Ayurvedic Medicine Constituent Withaferin A Causes G2 and M Phase Cell Cycle Arrest in Human Breast Cancer Cells (original) (raw)
. Author manuscript; available in PMC: 2009 Jan 1.
Published in final edited form as: Nutr Cancer. 2008;60(Suppl 1):51–60. doi: 10.1080/01635580802381477
Abstract
Withaferin A (WA) is derived from the medicinal plant Withania somnifera that has been safely used for centuries in the Indian Ayurvedic medicine for treatment of various ailments. We now demonstrate that WA treatment causes G2 and mitotic arrest in human breast cancer cells. Treatment of MDA-MB-231 (estrogen-independent) and MCF-7 (estrogen-responsive) cell lines with WA resulted in a concentration- and time-dependent increase in G2-M fraction, which correlated with a decrease in levels of cyclin-dependent kinase 1 (Cdk1), cell division cycle 25B (Cdc25B) and/or Cdc25C proteins leading to accumulation of Tyr15 phosphorylated (inactive) Cdk1. Ectopic expression of Cdc25C conferred partial yet significant protection against WA-mediated G2-M phase cell cycle arrest in MDA-MB-231 cells. The WA-treated MDA-MB-231 and MCF-7 cells were also arrested in mitosis as judged by fluorescence microscopy and analysis of Ser10 phosphorylated histone H3. Mitotic arrest resulting from exposure to WA was accompanied by an increase in the protein level of anaphase promoting complex/cyclosome substrate securin. In conclusion, the results of the present study suggest that G2-M phase cell cycle arrest may be an important mechanism in anti-proliferative effect of WA against human breast cancer cells.
Keywords: Withaferin A, Dietary Supplement, Breast Cancer, Cdc25C, Cell Cycle
Introduction
Breast cancer is a major health concern for women Worldwide. The American Cancer Society estimates diagnosis of nearly 182,460 new cases of invasive breast cancer in the United States alone in the year 2008. The known risk factors for breast cancer include family history, Li-Fraumeni syndrome, atypical hyperplasia of the breast, late-age at first full-term pregnancy, early menarche, and late menopause (1-3). Because some of these risk factors are not easily modifiable (e.g., genetic predisposition), other strategies for reduction of breast cancer risk must be considered. Even though selective estrogen-receptor (ER) modulators (e.g., tamoxifen) appear promising for prevention of breast cancer, this strategy is largely ineffective against ER negative breast cancers (4,5). Moreover, selective ER modulators have other side effects including increased risk of uterine cancer, thromboembolism, cataracts and perimenopausal symptoms (4,5). Therefore, identification and clinical development of agents that are relatively safe but can suppress growth of both ER positive and ER negative human breast cancers is highly desirable. Natural products have received increasing attention in recent years for identification of novel anticancer agents (6).
Withaferin A (WA) is derived from the medicinal plant Withania somnifera L. (commonly known as Ashwagandha or Indian winter cherry), which has been safely used for centuries in the traditional Indian (Ayurvedic) medicine practice for the treatment of various ailments. The known pharmacological effects of Withania somnifera include modulation of immune function (7), cardioprotection from ischemia reperfusion injury (8), protection against 6-hydroxydopamine-induced Parkinsonism in rats (9), anti-bacterial and anti-inflammatory effects (10,11). Ashwagandha is also recommended as a tonic for overall well being (12). The WA has attracted a great deal of research interest as a potential anticancer agent. For example, WA is a radiosensitizer and inhibitor of growth of mouse Ehrlich ascites carcinoma and transplantable sarcoma in vivo (13-16). The WA is a potent inhibitor of nuclear factor-κB activation (17,18). Crude ethanol extract of Withania somnifera Dunal suppressed the lipopolysaccharide-induced production of inflammatory cytokines in peripheral blood mononuclear cells (19). Suppression of angiogenesis, alteration of cytoskeletal architecture, and inhibition of proteasomal activity by WA has also been documented (20-22). More recent studies have demonstrated that WA suppresses growth of human cancer cells in culture and in vivo by causing apoptosis (23-25). For instance, WA treatment triggered a Par-4 dependent apoptosis in PC-3 human prostate cancer cells and inhibited growth of PC-3 xenografts in vivo (23). Another study documented generation of reactive oxygen species and mitochondrial dysfunction in apoptotic response to WA in HL-60 cells (24).
We have shown recently that WA treatment triggers FOXO-3a and Bim-mediated apoptosis in MDA-MB-231 and MCF-7 human breast cancer cells and WA administration suppresses growth of MDA-MB-231 xenografts in vivo in female athymic mice in association with apoptosis induction (25). The present study demonstrates that WA treatment causes irreversible G2-M phase cell cycle arrest in MDA-MB-231 and MCF-7 cell lines.
Materials and Methods
Reagents
The WA was purchased from ChromaDex. The Propidium iodide, RNaseA, 4′,6-diamidino-2-phenylindole (DAPI), phosphatase inhibitors, and the antibodies against α-tubulin and Tyr15 phosphorylated Cdk1 were purchased from Sigma. The protease inhibitor mixture was from BD Pharmingen. The antibiotic mixture, non-essential amino acids, sodium pyruvate, trypan blue, and fetal bovine serum (FBS) were purchased from Invitrogen. The 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was from Molecular Probes. The combined superoxide dismutase and catalase mimetic EUK134 was from Eukarion. The antibodies against Cdk1 and Cdc25C were from Santa Cruz Biotechnology, the antibodies against cyclinB1 and actin were from Oncogene Research Products, and the antibody against Cdc25B was from BD Pharmingen. The antibody specific for detection of Ser10 phosphorylated histone H3 was from Upstate Biotechnology and the anti-securin antibody was from Medical and Biological Laboratories.
Cell culture and analysis of cell cycle distribution
The human breast cancer cell lines MDA-MB-231 and MCF-7 and spontaneously immortalized normal mammary epithelial cell line MCF-10A were obtained from American Type Culture Collection and maintained as described by us previously (26). Stock solution of WA was prepared in DMSO and an equal volume of DMSO was added to the controls. The effect of WA treatment on cell cycle distribution was determined by flow cytometry after staining the cells with propidium iodide as described by us previously (26). Briefly, 1×106 cells were seeded, allowed to attach by overnight incubation, and exposed to DMSO (control) or desired concentrations of WA for specified time periods. Both floating and adherent cells were collected, washed with phosphate buffered saline (PBS), and fixed in 70% ethanol overnight at 4°C. The cells were then treated with 80 μg/mL RNaseA and 50 μg/mL propidium iodide for 30 min and analyzed using a Coulter Epics XL Flow cytometer. Percentage of cells in different phases of the cell cycle was computed for DMSO-treated control and WA-treated cultures. To determine if the WA-mediated cell cycle arrest was reversible, the cells were first treated with either DMSO (control) or 2 μM WA for 24 h and then washed with PBS and maintained in drug-free complete medium for an additional 24 h prior to the analysis of cell cycle distribution.
Immunoblotting
Desired cell line (1×106) was seeded in 100-mm culture dishes, allowed to attach by overnight incubation, and treated with 2 μM WA for specified time intervals. Both floating and attached cells were collected and lysed as described by us previously (27). The cell lysate was cleared by centrifugation at 14,000 rpm for 30 min. The lysate proteins were resolved by 10 or 12.5% sodium-dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto membrane. The membrane was incubated with Tris-buffered saline containing 0.05% Tween-20, and 5% (w/v) nonfat dry milk. The membrane was then treated with the desired primary antibody for 1 h at room temperature or overnight at 4°C. Following treatment with an appropriate secondary antibody, the immunoreactive bands were visualized using enhanced chemiluminescence method. The blots were stripped and re-probed with anti-actin antibody to normalize for differences in protein loading.
Determination of reactive oxygen species (ROS)
Intracellular ROS generation was determined by flow cytometry following staining with H2DCFDA as described by us previously (28). Briefly, cells were seeded, allowed to attach by overnight incubation, and exposed to DMSO (control) or specified concentrations of WA for the indicated time intervals. The cells were then stained with 5 μM H2DCFDA for 30 min at 37°C. The cells were collected and the fluorescence was measured using a Coulter Epics XL flow cytometer.
Ectopic expression of Cdc25C by transient transfection
The MDA-MB-231 cells were transiently transfected with empty pcDNA3.1 vector or pcDNA3.1 vector encoding wild type Cdc25C (29). Briefly, the cells were seeded in 6-well plates and transfected with plasmid DNA using Fugene6 transfection reagent. After 24 h of transfection, the cells were treated with DMSO (control) or 2 μM WA for 8 h. Both floating and adherent cells were collected and processed for analysis of cell cycle distribution or immunoblotting as described above.
Immunofluorescence microscopy
The MDA-MB-231 or MCF-7 cells (5×104) were plated on coverslips and allowed to attach by overnight incubation. The cells were then exposed to DMSO (control) or 2 μM WA for specified time intervals at 37°C, washed with PBS, and fixed with 3% paraformaldehyde at 4°C overnight. The cells were permeabilized with 0.5% Triton X-100 for 15 min at room temperature, washed with PBS, and incubated with PBS containing 0.5% (w/v) bovine serum albumin (BSA), and 0.15% (w/v) glycine (BSA buffer) for 1 h at room temperature. Subsequently, the cells were treated with anti-α-tubulin antibody (1:4,000 dilution in BSA buffer) for 1 h at room temperature. The cells were washed with BSA buffer and incubated with 1 μg/mL Alexa Fluor 568-conjugated antibody for 1 h at room temperature followed by counterstaining with 500 ng/mL DAPI for 5 min. Slides were mounted and examined under a Leica fluorescence microscope at 100× magnification. At least 100 cells were analyzed for mitotic figures with condensed chromatin.
Flow cytometric analysis of Ser10 phosphorylated histone H3
The effect of WA treatment on level of Ser10 phosphorylated histone H3 was determined by flow cytometry as previously described (30). Briefly, the cells were treated with DMSO (control) or WA, fixed with 70% ethanol at 4°C overnight, and incubated on ice for 15 min with PBS containing 0.5% Triton X-100. The cells were collected, suspended in 100 μL of PBS containing 1% BSA and 0.75 μg polyclonal anti-phospho-(Ser10)-histone H3 antibody, and incubated at 4°C overnight. The cells were then rinsed with PBS containing 1% BSA and incubated in dark with FITC-conjugated goat anti-rabbit antibody (1:50 dilution in PBS containing 1% BSA) for 30 min at room temperature. The fluorescence was examined using a Coulter Epics XL flow cytometer.
Statistical analysis
One-way ANOVA followed by Dunnett's or Bonferroni's multiple comparison test was used to determine statistical significance of difference in measured parameters between groups. Difference was considered significant at P<0.05.
Results
WA treatment caused G2-M phase cell cycle arrest in human breast cancer cells
We have shown previously that WA treatment suppresses viability of MDA-MB-231 and MCF-7 human breast cancer cells (25). In the present study, we used the same cell lines to test whether growth suppressive effect of WA against breast cancer cells was due to perturbations in the cell cycle progression. The MDA-MB-231 cell line, which was originally derived from a stage IV invasive ductal carcinoma, is estrogen receptor negative and expresses mutant p53. The MCF-7 cell line, which is estrogen receptor positive, was isolated from pleural effusion of a stage IV invasive ductal carcinoma. The MCF-7 cells are aneuploid with high chromosomal instability and express normal p53. Fig. 1A depicts flow histograms for cell cycle distribution in DMSO-treated control and WA-treated MDA-MB-231 cells. As can be seen in Fig. 1B, the WA treatment (24 h exposure) resulted in statistically significant enrichment of G2-M phase cells at 2 and 3 μM concentrations compared with DMSO-treated control. The WA-mediated G2-M phase cell cycle arrest was accompanied by a decrease in G0-G1 and/or S phase cells (Fig. 1B). Similar to the MDA-MB-231 cell line, the WA treatment caused G2-M phase cell cycle arrest in MCF-7 cells that was accompanied by a decrease in G0-G1 and S fractions (Fig. 1C). However, the WA-mediated G2-M phase cell cycle arrest in MCF-7 cells was evident even at 1 μM concentration (Fig. 1D). In time course experiments using 2 μM WA, the G2-M phase cell cycle arrest in both MDA-MB-231 (Fig. 2A) and MCF-7 (Fig. 2B) cell lines was evident as early as 8 h after treatment and persisted for the duration of the experiment (30 h post-treatment). Collectively, these results indicated that WA treatment caused sustained G2-M phase cell cycle arrest in human breast cancer cells regardless of their estrogen responsiveness or p53 status.
Figure 1.
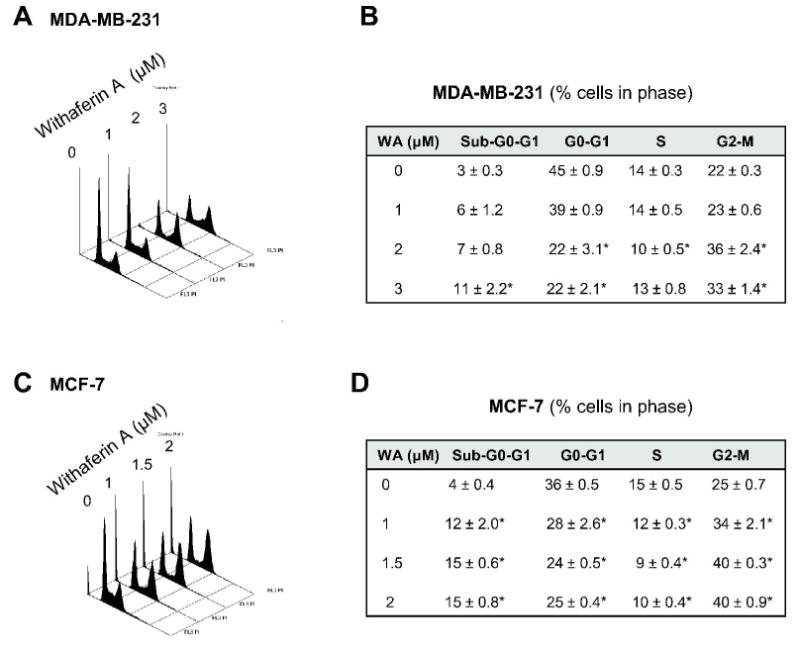
Representative histograms depicting cell cycle distribution in (A) MDA-MB-231 and (C) MCF-7 cultures following 24 h treatment with DMSO (control) or the indicated concentrations of withaferin A (WA). Percentage of cells in different phases of the cell cycle in (B) MDA-MB-231 and (D) MCF-7 cultures following 24 h treatment with DMSO (control) or the indicated concentrations of WA. Results are mean ± SE (n=3). *, P<0.05, significantly different compared with DMSO-treated control by one-way ANOVA followed by Dunnett's test. Similar results were observed in two independent experiments.
Figure 2.
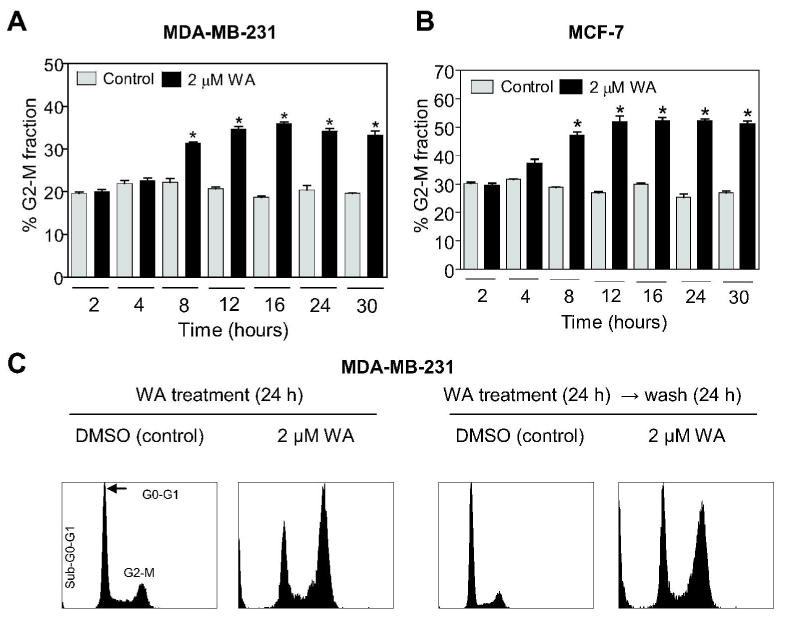
Percentage of G2-M fraction in (A) MDA-MB-231 and (B) MCF-7 cultures following treatment with DMSO (control) or 2 μM WA for the indicated time periods. C, left panels, cell cycle distribution in MDA-MB-231 cultures treated for 24 h with DMSO (control) or 2 μM WA; right panels, cell cycle distribution in MDA-MB-231 cells treated for 24 h with either DMSO (control) or 2 μM WA and then cultured in drug-free media for 24 h prior to the analysis of cell cycle distribution. Columns, mean (n=3); bars, SE. *, P<0.05, significantly different compared with the corresponding control by one-way ANOVA. Similar results were observed in two independent experiments.
WA-mediated G2-M phase cell cycle arrest in human breast cancer cells was irreversible
The cell cycle checkpoints are normally activated in response to external stimuli (e.g., DNA damage) to allow the cells time to repair damage prior to resumption of the cell cycle progression. We raised the question of whether WA-mediated G2-M phase cell cycle arrest in breast cancer cells was reversible. To test this possibility, we treated the MDA-MB-231 and MCF-7 cells with either DMSO (control) or 2 μM WA for 24 h. The cells were then either processed for analysis of cell cycle distribution or washed with PBS and incubated in drug-free medium for an additional 24 h prior to flow cytometry for cell cycle analysis. A twenty-four hour exposure of MDA-MB-231 (Fig. 2C, left panels) and MCF-7 cells (results not shown) to 2 μM WA resulted in statistically significant enrichment of G2-M fraction relative to DMSO-treated control. The G2-M phase cell cycle arrest in WA-treated MDA-MB-231 (Fig. 2C, right panels) and MCF-7 cells (results not shown) was maintained even after culture of these cells in drug free medium for an additional 24 h. However, the sub-diploid apoptotic fraction was relatively higher in WA-treated cells that were cultured in drug-free medium for an additional 24 h compared with cells treated with WA for 24 h. Collectively, these results indicated that the WA-mediated G2-M phase cell cycle arrest was irreversible in both cell lines and a fraction of G2-M arrested cells were driven to apoptotic cell death.
Next, we raised the question of whether WA-mediated cell cycle arrest was selective towards breast cancer cells, which is a highly desirable feature of potential anticancer agents. We addressed this question by determining the effect of WA treatment on cell cycle distribution of MCF-10A cells. The MCF-10A cell line is non-tumorigenic in athymic mice and has been used extensively as a representative normal mammary epithelial cell line. The MCF-10A cell line was originally isolated from fibrocystic breast disease and was spontaneously immortalized. The MCF-10A cells have intact cell cycle checkpoints and normal proliferation controls. The MCF-10A cell line was resistant to G2-M phase cell cycle arrest by WA treatment compared with MDA-MB-231 or MCF-7 cells (results not shown).
WA treatment altered levels of proteins involved in regulation of G2-M transition
To gain insight into the mechanism of G2-M phase cell cycle arrest in our model, we determined the effect of WA treatment on levels of proteins involved in regulation of G2-M transition including cyclinB1, Cdk1, Cdc25B and Cdc25C (31,32). The WA treatment caused an increase in the level of cyclinB1 protein in both cell lines (Fig. 3). The level of Cdk1 protein was decreased markedly upon treatment with WA in both MDA-MB-231 (up to 60% decrease compared with control) and MCF-7 cell lines (up to 80% decrease compared with control), which was clearly evident at 16 and 24 h time points. The WA treatment exhibited a differential response on Cdc25B protein level in MDA-MB-231 (induction of Cdc25B protein level) versus MCF-7 cells (moderate induction of Cdc25B protein level at 2-4 h time points followed by a decline in its level at 8-24 time points). Furthermore, the WA treatment caused a sustained decrease in protein level of Cdc25C for up to 30 h in MDA-MB-231 cells (Fig. 3). Interestingly, the WA-mediated decline in Cdc25C protein level was transient (2-8 h time points) in MCF-7 cells (Fig. 3). The net result of these effects was accumulation of Tyr15 phosphorylated (inactive) Cdk1, which persisted for up to 12-16 h (Fig. 3). These results indicated that the WA treatment caused inactivation of Cdk1/cyclinB1 kinase complex.
Figure 3.
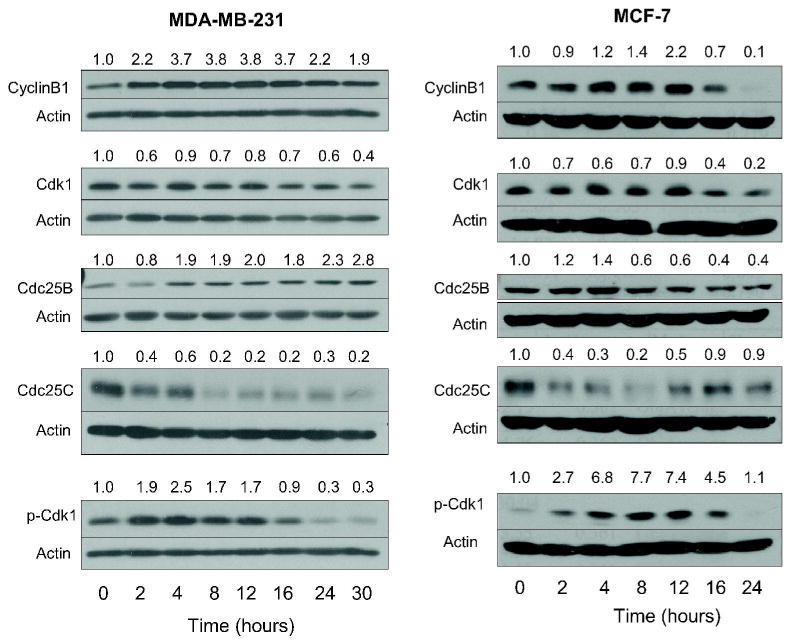
Immunoblotting for cyclinB1, Cdk1, Cdc25B, Cdc25C, and Tyr15 phosphorylated Cdk1 using lysates from MDA-MB-231 and MCF-7 cells treated with 2 μM withaferin A (WA) for the indicated time periods. Membranes were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Numbers on top of the bands represent changes in protein levels as determined by densitometric analysis of the immunoreactive bands and corrected for actin loading control. Immunoblotting for each protein was performed at least twice using independently prepared lysates.
Ectopic expression of Cdc25C conferred protection against WA-mediated G2-M phase cell cycle arrest
Next, we designed experiments using MDA-MB-231 cell line to explore a possibility that the WA-mediated G2-M phase cell cycle arrest was due to suppression of Cdc25C protein level. Previous studies have indicated that Cdc25C protein contains two cysteine residues that are susceptible to redox modification (29). The redox modification of Cdc25C cysteine controls proteasomal degradation and hence stability of this protein (29). Initially, we designed experiments to test whether WA treatment caused oxidative stress in breast cancer cells, an effect previously documented in WA-treated HL-60 cells (24). Intracellular ROS generation in control and WA-treated cells was determined by flow cytometry following staining with H2DCFDA, which is oxidized in the presence of peroxides (e.g., hydrogen peroxide) to yield fluorescent 2′,7′-dichlorofluorescein (DCF). As can be seen in Fig. 4A, the WA treatment caused oxidation of H2DCFDA in MDA-MB-231 cells, which was evident as early as 1 h after treatment. Similar results were observed in MCF-7 cells (results not shown). To test contribution of ROS generation in cell cycle arrest caused by WA, we used a combined mimetic of superoxide dismutase and catalase (EUK134). As shown in Fig. 4B, the WA-mediated ROS generation was significantly attenuated in the presence of EUK134. On the other hand, EUK134 treatment did not have any appreciable effect on WA-induced G2-M phase cell cycle arrest (Fig. 4C). As shown in Fig. 4D, similar to un-transfected cells (Fig. 1A), the WA treatment caused enrichment of G2-M fraction in MDA-MB-231 cells transiently transfected with the empty-vector. Ectopic expression of Cdc25C protein in MDA-MB-231 cells [about 2-fold overexpression (Fig. 4D, inset, lane 2) compared with empty-vector-transfected cells (Fig. 4D, inset, lane 1)] conferred partial yet statistically significant protection against WA-mediated G2-M arrest (Fig. 4D). These results indicated that WA-mediated G2-M phase cell cycle arrest in our model was, at least in part, caused by a decline in the level of Cdc25C protein.
Figure 4.
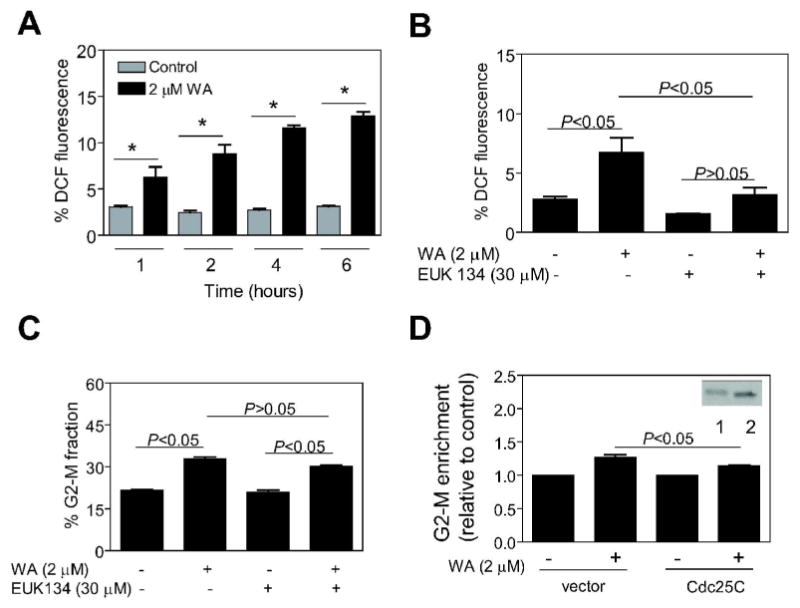
(A) Percentage of 2′,7′-dichlorofluorescein (DCF) positive (a measure of reactive oxygen species production) MDA-MB-231 cells following treatment with DMSO (control) or 2 μM withaferin A (WA) for the indicated time periods. Columns, mean (n=3); bars, SE. *, P<0.05, significantly different compared with the corresponding control by one-way ANOVA. (B) Percentage of DCF fluorescence in MDA-MB-231 cells treated with 2 μM WA for 8 h with or without a 2 h pre-treatment with 30 μM EUK134. (C) Percentage of G2-M fraction in MDA-MB-231 cells treated with 2 μM WA for 8 h with or without a 2 h pretreatment with 30 μM EUK134. (D) Enrichment of G2-M fraction relative to corresponding DMSO-treated control in MDA-MB-231 cells transiently transfected with either empty-vector or vector encoding Cdc25C and then treated with DMSO (control) or 2 μM WA for 8 h. Inset, immunoblotting for Cdc25C protein using lysates from MDA-MB-231 cells transiently transfected with empty-vector (lane 1) or vector encoding Cdc25C (lane 2). In panels B-D, columns, mean (n=3); bars, SE; *, P<0.05, significantly different between the indicated groups by one-way ANOVA followed by Bonferroni's multiple comparison test.
WA treatment caused mitotic arrest in human breast cancer cells
It is interesting to note that the WA-treated MDA-MB-231 and MCF-7 cells exhibit an increase in cyclinB1 protein levels (Fig. 3). In cycling cells, the level of cyclinB1 protein increases abruptly as the cells acquire 4N DNA content (G2 phase), peaks during the metaphase/anaphase transition, and declines precipitously upon completion of mitosis (33,34). Degradation of cyclinB1 protein by APC/C is believed to be necessary for exit from mitosis (33,34). Because WA treatment resulted in accumulation of cyclinB1 protein (Fig. 3), we reasoned that the WA-treated breast cancer cells might be arrested in mitosis. We explored this possibility by immunofluorescence microscopy following staining of the control (DMSO-treated) and the WA-treated cells with anti-α-tubulin antibody and DAPI. The DMSO-treated control MDA-MB-231 (Fig. 5A, left panel) and MCF-7 cells (results not shown) exhibited intact microtubule network. The α-tubulin staining in a large fraction of WA-treated MDA-MB-231 (Fig. 5A, left panel) and MCF-7 cells (results not shown) was characterized by porous appearance suggesting disruption of the microtubule network. Furthermore, DAPI staining revealed abundance of mitotic figures with condensed chromatin in WA-treated MDA-MB-231 cells, which were much less frequent in the DMSO-treated control cells. The mitotic figures with condensed chromatin were scored from cultures of control and WA-treated MDA-MB-231 cells, and the results are presented in Fig. 5A (right panel). The mitotic arrest was evident as early as 12 h post-treatment with 2 μM WA (Fig. 5A, right panel).
Figure 5.
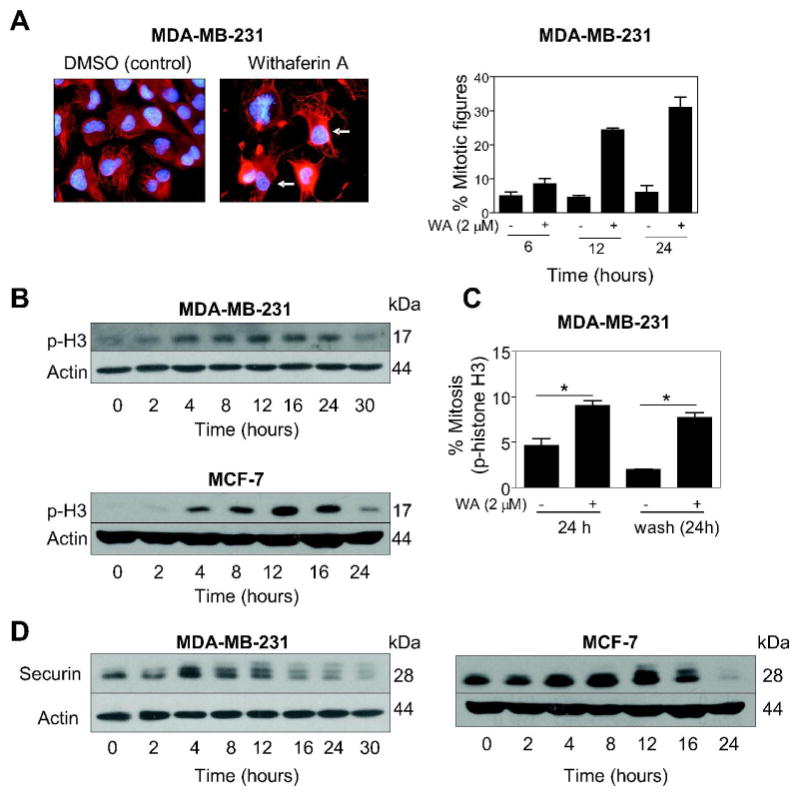
(A) left panels, fluorescence microscopic images depicting α-tubulin (red) and DAPI (blue) staining in MDA-MB-231 cells treated for 24 h with either DMSO or 2 μM withaferin A (WA). Note abundance of mitotic figures with condensed chromatin (identified by arrows) in WA-treated MDA-MB-231 cells, which were rarely seen in DMSO-treated controls. Right panel, percentage of mitotic figures with condensed chromatin in MDA-MB-231 cells treated with DMSO (control) or 2 μM WA for 6, 12 or 24 h. Columns, mean (n= 2); error bars are included to indicate the range of values. (B) Immunoblotting for Ser10 phosphorylated histone H3 using lysates from MDA-MB-231 and MCF-7 cells treated with 2 μM WA for the indicated time intervals. (C) Percentage of mitotic cells, determined by flow-cytometric analysis of Ser10 phosphorylated histone H3, in MDA-MB-231 cells treated for 24 h with DMSO (control) or 2 μM WA or maintained in drug-free complete medium after DMSO or WA treatment for an additional 24 h. Columns, mean (n=3); bars, SE. *, P<0.05, significantly different between the indicated groups by one-way ANOVA followed by Bonferroni's multiple comparison test. (D) Immunoblotting for securin using lysates from MDA-MB-231 and MCF-7 cells treated with 2 μM WA for the indicated time periods. In panels B and D, membranes were stripped and re-probed with anti-actin antibody to ensure equal protein loading. Immunoblotting for each protein was performed at least twice using independently prepared lysates.
WA treatment caused Ser10 phosphorylation of histone H3
The mitotic arrest caused by WA was confirmed by analysis of Ser10 phosphorylated histone H3, which is a highly sensitive marker for mitotic cells (35). As can be seen in Fig. 5B, the WA treatment (2 μM) caused an increase in the levels of Ser10 phosphorylated histone H3 in both cell lines. The WA-mediated Ser10 phosphorylation of histone H3 was more pronounced in MCF-7 cells than in the MDA-MB-231 cell line (Fig. 5B) and decreased markedly by 24 (MCF-7) to 30 h (MDA-MB-231). Next, we proceeded to test whether the WA-mediated mitotic arrest was reversible. The fraction of mitotic cells was only slightly decreased even after a 24 h culture of WA-treated MDA-MB-231 cells (2 μM for 24 h) in drug-free medium as judged by flow cytometric analysis of Ser10 phosphorylated histone H3 (Fig. 5C). Collectively, these results indicated that the WA-treated MDA-MB-231 and MCF-7 cells were arrested in mitosis.
Initiation of anaphase as well as mitotic exit depends on active APC/C, which is a multi-subunit ubiquitin protein ligase complex (36,37). To determine if the WA-mediated mitotic arrest was due to inactivation of APC/C, we performed immunoblotting for a known APC/C substrate securin. As can be seen in Fig. 5D, the WA-treated MDA-MB-231 and MCF-7 cells exhibited an increase in protein level of securin, which was maintained for at least up to 12 h post-exposure (Fig. 5D). Interestingly, securin protein accumulation was accompanied by appearance of slower migrating bands suggesting post-translational modification (possibly phosphorylation) of this protein by WA treatment. Nonetheless, it is reasonable to conclude that WA treatment causes inactivation of APC/C as evidenced by accumulation of its substrate securin.
Discussion
We used MDA-MB-231 (estrogen-independent) and MCF-7 (estrogen-responsive) cell lines to determine the effect of WA treatment on cell cycle distribution. We found that WA treatment causes G2-M phase cell cycle arrest in both cell lines. The cell cycle checkpoints are normally activated when a cell experiences damage. Activation of checkpoints helps maintain genomic stability by allowing the cell time to repair the damage prior to resumption of the cell cycle progression. The WA-mediated G2-M phase cell cycle arrest in both cell lines is maintained for at least up to 24 h even after removal of the drug. These results suggest that the cells are probably unable to fully repair the damage caused by WA treatment leading to sustained cell cycle arrest. Further studies are needed to determine whether the low μM concentrations of WA required for cancer cell growth inhibition and cell cycle arrest are achievable in vivo because pharmacokinetic parameters for this agent have not yet been measured.
Eukaryotic cell cycle progression involves sequential activation of Cdks whose activation is dependent upon their association with corresponding cyclins (31). A complex between Cdk1 (also known as p34cdc2) and cyclinB1 plays an important role in regulation of G2-M transition (31). Activity of Cdk1/cyclinB1 kinase complex is negatively regulated by reversible phosphorylations at Thr14 and Tyr15 of Cdk1 (31). Dephosphorylation of Thr14 and Tyr15 of Cdk1 and hence activation of the Cdk1/cyclinB1 kinase is catalyzed by Cdc25 family of dual-specificity phosphatases (32).We found that the WA-mediated G2-M phase cell cycle arrest in MDA-MB-231 and MCF-7 cells correlates with a decrease in the protein levels of Cdk1 and Cdc25C, which is likely to inhibit activity of the Cdk1/cyclinB1 kinase complex. Consistent with this prediction, the WA treatment causes an increase in the levels of Tyr15 phosphorylated (inactive) Cdk1 in both cell lines.
The present study reveals that p53 tumor suppressor, which is implicated in the regulation of G2-M transition (38), is not necessary for the WA-induced cell cycle arrest because the G2-M fraction enrichment is observed in both MDA-MB-231 (mutant p53) and MCF-7 cells (normal p53). However, it is reasonable to postulate that the p53 status may be a contributing factor in differential sensitivity of MDA-MB-231 and MCF-7 cells to WA-induced G2-M phase cell cycle arrest. This conclusion is based on the following observations: (a) the WA-mediated G2-M phase cell cycle arrest in the MCF-7 cell line is evident at a relatively lower concentration than in the MDA-MB-231; (b) both cdk1 and cyclinB1 genes are transcriptionally repressed by p53 (reviewed in Ref. 38); (c) the WA-mediated down-regulation of Cdk1 protein level is relatively more severe in the MCF-7 cell line than in the MDA-MB-231; and (d) WA causes a near complete loss in cyclinB1 protein level by 24 h of treatment in MCF-7 cells but not in the MDA-MB-231 cell line. At the same time, the possibility that difference in sensitivity of MDA-MB-231 and MCF-7 cells to G2-M phase cell cycle arrest by WA is related to its differential effect on Cdc25B cannot be ignored. In MDA-MB-231 cells, the WA treatment causes a sustained increase in protein level of Cdc25B. On the other hand, WA treatment results in down-regulation of Cdc25B protein level in MCF-7 cells (up to 60% decrease compared with control). Further studies are needed to systematically explore whether differential sensitivity of MDA-MB-231 and MCF-7 cells to WA-mediated G2-M phase cell cycle arrest is linked to p53 and/or Cdc25B.
The present study suggests involvement of Cdc25C protein in the WA-induced G2-M phase cell cycle arrest. The cell cycle arrest caused by WA treatment in both MDA-MB-231 and MCF-7 cell lines correlates with a marked decrease in the protein level of Cdc25C. The WA-mediated down-regulation of Cdc25C protein precedes onset of G2-M arrest in both cell lines. Moreover, the WA-mediated G2-M phase cell cycle arrest in MDA-MB-231 cells is partially but statistically significantly attenuated by ectopic expression of Cdc25C. Previous studies have shown that hydrogen peroxide treatment causes ubiquitin-proteasome-mediated degradation of Cdc25C protein due to redox-modification of its critical cysteine residues (29). Even though WA treatment causes ROS generation in breast cancer cells, the cell cycle arrest is probably independent of ROS generation because attenuation of ROS generation has minimal effect on G2-M phase cell cycle arrest in our model. Collectively, these results suggest that (a) the WA-mediated decrease in Cdc25C protein level is probably not due to redox-modification of its cysteine residues, and (b) the WA-mediated G2-M phase cell cycle arrest is only partially caused by the decline of Cdc25C protein level.
We also found that the WA-treated MDA-MB-231 and MCF-7 cells are arrested in mitosis. Similar to G2 arrest, the mitotic block caused by WA treatment is maintained for at least up to 24 h even after removal of the drug. The WA-induced mitotic arrest in both cell lines correlates with accumulation of securin, which is a substrate of APC/C. The APC/C ubiquitin-ligase complex plays an important role in the initiation of anaphase as well as mitotic exit by sequentially degrading key cell cycle regulators including securin (36,37). Securin regulates mitotic progression by functioning as inhibitor of sister-chromatid separation (39,40). Our data suggest that the WA-mediated mitotic arrest in human breast cancer cells is probably caused by inhibition of APC/C. In conclusion, the present study reveals engagement of complex signal transduction leading to sustained G2 and mitotic arrest by WA in breast cancer cells.
Acknowledgments
This investigation was supported by USPHS grant RO1 CA129347 awarded by the National Cancer Institute.
References
- 1.Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- 2.Hulka BS, Stark AT. Breast cancer: cause and prevention. Lancet. 1995;346:883–887. doi: 10.1016/s0140-6736(95)92713-1. [DOI] [PubMed] [Google Scholar]
- 3.Kelsey JL, Bernstein L. Epidemiology and prevention of breast cancer. Annu Rev Public Health. 1996;17:47–67. doi: 10.1146/annurev.pu.17.050196.000403. [DOI] [PubMed] [Google Scholar]
- 4.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, et al. Tamoxifen for prevention of breast cancer: report of the national surgical adjuvant breast and bowel project P-1 study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 5.Cuzick J, Forbes J, Edwards R, Baum M, Cawthorn S, et al. First results from the International Breast Cancer Intervention study (IBIS-I): a randomized prevention trial. Lancet. 2002;360:817–824. doi: 10.1016/s0140-6736(02)09962-2. [DOI] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg CM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal R, Diwanay S, Patki P, Patwardhan B. Studies on immunomodulatory activity of Withania somnifera (Ashwagandha) extracts in experimental immune inflammation. J Ethnopharmacol. 1999;67:27–35. doi: 10.1016/s0378-8741(99)00065-3. [DOI] [PubMed] [Google Scholar]
- 8.Gupta SK, Mohanty I, Talwar KK, Dinda A, Joshi S, et al. Cardioprotection from ischemia reperfusion injury by Withania somnifera: a hemodynamic, biochemical and histopathological assessment. Mol Cell Biochem. 2004;260:39–47. doi: 10.1023/b:mcbi.0000026051.16803.03. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad M, Saleem S, Ahmad AS, Ansari MA, Yousuf S, et al. Neuroprotective effects of Withania somnifera on 6-hydroxydopamine induced Parkinsonism in rats. Hum Exp Toxicol. 2005;24:137–147. doi: 10.1191/0960327105ht509oa. [DOI] [PubMed] [Google Scholar]
- 10.Owais M, Sharad KS, Shehbaz A, Saleemuddin M. Antibacterial efficacy of Withania somnifera (ashwagandha) an indigenous medicinal plant against experimental murine salmonellosis. Phytomedicine. 2005;12:229–235. doi: 10.1016/j.phymed.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Rasool M, Varalakshmi P. Immunomodulatory role of Withania somnifera root powder on experimental induced inflammation: An in vivo and in vitro study. Vascul Pharmacol. 2006;44:406–410. doi: 10.1016/j.vph.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (ashwagandha): a review. Altern Med Rev. 2000;5:334–346. [PubMed] [Google Scholar]
- 13.Devi PU, Akagi K, Ostapenko V, Tanaka Y, Sugahara T. Withaferin A: a new radiosensitizer from the Indian medicinal plant Withania somnifera. Int J Radiat Biol. 1996;69:193–197. doi: 10.1080/095530096146020. [DOI] [PubMed] [Google Scholar]
- 14.Devi PU, Kamath R, Rao BS. Radiosensitization of a mouse melanoma by withaferin A: in vivo studies. Indian J Exp Biol. 2000;38:432–437. [PubMed] [Google Scholar]
- 15.Devi PU, Sharada AC, Solomon FE. In vivo growth inhibitory and radiosensitizing effects of withaferin A on mouse Ehrlich ascites carcinoma. Cancer Lett. 1995;95:189–193. doi: 10.1016/0304-3835(95)03892-z. [DOI] [PubMed] [Google Scholar]
- 16.Devi PU, Sharada AC, Solomon FE, Kamath MS. In vivo growth inhibitory effect of Withania somnifera (Ashwagandha) on a transplantable mouse tumor sarcoma 180. Ind J Exp Biol. 1992;30:169–172. [PubMed] [Google Scholar]
- 17.Ichikawa H, Takada Y, Shishodia S, Jayaprakasam B, Nair MG, et al. Withanolides potentiate apoptosis, inhibit invasion, and abolish osteoclastogenesis through suppression of nuclear factor-kappaB (NF-kappaB) activation and NF-kappaB-regulated gene expression. Mol Cancer Ther. 2006;5:1434–1445. doi: 10.1158/1535-7163.MCT-06-0096. [DOI] [PubMed] [Google Scholar]
- 18.Kaileh M, Vanden Berghe W, Heyerick A, Horion J, Piette J, et al. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J Biol Chem. 2007;282:4253–4264. doi: 10.1074/jbc.M606728200. [DOI] [PubMed] [Google Scholar]
- 19.Singh D, Aggarwal A, Maura R, Naik S. Withania somnifera inhibits NF-κB and AP-1 transcription factors in human peripheral blood and synovial fluid mononuclear cells. Phytother Res. 2007;21:905–913. doi: 10.1002/ptr.2180. [DOI] [PubMed] [Google Scholar]
- 20.Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7:115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 21.Falsey RR, Marron MT, Gunaherath GM, Shirahatti N, Mahadevan D, et al. Actin microfilament aggregation induced by withaferin A is mediated by annexin II. Nat Chem Biol. 2006;1:33–38. doi: 10.1038/nchembio755. [DOI] [PubMed] [Google Scholar]
- 22.Yang H, Shi G, Dou QP. The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry”. Mol Pharmacol. 2007;71:426–437. doi: 10.1124/mol.106.030015. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan S, Ranga R, Burikhanov R, Han S, Chendil D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res. 2007;67:246–253. doi: 10.1158/0008-5472.CAN-06-2430. [DOI] [PubMed] [Google Scholar]
- 24.Malik F, Kumar A, Bhushan S, Khan S, Bhatia A, et al. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound withaferin A with concomitant protection by N-acetyl cysteine. Apoptosis. 2007;12:2115–2133. doi: 10.1007/s10495-007-0129-x. [DOI] [PubMed] [Google Scholar]
- 25.Stan S, Hahm E, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008 doi: 10.1158/0008-5472.CAN-08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao D, Vogel V, Singh SV. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–2945. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 27.Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–897. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 28.Kim YA, Xiao D, Xiao H, Powolny AA, Lew KL, et al. Mitochondria-mediated apoptosis by diallyl trisulfide in human prostate cancer cells is associated with generation of reactive oxygen species and regulated by Bax/Bak. Mol Cancer Ther. 2007;6:1599–1609. doi: 10.1158/1535-7163.MCT-06-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savitsky PA, Finkel T. Redox regulation of Cdc25C. J Biol Chem. 2002;277:20535–20540. doi: 10.1074/jbc.M201589200. [DOI] [PubMed] [Google Scholar]
- 30.Herman-Antosiewicz A, Singh SV. Checkpoint kinase 1 regulates diallyl trisulfide-induced mitotic arrest in human prostate cancer cells. J Biol Chem. 2005;280:28519–28528. doi: 10.1074/jbc.M501443200. [DOI] [PubMed] [Google Scholar]
- 31.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 32.Draetta G, Eckstein J. Cdc25 protein phosphatase in cell proliferation. Biochim Biophys Acta. 1997;1322:M53–M63. doi: 10.1016/s0304-419x(96)00049-2. [DOI] [PubMed] [Google Scholar]
- 33.Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle dependent nuclear transport. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Widrow RJ, Rabinovitch PS, Cho K, Laird CD. Separation of cells at different times within G2 and mitosis by cyclin B1 flow cytometry. Cytometry. 1997;27:250–254. doi: 10.1002/(sici)1097-0320(19970301)27:3<250::aid-cyto6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 35.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, et al. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 36.Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, et al. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–943. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- 38.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 39.Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto A, Guacci V, Koshland D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]