR-mediated stimulation. Our results using KC’s immature myeloid precursors, the monocytes, and their mature forms, the _in vitro_-differentiated human macrophages, as well as mouse KCs lend support to the contention that human KCs in cHCV infection may present a similar pro-inflammatory-activated phenotype.
In conclusion, we propose that IFNγ, endotoxin, and HCV core protein collectively contribute to functional modulation of monocytes and macropages during cHCV infection. Due to the common cellular origin, it is possible that similar changes may occur in Kupffer cells locally in the liver. Our results suggest novel mechanisms for persistent, immune-mediate inflammation in patients with chronic hepatitis C virus infection.
Acknowledgments
The authors thank Drs. Rudy Bayaert, Mark Goggeshall, and Paul Bonjanen for technical advises. We are grateful to the members of Liver Center at UMass for their help with patient recruitment, to patients and blood donors for cooperation, and to UMMass Flow Cytometry Facility and CFAR for technical support.
Grant support: AA11576 from NIAAAA to G.S., PHS grant AA12862, UMass CFAR grant 5P30 AI42845 and the PHS grant DK32520.
Footnotes
The authors have no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim WR, Brown RS, Jr, Terrault NA, El-Serag H. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–242. doi: 10.1053/jhep.2002.34734. [DOI] [PubMed] [Google Scholar]
- 2.Ward S, Lauer G, Isba R, Walker B, Klenerman P. Cellular immune responses against hepatitis C virus: the evidence base 2002. Clin Exp Immunol. 2002;128:195–203. doi: 10.1046/j.1365-2249.2002.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neuman MG, Benhamou JP, Malkiewicz IM, Ibrahim A, Valla DC, Martinot-Peignoux M, Asselah T, Bourliere M, Katz GG, Shear NH, Marcellin P. Kinetics of serum cytokines reflect changes in the severity of chronic hepatitis C presenting minimal fibrosis. J Viral Hepat. 2002;9:134–140. doi: 10.1046/j.1365-2893.2002.00343.x. [DOI] [PubMed] [Google Scholar]
- 4.Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 5.Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res. 2004;63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 6.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 7.Duesberg U, von dem Bussche A, Kirschning C, Miyake K, Sauerbruch T, Spengler U. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)--core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol Lett. 2002;84:89–95. doi: 10.1016/s0165-2478(02)00178-5. [DOI] [PubMed] [Google Scholar]
- 8.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR) 2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 9.Adib-Conquy M, Cavaillon JM. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J Biol Chem. 2002;277:27927–27934. doi: 10.1074/jbc.M200705200. [DOI] [PubMed] [Google Scholar]
- 10.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 11.Naito M, Hasegawa G, Ebe Y, Yamamoto T. Differentiation and function of Kupffer cells. Med Electron Microsc. 2004;37:16–28. doi: 10.1007/s00795-003-0228-x. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka M, Ishibashi H, Hirata Y, Miki K, Kudo J, Niho Y. Tumor necrosis factor production by rat Kupffer cells-regulation by lipopolysaccharide, macrophage activating factor and prostaglandin E2. J Clin Lab Immunol. 1996;48:17–31. [PubMed] [Google Scholar]
- 13.Wheeler MD. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health. 2003;27:300–306. [PMC free article] [PubMed] [Google Scholar]
- 14.Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 15.Adams DH, Hubscher SG. Systemic viral infections and collateral damage in the liver. Am J Pathol. 2006;168:1057–1059. doi: 10.2353/ajpath.2006.051296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgio VL, Ballardini G, Artini M, Caratozzolo M, Bianchi FB, Levrero M. Expression of co-stimulatory molecules by Kupffer cells in chronic hepatitis of hepatitis C virus etiology. Hepatology. 1998;27:1600–1606. doi: 10.1002/hep.510270620. [DOI] [PubMed] [Google Scholar]
- 17.Royer C, Steffan AM, Navas MC, Fuchs A, Jaeck D, Stoll-Keller F. A study of susceptibility of primary human Kupffer cells to hepatitis C virus. J Hepatol. 2003;38:250–256. doi: 10.1016/s0168-8278(02)00418-x. [DOI] [PubMed] [Google Scholar]
- 18.Ray RB, Ray R. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol Lett. 2001;202:149–156. doi: 10.1111/j.1574-6968.2001.tb10796.x. [DOI] [PubMed] [Google Scholar]
- 19.Holtmann MH, Neurath MF. Differential TNF-signaling in chronic inflammatory disorders. Curr Mol Med. 2004;4:439–444. doi: 10.2174/1566524043360636. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Sidiropoulos P, Song G, Pagliari LJ, Birrer MJ, Stein B, Anrather J, Pope RM. TNF-alpha gene expression in macrophages: regulation by NF-kappa B is independent of c-Jun or C/EBP beta. J Immunol. 2000;164:4277–4285. doi: 10.4049/jimmunol.164.8.4277. [DOI] [PubMed] [Google Scholar]
- 21.Hiraoka A, Horiike N, Akbar SM, Michitaka K, Matsuyama T, Onji M. Expression of CD163 in the liver of patients with viral hepatitis. Pathol Res Pract. 2005;201:379–384. doi: 10.1016/j.prp.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Rehermann B. Cellular immune response to the hepatitis C virus. J Viral Hepat. 1999;6:31–35. doi: 10.1046/j.1365-2893.1999.00008.x. [DOI] [PubMed] [Google Scholar]
- 23.Tilg H. Cytokines and liver diseases. Can J Gastroenterol. 2001;15:661–668. doi: 10.1155/2001/746736. [DOI] [PubMed] [Google Scholar]
- 24.Ramos-Casals M, Garcia-Carrasco M, Cervera R, Filella X, Trejo O, de la Red G, Gil V, Sanchez-Tapias JM, Font J, Ingelmo M. Th1/Th2 cytokine imbalance in patients with Sjogren syndrome secondary to hepatitis C virus infection. Semin Arthritis Rheum. 2002;32:56–63. doi: 10.1053/sarh.2002.33724. [DOI] [PubMed] [Google Scholar]
- 25.Nelson DR, Lim HL, Marousis CG, Fang JW, Davis GL, Shen L, Urdea MS, Kolberg JA, Lau JY. Activation of tumor necrosis factor-alpha system in chronic hepatitis C virus infection. Dig Dis Sci. 1997;42:2487–2494. doi: 10.1023/a:1018804426724. [DOI] [PubMed] [Google Scholar]
- 26.Caradonna L, Mastronardi ML, Magrone T, Cozzolongo R, Cuppone R, Manghisi OG, Caccavo D, Pellegrino NM, Amoroso A, Jirillo E, Amati L. Biological and clinical significance of endotoxemia in the course of hepatitis C virus infection. Curr Pharm Des. 2002;8:995–1005. doi: 10.2174/1381612024606983. [DOI] [PubMed] [Google Scholar]
- 27.Cribier B, Schmitt C, Rey D, Lang JM, Kirn A, Stoll-Keller F. Production of cytokines in patients infected by hepatitis C virus. J Med Virol. 1998;55:89–91. doi: 10.1002/(sici)1096-9071(199806)55:2<89::aid-jmv1>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 28.Bundschuh DS, Barsig J, Hartung T, Randow F, Docke WD, Volk HD, Wendel A. Granulocyte-macrophage colony-stimulating factor and IFNgamma restore the systemic TNFalpha response to endotoxin in lipopolysaccharide-desensitized mice. J Immunol. 1997;158:2862–2871. [PubMed] [Google Scholar]
- 29.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 30.Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133–150. doi: 10.1179/096805106X102255. [DOI] [PubMed] [Google Scholar]