The replisome uses mRNA as a primer after colliding with RNA polymerase (original) (raw)
. Author manuscript; available in PMC: 2009 Jun 11.
Published in final edited form as: Nature. 2008 Nov 19;456(7223):762–766. doi: 10.1038/nature07527
Abstract
Replication forks are impeded by DNA damage and protein-nucleic acid complexes such as transcribing RNA polymerase. For example, head-on collision of the replisome with RNA polymerase results in replication fork arrest. However, co-directional collision of the replisome with RNA polymerase has little or no effect on fork progression. The current study examines co-directional collisions between a replisome and RNA polymerase in vitro. Surprisingly, we find that the E. coli replisome utilizes the RNA transcript as a primer to continue leading strand synthesis following the collision with RNA polymerase which is displaced from the DNA. This action results in a discontinuity in the leading strand, yet the replisome remains intact and bound to DNA during the entire process. These findings underscore the remarkable plasticity by which the replisome operates to circumvent obstacles in its path and may explain why the leading strand is synthesized discontinuously in vivo.
DNA damage and high affinity protein-nucleic acid complexes, such as transcribing RNA polymerase (RNAP), act as impediments to bacterial and eukaryotic replication forks1-5. Arrest of the replication machinery can lead to mutagenesis and cell death. Thus, several pathways have evolved to repair and restart various types of collapsed replication forks. Mechanisms that facilitate replication past sites of DNA damage, such as recombinational repair and translesion synthesis, have been widely studied3,4,6-9. However, little is known regarding how the replisome proceeds through protein-nucleic acid blocks. In particular, replication forks often collide with transcription complexes that translocate in the same (co-directional) or opposite (head-on) direction as the replisome1,2,5. In bacteria, the rate of replication (∼600 nt · s-1) is 12-30 fold greater than transcription (20-50 nt · s-1) and there is no temporal separation between the two processes1,2,10,11. Thus, both head-on and co-directional collisions between the replisome and RNAP are likely to be frequent. We investigate here the mechanism by which the E. coli replisome passes a RNAP that is co-directional with replication fork movement.
Essential genes and the overall majority of transcription units in bacteria are encoded by the leading strand which suggests a natural selection for co-directional collisions in the cell1,5,12,13,14. It therefore seems likely that cell survival requires resolution of co-directional collisions in a manner that does not block fork progression. Indeed, in vivo studies in bacteria and eukaryotes indicate that co-directional transcription complexes do not impede replisome progression1,2,15-20. In contrast, head-on collisions predominately result in replication fork arrest and induce DNA recombination in bacteria and yeast1,2,15-21. In eukaryotes, replication fork barriers have evolved that prevent head-on collisions within highly expressed genes during S-phase1,2. Further, a recent study indicates that human cells also favor co-directional movement of the replisome with RNAP 22.
In the case of a co-directional collision, the leading strand DNA polymerase and RNAP utilize the same strand as a template (see Fig. 1a). The replicative helicase, DnaB, unwinds the DNA ahead of the E. coli replication fork by translocating on the opposite (lagging) strand. Thus, the helicase may continue past the RNAP in which case a physical interaction between the two co-directional polymerases upon collision is almost certain. How then does the replication fork bypass a co-directional RNAP without collapsing? Previous in vitro studies of the bacteriophage T4 replisome indicate that a co-directional transcription complex poses no obstacle to the progression of the T4 replication fork23-25. These studies indicated that RNAP remains bound to the DNA during passage of the T4 replisome.
Figure 1. Leading strand synthesis is interrupted by a co-directional RNA polymerase.

a, Schematic of replisome components and a co-directional RNAP. Replisome proteins include: Pol III core (orange), β-clamp (dark blue), DnaB (yellow), and the clamp-loader (light blue). Primase was omitted from reactions and the lagging strand polymerase is not pictured. b, A 2.2 kb template was constructed which supports leading strand synthesis and co-directional transcription. c,d Leading strand synthesis was performed in the presence of increasing concentrations of a RNAP open and halted elongation complex, respectively. Radio-labeled DNA products were analyzed in alkaline agarose gels (c,d).
In this report we elucidate a novel mechanism by which the E. coli replisome bypasses a co-directional transcription complex in vitro. We have used T7 RNAP as well as E. coli RNAP and find that the leading strand terminates upon collision with RNAP, but in a remarkable transaction the replisome uses the mRNA as a primer to continue the leading strand. This process results in a discontinuity in the leading strand and therefore may explain why leading strand synthesis is performed discontinuously in vivo26,27.
Observation of co-directional collisions
The E. coli replicase, referred to as DNA polymerase III (Pol III) holoenzyme (HE), is a multicomponent protein complex that performs rapid and highly processive replication of chromosomal DNA28,29. A co-directional RNAP may block the leading strand, and the current report focuses on leading strand synthesis by omitting primase. The proteins that perform leading strand synthesis are illustrated in Fig. 1a and include the following components: Pol III; the β-clamp which confers processivity to Pol III; the clamp-loader that assembles clamps at primed sites; and the DnaB helicase which unwinds duplex DNA ahead of the replication fork.
We first investigated the effect of a co-directional bacteriophage T7 transcription complex on the progression of the E. coli replication fork. We constructed a 2.2 kb linear forked DNA template that supports replication from one end and includes a co-directional T7 RNAP promoter 1 kb downstream from the replication fork (Fig. 1b). T7 RNAP serves as a model system for multisubunit RNAPs such as E. coli RNAP and the basic mechanisms of transcription are identical between these enzymes30.
During transcription initiation RNAP binds the promoter and unwinds DNA to form an open promoter complex. In Fig. 1c we asked whether a co-directional T7 RNAP open promoter complex affects progression of the replication fork. Pol III HE, DnaB and T7 RNAP were first pre-incubated with the 2.2 kb linear forked DNA in the presence of ATP which results in the assembly of the replisome at the fork and a T7 RNAP open promoter complex. Leading strand synthesis was initiated by the addition of α-32P labeled deoxyribonucleoside triphosphates (dNTPs) and DNA products were analyzed by electrophoresis in denaturing alkaline agarose gels. The results show that replisome progression is unaffected by the open promoter complex, as indicated by the appearance of only full-length product (2.2 kb; Fig. 1c).
Once RNAP synthesizes a transcript ∼10-12 nt in length it leaves the promoter and enters into a highly processive elongation complex30-32. Elongating RNAP often pauses or is arrested due to regulatory signals or lesions in the DNA2,33. Halted elongation complexes increase the probability of replisome-RNAP collisions in the cell, especially in strains that lack factors which revive or displace a halted RNAP34. To examine whether a halted co-directional T7 RNAP affects fork progression we added ATP and GTP which enables RNAP to synthesize a 22 nt transcript (Fig. 1d). If replisome advance is not blocked by a co-directional transcription complex, as indicated by in vivo studies, full-length 2.2 kb product should still be observed. However, the result indicates that RNAP prevents formation of full-length leading strand product and yields instead a 1 kb product, the distance to the halted RNAP (Fig. 1d). Surprisingly, we also observe a 1.2 kb product which corresponds to the length of the DNA template downstream from the promoter. The formation of the 1 kb product suggests that leading strand synthesis is terminated by the halted RNAP, but the 1.2 kb product suggests the unexpected possibility that the leading strand is reinitiated using the mRNA as a primer. This hypothesis predicts that the position of the RNAP along the template dictates the length of the two leading strand products. Indeed, moving the promoter to a different position changes the lengths of the upstream and downstream leading strand products accordingly (Supplementary Fig. 1).
Pol III uses a RNA transcript as a primer
To gain further evidence that Pol III uses the RNA transcript as a primer we terminated the mRNA prior to initiating replication by adding 3′-deoxy-cytidineribonucleoside-triphosphate (3′dCTP), a RNA chain terminator that is incorporated by RNAP (Fig. 2a). Addition of 3′dCTP prevented synthesis of the downstream portion of the leading strand (1.2 kb DNA), but did not affect synthesis of the initial 1 kb product (compare lanes 1 and 2). Similar results were obtained using a template that includes the promoter at a different position (Supplementary Fig. 2). Next, we observed extension of the transcript by Pol III directly by labeling the RNA instead of the DNA (Fig. 2b). In this case 32P-α labeled GTP and ATP were added which are incorporated into the 22 nt transcript by RNAP prior to replication (lane 1). Initiating replication results in extension of the transcript to 1.2 kb which corresponds to the length of the DNA downstream from the halted RNAP (lane 2). These results confirm that the mRNA is extended by Pol III.
Figure 2. The replisome extends the transcript of a co-directional RNA polymerase.

a, A co-directional collision of the replisome with a halted RNAP was performed. Extension of the RNA was permitted (lane 1) or blocked (lane 2) by the addition of RNA chain terminator 3′dCTP. RNA sequences are indicated. b, The transcript was radio-labeled by the addition of 32P-α-GTP and 32P-α-ATP and analyzed by urea-PAGE prior to (lane 1) and following (lane 2) replication. c, A co-directional collision was performed on a template that either includes (lane 1) or lacks (lane 2) a fork structure. Radio-labeled DNA products were analyzed in alkaline agarose gels (a,c).
Next we used a 2.2 kb linear duplex without a forked junction to determine whether the replication proteins could assemble at the transcription bubble of a halted RNAP and extend the RNA to form a 1.2 kb product (Fig. 2c). However, no products are observed in the absence of a replication fork (lane 2). Therefore, collision of the replisome with the RNAP is required for Pol III extension of the transcript. The result in lane 2 also demonstrates that RNAP is unable to form the 1.2 kb downstream product by misincorporating dNTPs.
Fate of the replisome and RNA polymerase
Since the replisome must collide with the transcription complex in order to gain access to the RNA, it is likely that the collision results in displacement of RNAP from the DNA. To test this we immobilized a His-tagged T7 RNAP halted elongation complex to Ni2+ beads and asked whether the DNA remains bound to the RNAP (pellet) or is released into solution (supernatant) following a co-directional collision (Fig. 3a). First, the transcription complex was immobilized, then unbound DNA and RNAP were removed by washing followed by initiation of replication. Upstream (1 kb) and downstream (1.2 kb) products are observed only in the supernatant indicating that RNAP is displaced by the replisome (left). Some full-length product is also observed presumably due to a fraction of transcription complexes that dissociated prior to replication. In the absence of replication, the DNA was analyzed in a native agarose gel stained with ethidium bromide (right). In this case most of the DNA remains bound to RNAP (pellet), whereas only a small fraction of the DNA is released into the supernatant. These data support the conclusion that the replisome displaces a co-directional RNAP from the DNA.
Figure 3. The replisome remains intact and displaces a co-directional RNA polymerase from the DNA.

a, A His-tagged RNAP halted elongation complex was assembled and immobilized to Ni2+ beads. Excess RNAP and DNA were removed by washing followed by replication initiation. Supernatant and pellet fractions were analyzed in an alkaline agarose gel (left). A His-tagged RNAP halted elongation complex was assembled and immobilized as in the left panel, however, replication was not initiated. Supernatant and pellet fractions were analyzed in a non-denaturing agarose gel stained with ethidium bromide (right). b, Leading strand synthesis was performed in solid-phase following the removal of excess Pol III* and DnaB (lanes 1 and 2) either in the presence (lanes 2 and 3) or absence (lane 1) of a co-directional halted RNAP. Radio-labeled DNA products were analyzed in an alkaline agarose gel.
Studies in vivo indicate that replication forks are not impeded by collisions with co-directional transcription complexes, suggesting that the replisome remains intact during bypass of a co-directional RNAP 1,2,15,16,20. An important factor that determines the integrity of the replication fork is whether the replicative helicase, DnaB, remains associated with the lagging strand (see Fig. 1a). To determine whether DnaB dissociates from the replisome during bypass of a co-directional RNAP we assembled the replisome and a halted T7 RNAP on a biotinylated template in the presence of ATP and GTP and then immobilized the DNA to streptavidin beads (Fig. 3b). Excess unbound DnaB and Pol III HE were then removed by washing. Replication was then initiated upon addition of dNTPs, the β-clamp and SSB, and radio-labeled DNA products were analyzed in an alkaline agarose gel. The results show that both 1 and 1.2 kb products were formed, indicating that the replisome can bypass a co-directional RNAP without dissociating from DNA (lane 2). In a control reaction RNAP was omitted which results in only full-length product (lane 1). A further control reaction demonstrates that replication proteins do not adhere to the beads following washing (Supplementary Fig. 3). To ensure that DnaB is a necessary participant in these reactions, the experiment was repeated but the helicase was omitted (lane 3). The absence of products in lane 3 indicates that DnaB is required for leading strand synthesis as expected. These results indicate that the only proteins required for replisome bypass of a co-directional RNAP are those that are present at the replication fork, and that the replisome bypasses RNAP without collapsing.
Replisome bypass of E. coli RNA polymerase
Although T7 RNAP serves as an important model enzyme, the multisubunit E. coli RNAP could conceivably behave somewhat different. Therefore, we examined the replisome for the ability to bypass a halted E. coli RNAP (Fig. 4). We constructed a linear 3.5 kb DNA that includes the strong E.coli RNAP T7A1 promoter 1.1 kb downstream from the replication fork and a biotin at the downstream edge. A halted E. coli RNAP elongation complex was first assembled by the addition of E. coli RNAP σ70 HE, ApU, GTP, CTP, and ATP which limits RNA synthesis to 20 nt. The DNA was then immobilized to streptavidin beads and washed with high salt to remove non-specific RNAP-DNA complexes. The fork was then ligated to the DNA followed by initiation of replication. Similar to experiments using a halted T7 RNAP, we observe replication products corresponding to the lengths of the template upstream (1.1 kb) and downstream (2.4 kb) from the promoter as well as some full-length product (lane 2). The percentage of full-length product (33 %) corresponds relatively well with the number of promoters unoccupied by RNAP (24 %; Supplementary Fig. 4). Omitting RNAP from the reaction results in only full-length product (lane 1). Lastly, we observe Pol III extension of the 20 nt E. coli RNAP transcript directly by labelling the RNA (Supplementary Fig. 5). These data indicate that the replisome can bypass a halted co-directional E. coli RNAP by using the transcript as a primer to continue the leading strand as observed using the T7 RNAP.
Figure 4. Replisome bypass of a co-directional E. coli RNAP elongation complex.

Leading strand synthesis was performed in solid-phase on a 3.5 kb template that either includes (lane 2) or lacks (lane 1) a co-directional halted E. coli RNAP elongation complex. Radio-labeled DNA products were analyzed in an alkaline agarose gel. Percentage of full-length product is indicated and was calculated as described in the methods section.
Discussion
We demonstrate herein that leading strand synthesis is terminated upon colliding with a co-directional RNAP, but can then be reinitiated by using the mRNA as a primer. A model of this mechanism is presented in Fig. 5. We propose that RNAP is displaced from the DNA by the leading strand polymerase, whereas DnaB remains bound to the lagging strand. The leading strand polymerase hops over the mRNA by remaining bound to the clamp-loader which assembles a new clamp at the 3′ terminus of the RNA-DNA hybrid. Pol III then binds to the newly assembled clamp and extends the transcript, leaving behind a nick or gap in the leading strand. The RNA may then be excised and replaced by DNA in a similar repair reaction as occurs during maturation of Okazaki fragments.
Figure 5. Model of replisome bypass of a co-directional RNA polymerase.
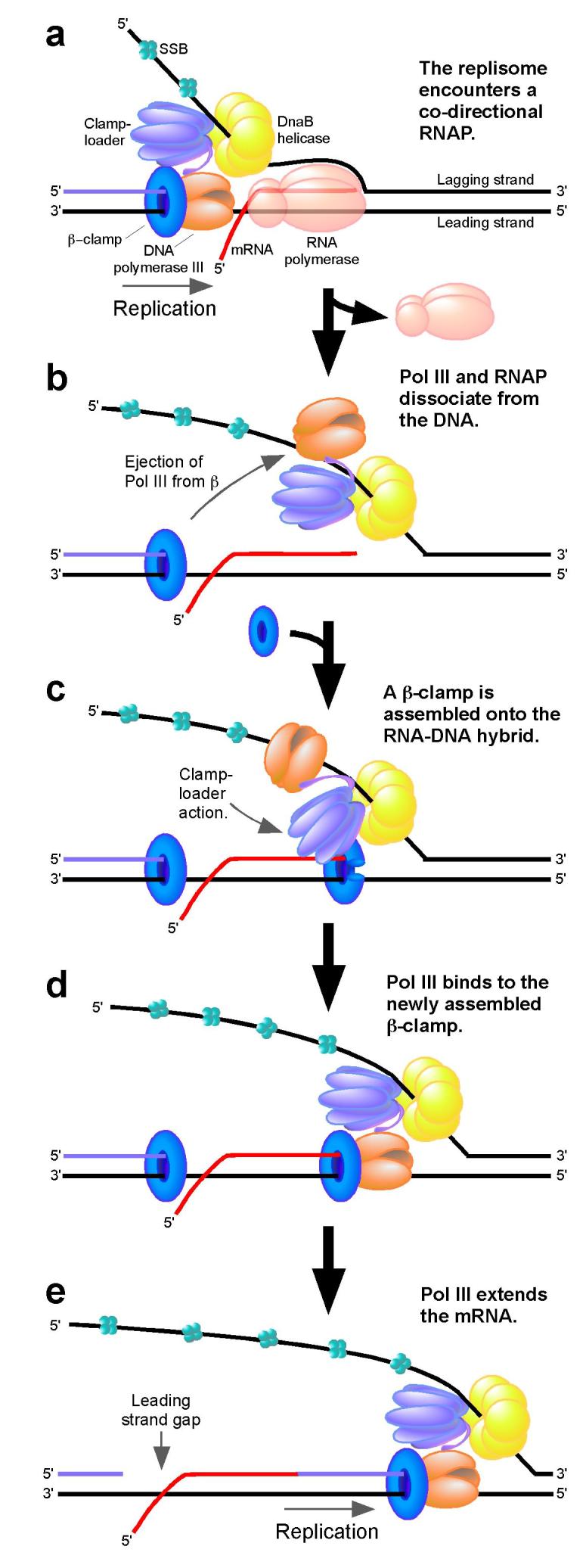
a, The replisome encounters a co-directional RNAP. b, RNAP is displaced from the DNA. The lagging strand polymerase dissociates from the β-clamp and DNA while remaining bound to the clamp-loader. DnaB remains bound to the lagging strand. c, The clamp-loader assembles a new β-clamp at the 3′ terminus of the RNA-DNA hybrid. d, The leading strand polymerase binds to the newly assembled β-clamp. e, The leading strand polymerase extends the mRNA leaving behind a nick or gap in the leading strand.
The scheme hypothesized in Fig. 5 has precedent in synthesis of the lagging strand in which Pol III rapidly hops from a clamp on a completed Okazaki fragment to a newly assembled clamp on a RNA-DNA hybrid every few seconds. Collision of the lagging strand polymerase with the 5′ terminus of an Okazaki fragment triggers the release of Pol III from the clamp35. Thus, hopping of the leading strand polymerase proposed in Fig. 5 may be initiated by a similar collision mechanism. During lagging strand synthesis RNA primers are made by primase. In the current report RNA primers are provided by RNAP on the leading strand. Primase activity on the leading strand is probably low since it requires stimulation by DnaB on the lagging strand.
In vivo, replication forks likely encounter co-directional RNAPs that have synthesized long transcripts. We are currently investigating the consequence of replisome collision with co-directional transcription complexes farther downstream from the promoter. Replisome takeover of long transcripts in the cell might trigger translational regulatory mechanisms such as the tmRNA system which removes stalled ribosomes from truncated mRNA and targets the mRNA for degradation36.
Synthesis of the leading strand is predominately viewed as a continuous process. This view is mostly based on in vitro studies that lack impediments to the replication fork. In contrast, several in vivo studies demonstrate that the leading strand is synthesized in a discontinuous fashion even as far back as Okazaki’s original work26,27,37-45. One source of leading strand interruptions may be due to replication fork collapse, since restart mechanisms that reactivate the fork involve new primers and thus produce single-strand gaps4,46,47. The current report provides a new explanation for leading strand interruptions in which a replication fork simply recruits the 3′ terminus of the mRNA to continue leading strand synthesis following a collision with RNAP. These protein dynamics emphasize the remarkable plasticity of the moving replisome apparatus, and underscore a driving force during evolution that has enabled replication machines to efficiently deal with obstacles along the path of chromosome duplication.
METHODS SUMMARY
DNA templates
Linear forked DNA was prepared in a similar fashion to a previous study48. 2.2 kb DNA: pPK731 was digested with BsaI followed by ligation in the presence of excess complementary forked DNA that was pre-annealed by mixing oligos RP25, RP26, and RP33 together followed by boiling and slow cooling to room temp. The 2.2 kb DNA without a fork was prepared by digesting pPK7 with BsaI. 10.5 kb DNA: pRSF2 was digested with SapI and then ligated in the presence of excess forked DNA (RP25, RP26, RP10). 3.5 kb DNA: PCR was performed using pRP50 as a template and primers RP64B and RP65. PCR product was purified, digested with SapI and then ligated in the presence of excess forked DNA (RP25, RP26, RP10). Ligation products were purified by gel filtration followed by phenol extraction and ethanol precipitation.
Leading strand synthesis
44.8 pmol of DnaB (as hexamer) was incubated with 1.5 nM final concentration of linear forked DNA in 15 μl of buffer A (20 mM Tris-Cl (pH 7.5), 8 mM MgCl2, 0.5 mM EDTA, 5 mM DTT, 10% glycerol) for 15 s at 23 °C. 488 fmol of Pol III* (Pol III HE minus β), 1.5 pmol of β, 2 mM ATP, and 60 μM each dGTP and dATP were added to a volume of 20 μl and incubated a further 5 min at 23 °C. Replication was initiated upon adding 1 μg SSB and [α-32P]dTTP and [α-32P]dCTP (specific activity, 3,000-5,000 cpm/pmol) to a final volume of 25 μL. Reactions were terminated after 10 min upon adding 5 μl of 120 mM EDTA and 3% SDS. All experiments except where indicated used Pol III* reconstituted from pure subunits and using an ε mutant that abolishes 3′-5′ exonuclease activity49. Radio-labeled products were analyzed in alkaline agarose gels.
Other experimental methods are available online.
METHODS
Co-directional collision of the replisome with T7 RNAP
Leading strand synthesis was performed as described in the methods summary except for the following additions. 20 nM (or as specified) T7 RNAP was added along with Pol III* and β. Then either 3 μM GTP, or a mixture of 3 μM GTP and 1 μM UTP, was added along with DnaB to assemble a T7 RNAP halted elongation complex on the 2.2 kb and 10.5 kb templates, respectively. Assembly of the T7 RNAP open promoter complex required no additional NTPs. 20 μM 3′dCTP (Tri-link) was added along with DnaB in Fig. 2a and Supplementary Fig. 2. Supplementary Fig. 1 and 2 included a 10.5 kb forked DNA template instead of the 2.2 kb DNA substrate and a mixture of 3 μM GTP and 1 μM UTP was added along with DnaB.
In the experiment of Fig. 2b, 20 reactions were pooled, dNTPs were unlabeled, and [α-32P]GTP and [α-32P]ATP were added along with DnaB and Pol III*, respectively. Reactions were then terminated by removing nucleotides through centrifugation over G-25 spin columns (Roche) followed by phenol extraction and ethanol precipitation with 10 μg of carrier DNA. Precipitated nucleic acid was resuspended in 10 μl of 20 mM Tris-Cl (pH 8.5) and then mixed with 10 μl of 90% w/v formamide and 50 mM EDTA. Samples were boiled and analyzed in a 8% urea-polyacrylamide gel.
Co-directional collision of the replisome with a His-tagged T7 RNAP elongation complex immobilized to nickel beads
130 nM final concentration of His-tagged T7 RNAP was incubated with 10 nM final concentration of 2.2 kb linear forked DNA along with 300 μM GTP and 100 μM ATP in 25 μl of buffer A for 5 min at room temp. 30 μl of Ni2+ magnetic coated beads (Promega) were added for an additional 5 min. Next, the beads were washed 3 times with 100 μl of buffer A. Leading strand synthesis was then performed as described in the methods summary except for the following modifications. 1 and 3 pmol of Pol III* and β were added, respectively. After the reaction was terminated the supernatant (25 μl total volume) was removed for analysis. The beads were then washed 2 times with 100 μl of buffer A. The pellet fraction was then removed from the beads by the addition of 0.5 M imidazole and 100 mM EDTA in a total volume of 25 μl for 5 min at room temp. Equal volumes of supernatant and pellet fractions were analyzed in an alkaline agarose gel. In the absence of leading strand synthesis (Fig. 3a, right), the supernatant and pellet fractions were analyzed in a native agarose gel stained with ethidium bromide.
Co-directional collision of single replisome particles with a T7 RNAP elongation complex on DNA immobilized to streptavidin beads
44.8 pmol of DnaB (where indicated) was incubated with 5 nM final concentration of 2.2 kb linear forked DNA, which was biotinylated at the 5′ terminus of the lagging strand, in 15 μl of buffer A for 15 s at 23 °C. 20 nM T7 RNAP and 3 μM GTP were added (where indicated) along with DnaB. 841 fmol of Pol III* (including wild-type ε) and 5 pmol of β were then added along with 2 mM ATP and 60 μM each of dGTP and dATP to a volume of 20 μl for a further 5 min. Reactions were then mixed with 20 μl of streptavidin coated magnetic beads (Invitrogen) pre-washed with buffer A for 10 min at 23 °C. Beads were washed 3 times with 100 μl of buffer A along with 60 μM each of dGTP and dATP, 2 mM ATP, 5 pmol of β and, where indicated, 20 nM T7 RNAP along with 3 μM GTP. Beads were resuspended in 20 μl of their respective wash buffers (with or without T7 RNAP and GTP) and replication was initiated as described in the methods summary. Reactions were terminated after 20 min by the addition of 5 μl of 120 mM EDTA and 3% SDS. Beads were boiled and the supernatant was removed for gel analysis. Beads were then treated with proteinase K in 10 μl of 10 mM Tris-Cl (pH 7.5), 5 mM EDTA, 1% SDS for 30 min at 50 °C to remove residual DNA from the solid support. The supernatant was pooled and radio-labeled DNA was analyzed in a 1.2% alkaline agarose gel. Supplementary Fig. 2 was performed in a similar fashion, however, T7 RNAP and GTP were omitted and biotinylated DNA was either pre-incubated with DnaB or added along with SSB and dNTPs as indicated.
Co-directional collision of the replisome with an E. coli RNAP elongation complex
500 nM final concentration of E. coli RNAP σ70 HE was mixed with 5 nM final concentration of a 3.5 kb DNA in 100 μl of buffer A for 10 min at 37 °C. 100 μM of ApU and 40 μM each of GTP and ATP were added for an additional 10 min at 37 °C. 200 μl of streptavidin magnetic coated beads (Invitrogen) were added for a further 10 min at room temp. The beads were then washed 5 times with 0.9 ml of buffer A containing 0.75 M NaCl, 200 μg/ml heparin, and 20 μg/ml ssDNA. Next, the beads were washed 2 times with 0.9 ml of buffer A. The beads were resuspended in 100 μl of New England Biolabs buffer 4 and 10 units of Sap I (New England Biolabs) was added for 10 min at 37 °C. The beads were washed 3 times with 0.9 ml of buffer A and then resuspended in 50 μl of Quick Ligation reaction buffer (New England Biolabs). 2 μl of Quick T4 ligase (New England Biolabs) was added along with 6 nM final concentration of pre-annealed forked DNA (RP10, RP22, RP25) for 10 min at room temp. The beads were washed 3 times with 0.9 ml of buffer A. Next, leading strand synthesis was performed as described in the methods summary except 10 reactions were pooled. The beads were boiled after the reaction was terminated and the supernatant was purified using the Qiagen PCR Cleanup kit. Purified radio-labeled DNA products were analyzed in an alkaline agarose gel. The percentage of full-length product was calculated using the following equation: IFL ● [IFL + (IB ● 3.18)]-1 ● (100), where IFL = intensity of full-length product and IB = intensity of replication block. The factor 3.18 corrects for the amount of full-length product that would have been formed relative to the intensity of the replication block (IB) and was calculated by dividing the length of the full-length product (3.5 kb) by the length of the blocked product (1.1 kb). The occupancy of promoters bound by E. coli RNAP in Supplementary Fig. 4 was determined by XhoI restriction digest of the immobilized 3.5 kb DNA in the absence of leading strand synthesis either with or without the addition of E. coli RNAP.
Pol III extension of a co-directional E. coli RNAP transcript
Leading strand synthesis was performed as described in the methods summary except for the following modifications. 30 reactions were pooled and performed at 37 °C. 40 μM each of ApU, GTP and CTP were added along with DnaB which was incubated with DNA for 30 s rather than 15 s. 50 nM final concentration of E. coli RNAP σ70 HE was added 2 min after the addition of Pol III* and β. [α-32P]dNTPs were omitted and [α-32P]GTP and [α- 32P]ATP were added along with DnaB and Pol III*, respectively. Reactions were terminated by removing nucleotides through centrifugation over G-25 spin columns (Roche) followed by phenol extraction and ethanol precipitation with 10 μg of carrier DNA and 30 μg of glycogen. Precipitated nucleic acid was resuspended in 5 μl of 20 mM Tris-Cl (pH 8.5) and then mixed with 5 μl of 90% w/v formamide and 50 mM EDTA. Samples were boiled and analyzed in an 8% urea-polyacrylamide gel.
Proteins
Replication proteins were expressed, purified and reconstituted as previously described49. T7 RNAP and E. coli RNAP core were gifts of William T. McAllister and Seth Darst, respectively. Sigma-70 was expressed from pET21aEc(His)6PPXsigma70 which was a gift from Seth Darst. His-tagged sigma-70 was purified on a Ni2+ column and then concentrated on a Mono-Q column.
DNA
pPK731 was a gift of William T. McAllister. pRP50 was derived from pRL70650 which includes the rpoB gene of E. coli. The T7A1 promoter sequence was inserted into the rpoB gene by ligation of pre-annealed oligos RP35 and RP36 to ClaI digested pRL706 to form pRP50. pRSF2 was constructed by inserting a 6.6 kb synthetic gene into a pRSFDuet-1 vector digested with NdeI and BglII. Oligo sequences were (5′-3′):RP10, Phosphate-AGCTGAGACCGCAATACGGATAAGGGCTGAGCACGTCCTGCGATCTGCAGCCTGCCAGAATCTGTG; RP25, OH-CACAGATTCTGGCAGGCTGCAGATCGC; RP22, Phosphatase-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAGCCCTTATCCGTATTGCGGTCTCA; RP26, Biotin-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAGCCCTTATCCGTATTGCGGTCTCA; RP33, Phosphate CGGTTGAGACCGCAATACGGATAAGGGCTGAGCACGTCCTGCGATCTGCAGCCTGCCAGAATCTGTG; RP35, OHCGGACGTTGACTTAAAGTCTAACCTATAGGATACTTACAGCCATCGAGAGGGACACGGCGAATTCTCGAG; RP36, OHCGCTCGAGAATTCGCCGTGTCCCTCTCGATGGCTGTAAGTATCCTATAGGTTAGACTTTAAGTCAACGTC; RP64B, Biotin-AACCGGTGGAACGCGCGTGC; RP65, OH-TTTCATCTGCTCTTCCGCTTCCACCGCCTTGGCGAACCGGTG.
Equipment and settings
All gels with the exception of supplementary figure 2, were analyzed by phosphorimager using a 200 pixel per inch resolution setting. Gel images were then converted to tiff format and adjusted for contrast using Adobe Photoshop software version 9. Image sections were then selected, copied, and pasted into a Canvas version 9 file. Pasted selections were then converted into images and cropped further using Canvas. The gel in supplementary figure 2 was photographed while exposed to ultraviolet light. The digital image was then cropped and adjusted for contrast using Adobe Photoshop version 9. The image was then selected, copied, and pasted into a Canvas file. All other image art was produced using Canvas with the exception of supplementary figure 2 which includes a digital graph that was created using Excel.
Supplementary Material
1
Acknowledgements
The authors are grateful to William T. McAllister and Ray Castagna for providing T7 RNAP and Seth Darst and Lars Westblade for providing E. coli RNAP proteins and plasmids. This work was supported by a grant from the NIH (M.O.D.) and from the Anderson Cancer Center (R.T.P.).
References
- 1.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71(1):13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudolph CJ, Dhillon P, Moore T, Lloyd RG. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst) 2007;6(7):981–993. doi: 10.1016/j.dnarep.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Cox MM, et al. The importance of repairing stalled replication forks. Nature. 2000;404(6773):37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 4.Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet. 2001;35:53–82. doi: 10.1146/annurev.genet.35.102401.090016. [DOI] [PubMed] [Google Scholar]
- 5.Brewer BJ. When polymerases collide: replication and the transcriptional organization of the E. coli chromosome. Cell. 1988;53(5):679–686. doi: 10.1016/0092-8674(88)90086-4. [DOI] [PubMed] [Google Scholar]
- 6.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15(2):70–77. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 8.Tippin B, Pham P, Goodman MF. Error-prone replication for better or worse. Trends Microbiol. 2004;12(6):288–295. doi: 10.1016/j.tim.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 10.Kornberg A, Baker TA. DNA Replication. 2nd New York: 1992. DNA Replication; p. 931. [Google Scholar]
- 11.Breier AM, Weier HU, Cozzarelli NR. Independence of replisomes in Escherichia coli chromosomal replication. Proc Natl Acad Sci U S A. 2005;102(11):3942–3947. doi: 10.1073/pnas.0500812102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 13.Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34(4):377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- 14.Rocha EP, Danchin A. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res. 2003;31(22):6570–6577. doi: 10.1093/nar/gkg859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirkin EV, Mirkin SM. Mechanisms of transcription-replication collisions in bacteria. Mol Cell Biol. 2005;25(3):888–895. doi: 10.1128/MCB.25.3.888-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JD, Berkmen MB, Grossman AD. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104(13):5608–5613. doi: 10.1073/pnas.0608999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prado F, Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. Embo J. 2005;24(6):1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi T. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol. 2003;23(24):9178–9188. doi: 10.1128/MCB.23.24.9178-9188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272(5264):1030–1033. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- 20.French S. Consequences of replication fork movement through transcription units in vivo. Science. 1992;258(5086):1362–1365. doi: 10.1126/science.1455232. [DOI] [PubMed] [Google Scholar]
- 21.Vilette D, Ehrlich SD, Michel B. Transcription-induced deletions in plasmid vectors: M13 DNA replication as a source of instability. Mol Gen Genet. 1996;252(4):398–403. doi: 10.1007/BF02173004. [DOI] [PubMed] [Google Scholar]
- 22.Huvet M, et al. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007;17(9):1278–1285. doi: 10.1101/gr.6533407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B, Alberts BM. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995;267(5201):1131–1137. doi: 10.1126/science.7855590. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, et al. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature. 1993;366(6450):33–39. doi: 10.1038/366033a0. [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Wong ML, Alberts B. A transcribing RNA polymerase molecule survives DNA replication without aborting its growing RNA chain. Proc Natl Acad Sci U S A. 1994;91(22):10660–10664. doi: 10.1073/pnas.91.22.10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogawa T, Okazaki T. Discontinuous DNA replication. Annu Rev Biochem. 1980;49:421–457. doi: 10.1146/annurev.bi.49.070180.002225. [DOI] [PubMed] [Google Scholar]
- 27.Wang TC. Discontinuous or semi-discontinuous DNA replication in Escherichia coli? Bioessays. 2005;27(6):633–636. doi: 10.1002/bies.20233. [DOI] [PubMed] [Google Scholar]
- 28.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 29.Pomerantz RT, O’Donnell M. Replisome mechanics: insights into a twin DNA polymerase machine. Trends Microbiol. 2007;15(4):156–164. doi: 10.1016/j.tim.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Steitz TA. The structural basis of the transition from initiation to elongation phases of transcription, as well as translocation and strand separation, by T7 RNA polymerase. Current opinion in structural biology. 2004;14(1):4–9. doi: 10.1016/j.sbi.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Mentesana PE, Chin-Bow ST, Sousa R, McAllister WT. Characterization of halted T7 RNA polymerase elongation complexes reveals multiple factors that contribute to stability. Journal of molecular biology. 2000;302(5):1049–1062. doi: 10.1006/jmbi.2000.4114. [DOI] [PubMed] [Google Scholar]
- 32.Jiang M, Rong M, Martin C, McAllister WT. Interrupting the template strand of the T7 promoter facilitates translocation of the DNA during initiation, reducing transcript slippage and the release of abortive products. J Mol Biol. 2001;310(3):509–522. doi: 10.1006/jmbi.2001.4793. [DOI] [PubMed] [Google Scholar]
- 33.Uptain SM, Kane CM, Chamberlin MJ. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 34.Trautinger BW, Jaktaji RP, Rusakova E, Lloyd RG. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol Cell. 2005;19(2):247–258. doi: 10.1016/j.molcel.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Stukenberg PT, Turner J, O’Donnell M. An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell. 1994;78(5):877–887. doi: 10.1016/s0092-8674(94)90662-9. [DOI] [PubMed] [Google Scholar]
- 36.Keiler KC. Biology of trans-Translation. Annu Rev Microbiol. 2008 doi: 10.1146/annurev.micro.62.081307.162948. [DOI] [PubMed] [Google Scholar]
- 37.Okazaki R, et al. Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proc Natl Acad Sci U S A. 1968;59(2):598–605. doi: 10.1073/pnas.59.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sternglanz R, Wang HF, Donegan JJ. Evidence that both growing DNA chains at a replication fork are synthesized discontinuously. Biochemistry. 1976;15(9):1838–1843. doi: 10.1021/bi00654a008. [DOI] [PubMed] [Google Scholar]
- 39.Pauling C, Hamm L. Properties of a temperature-sensitive, radiation-sensitive mutant of Escherichia coli. II. DNA replication. Proc Natl Acad Sci U S A. 1969;64(4):1195–1202. doi: 10.1073/pnas.64.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gottesman MM, Hicks ML, Gellert M. Genetics and function of DNA ligase in Escherichia coli. Journal of molecular biology. 1973;77(4):531–547. doi: 10.1016/0022-2836(73)90221-0. [DOI] [PubMed] [Google Scholar]
- 41.Konrad EB, Modrich P, Lehman IR. Genetic and enzymatic characterization of a conditional lethal mutant of Escherichia coli K12 with a temperature-sensitive DNA ligase. Journal of molecular biology. 1973;77(4):519–529. doi: 10.1016/0022-2836(73)90220-9. [DOI] [PubMed] [Google Scholar]
- 42.Okazaki R, Arisawa M, Sugino A. Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc Natl Acad Sci U S A. 1971;68(12):2954–2957. doi: 10.1073/pnas.68.12.2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olivera RM, Bonhoeffer E. Replication of Escherichia coli requires DNA polymerase I. Nature. 1974;250(5466):513–514. doi: 10.1038/250513a0. [DOI] [PubMed] [Google Scholar]
- 44.Wang TC, Smith KC. Discontinuous DNA replication in a lig-7 strain of Escherichia coli is not the result of mismatch repair, nucleotide-excision repair, or the base-excision repair of DNA uracil. Biochemical and biophysical research communications. 1989;165(2):685–688. doi: 10.1016/s0006-291x(89)80020-8. [DOI] [PubMed] [Google Scholar]
- 45.Wang TC, Chen SH. Okazaki DNA fragments contain equal amounts of lagging-strand and leading-strand sequences. Biochemical and biophysical research communications. 1994;198(3):844–849. doi: 10.1006/bbrc.1994.1120. [DOI] [PubMed] [Google Scholar]
- 46.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439(7076):557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 47.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7(12):932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 48.Heller RC, Marians KJ. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol Cell. 2005;17(5):733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 49.McInerney P, O’Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J Biol Chem. 2004;279(20):21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- 50.Severinov K, Mooney R, Darst SA, Landick R. Tethering of the large subunits of Escherichia coli RNA polymerase. J Biol Chem. 1997;272(39):24137–24140. doi: 10.1074/jbc.272.39.24137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1