Pericytes and Perivascular Fibroblasts Are the Primary Source of Collagen-Producing Cells in Obstructive Fibrosis of the Kidney (original) (raw)
Abstract
Understanding the origin of scar-producing myofibroblasts is vital in discerning the mechanisms by which fibrosis develops in response to inflammatory injury. Using a transgenic reporter mouse model expressing enhanced green fluorescent protein (GFP) under the regulation of the collagen type I, α 1 (coll1a1) promoter and enhancers, we examined the origins of coll1a1-producing cells in the kidney. Here we show that in normal kidney, both podocytes and pericytes generate coll1a1 transcripts as detected by enhanced GFP, and that in fibrotic kidney, **coll1a1**-GFP expression accurately identifies myofibroblasts. To determine the contribution of circulating immune cells directly to scar production, wild-type mice, chimeric with bone marrow from coll-GFP mice, underwent ureteral obstruction to induce fibrosis. Histological examination of kidneys from these mice showed recruitment of small numbers of fibrocytes to the fibrotic kidney, but these fibrocytes made no significant contribution to interstitial fibrosis. Instead, using kinetic modeling and time course microscopy, we identified **coll1a1**-GFP-expressing pericytes as the major source of interstitial myofibroblasts in the fibrotic kidney. Our studies suggest that either vascular injury or vascular factors are the most likely triggers for pericyte migration and differentiation into myofibroblasts. Therefore, our results serve to refocus fibrosis research to injury of the vasculature rather than injury to the epithelium.
The origin of scar-producing cell(s) in the kidney is of primary importance to understanding the mechanisms by which fibrosis develops in response to inflammatory injury. In the healthy kidney as in other organs, no clear endogenous precursor of the myofibroblast has been thought to exist.1 Derivation of myofibroblasts from bone marrow-derived CD34+ circulating cells known as fibrocytes has been proposed in several studies, and conflicting data have been published suggesting that a separate population of bone marrow-derived myofibroblasts contribute to interstitial fibroblasts in the kidney.2,3,4 Injured epithelial cells undergoing epithelial to mesenchymal transition (EMT) have been proposed to contribute to kidney myofibroblasts as well, with some studies suggesting epithelial cells are the major source of fibroblasts in the kidney and elsewhere.5,6 We have recently reported using robust, reproducible genetic fate mapping techniques in vivo that epithelial cells make no significant contribution to myofibroblasts in mouse kidney fibrosis (Humphreys et al, manuscript submitted for publication). Our recent report suggested that fresh investigation of the source of myofibroblasts in the kidney was merited.
Myofibroblasts are the cell type that generates and deposits collagen-I and collagen-III rich pathological extracellular matrix leading to irreversible fibrosis and causing organ dysfunction. Antibodies against the intermediate filament α-SMA has been widely used as a marker for collagen producing myofibroblasts, and other less-widely accepted markers such as the transcription factor S100A4 have been reported to label myofibroblasts.5 However none of these markers is unique to myofibroblasts and none labels proteins that define the cell functionally.
One unique characteristic of fibroblasts is that they generate and deposit into the extracellular matrix collagen-I. We have previously generated transgenic mice expressing enhanced green fluorescent protein (EGFP) in cells producing the collagen type I, α 1 (coll1a1) transcript (coll-GFP mice).7,8 These mice express a single copy of 3.5 kb of the 5′ coll1a1 promoter, 0.5 kb of the 3′ uncoded region and four upstream enhancers, driving EGFP expression, and have been shown to label with high sensitivity and specificity collagen-I α1 producing cells7 (Figure 1A). Collagen I, α1 is the most abundant protein in collagen-I fibrils.
Figure 1.

Pericytes and podocytes induce coll1a1 transcripts in normal coll-GFP mouse kidney. A: Coll-GFP transgenic mice were generated by linking the α1(I) collagen gene promoter (−3122 to + 111) and DNase I hypersensitive sites (HS) 4 & 5 (−7kb and −8kb) to GFP. The DNase I HS are position independent regulatory elements that enhance gene transcription in vivo. B: Image (magnification = original ×1000) of the normal glomerulus of coll-GFP mice colabeled with antibodies against podocin (red), and nuclei labeled with DAPI (blue). Right hand panel shows red and blue channels only. Podocytes exclusively express GFP in the glomerulus and colocalize with podocin. C: Coll1a1-GFP-expressing podocytes colocalize with synaptopodin, but do not localize with PDGFRβ (red) or CD31 (red). D: Periarteriolar _coll1a1-_GFP+ cells are identified surrounding and intimately associated with αSMA+ vascular smooth muscle cells (magnification = original × 1000). E: Interstitial _coll1a1-_GFP+ cells are identified interacting intimately throughout their length with CD31+ (red) endothelial cells of peritubular capillaries. The interacting _coll1a1-_GFP+ cells have intimate areas of adhesion with the endothelial cell suggestive of peg and socket processes (arrows), characteristic of pericytes (magnification = original ×1000). F: Three color labeling showing EBM (blue), CD31+ endothelial cells (red) and _coll1a1-_GFP+ pericytes (green). Note that the _coll1a1-_GFP+ pericytes are within EBM (magnification = original ×1000) in the cortical peritubular vascular plexus. G: In cortical interstitium labeled with antibodies against αSMA (red) many interstitial _coll1a1-_GFP+ cells can be seen but they do not express αSMA (magnification = original ×400).
We have used this mouse model to study collagen (collagen-I, α 1)-producing cells in kidney disease. Our studies indicate that a population of pericytes, subendothelial cells that regulate microvascular integrity in the peritubular capillary network, and also perivascular fibroblasts are a major source of myofibroblasts, and that transcription factors including Snail homologue 1 (Snai1) and inhibitor of DNA binding 1 (Id1) are induced during differentiation of pericytes into myofibroblasts in the ureteral obstruction model of fibrosis in the kidney.
Materials and Methods
Coll-GFP Mouse Model
Coll-GFP transgenic mice were generated and validated as previously described on the C57BL/6 background.7,8 In brief, 3.2 kb of the collagen-1 (α1) promoter was cloned as were DNase1 hypersensitivity sites (HS) with enhancer activity from 7 kb and 8 kb 5′ of the promoter. This construct, pCol9GFP-HS4,5 with hypersensitivity sites 4 and 5 positioned immediately 5′ of the promoter and the open reading frame of EGFP 3′ of the promoter yielded highest levels of GFP expression when coll1a1 transcripts were generated. Genotyping was performed as previously described.7,8 All studies were performed under a protocol approved by the Harvard Center for Animal Resources and Comparative Medicine.
Bone Marrow Chimerism
Bone marrow (BM) chimeric mice were generated as previously described.9 In brief, ten million BM cells from male donors in 200 μl of PBS were injected into the lateral tail vein of lethally irradiated (1000 Rads over 30 minutes) isogenic female recipients and chimerism was confirmed after 6 weeks by fluorescent in situ hybridization of buffy coat cells according to the method described previously.9 Briefly, methanol-acetic acid fixed buffy coat cells were incubated with 1 mol/L sodium thiocyanate (80°C, 10 minutes), then digested with proteinase K (37°C, 15 minutes), incubated with 0.1 mol/L HCl (37°C, 10 minutes), fixed with 4% paraformaldehyde, dehydrated, and air-dried. Fluorescent in situ hybridization mouse Y chromosome-specific probe Star*Fish (Cambio, Cambridge, UK) was added in the manufacturer’s buffer. Slides were heated (80°C, 10 minutes), incubated (16 hours, 37°C), then stringently washed in buffers finally mounted with Vectashield/DAPI (Vector Laboratories). In all cases 100% of leukocytes labeled positively for the Y chromosome.
Mouse Models of Fibrosis
Adult (12 to 20 weeks) or young (P12) mice (C57BL/6 or coll-GFP) were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.) before surgery. Unilateral ureteral obstruction (UUO) was performed as previously described (Humphreys et al, unpublished data),10 and kidneys, blood, spleen, and BM, were collected on days 0, 2, 3, 5, 7, 10, and 14. In some experiments unilateral ischemia-reperfusion of the left kidney was exposed through flank incisions was performed as previously described and kidneys harvested at days 0, 7, and 15. Full thickness skin wounds were created by flank incisions using a scalpel blade, and wounds held together with clips. Wounds were dissected on days 2, 3, 5, 7, 10, and 14 for analysis. To determine whether a second inflammatory signal could induce further fibrocyte differentiation, in some experiments, lipopolysaccharaide (6 μg/g, L-2880, Sigma) was injected i.v. for 3 days sequentially following UUO surgery and mice sacrificed on day 7 for analysis.
Tissue Preparation and Histology
Mouse tissues were prepared and stained as previously described.9,10 Wounded skin was dissected and cut transversely to expose the re-epithelialization of epidermis and the subepidermal granulation tissue and fixed as above. Primary antibodies against the following proteins were used for immunolabeling: αSMA-Cy3 (1:200, clone 1A4, Sigma), CD14, CD11b, CD11c, CD45, CD16/32, MHC class II (I-A/I-E) (1:200, eBioscience), CD34, Ly6C, (Pharmingen) 7/4, (ABD-Serotec), Ki-67 (1:200, clone SP6, Fisher), laminin (1:100, Sigma), NG2 (1:500), platelet-derived growth factor (PDGF) receptor beta (PDGFRβ [1:500], podocin [1:200]; S100A4 [1:200], DAKO, and also Abcam), and WT1 (1:100, Santa Cruz). Fluorescent conjugated affinity purified secondary antibody labeling (1:400 to 1:800, Jackson), mounting with Vectashield/4,6-diamidino-2-phenylindole (DAPI), image capture and processing were performed as previously described.9,10 To study proliferation of _coll1a1_-GFP cells, 5-bromo-2′-deoxyuridine (BrdU), 50 μg/g was injected i.p. 2 hours before sacrifice. Five-micron cryosections were labeled with chicken anti-GFP antibodies (1:500, Aves Labs), followed by anti-chicken-Cy2 (1:400), after postfixation (4% paraformaldehyde, 1 minute) and incubation with 2N HCl (15 minutes, 37°C), then 0.1 mol/L boric acid (5 minutes, room temperature) twice. Sections were incubated with sheep anti-BrdU (1:200, Abcam), then anti-sheep-Cy3 (1:400) and mounted as above. To study the relationship of coll1a1-producing cells with endothelial cells and capillary basement membrane (CBM), cryosections were labeled with antibody against CD31 (1:200, eBioscience), then Cy3-conjugated secondary antibodies, then anti-laminin antibodies, followed by anti-rabbit-Alexa Fluor350 (1:500, Molecular Probes), and were mounted with Prolong Gold. Quantification of specific cells in tissue sections was performed as previously described.9 In brief, sections were colabeled with DAPI, and _Coll1a1_-GFP+ cells were identified by blue and green nuclear colocalization; αSMA+, NG2+, or PDGFRβ + cells were identified by greater than 75% of the cell area immediately surrounding nuclei (detected by DAPI) staining positive with Cy3 fluorescence indicative of the antigen expression; Ki-67+ or BrdU+ cells were identified by positive nuclear staining for Cy3 fluorescence. Specific cells were counted in 10 cortical interstitial fields randomly selected at original magnification ×400 per mouse. To study the recruitment of fibrocytes into kidney, spleen, and skin wound, _CollIa1_-GFP+ cells were counted in six discontinuous whole sagittal sections per BM chimeric mouse studying three mice for each timepoint.
Single Cell Preparation from Blood, Spleen, and Kidney
Blood (500 μl) was collected from the inferior vena cava in sodium citrate (0.38%). Remaining blood flushed out as described above with ice cold PBS and spleen and kidneys were harvested. Peripheral blood mononuclear cells were isolated from citrated whole blood using Ficoll-Paque PLUS (GE Health care). Spleen was disaggregated by gentle pressure against glass slides, collected in PBS, and aggregates were removed (70 μm filter). Single cells were resuspended in fluorescence-activated cell sorting (FACS) buffer9 after centrifugation. Kidney was decapsulated, diced, incubated (37°C, 1 hour) with liberase (0.5 mg/ml, Roche) and DNase (100U/ml, Roche) in HBSS, and then resuspended in 10 ml of FACS buffer or PBS/0.1% bovine serum albumin, and cells were filtered (30 μm). In some cases leukocyte enrichment was performed by resuspending the single cell suspension in PBS, then overlaying it on a discontinuous percoll gradient (33%, 66% in PBS) and centrifuging (20 minutes, 620 ×g). Kidney fibroblasts or pericytes were purified from the single cell suspension of normal or d7 UUO Coll-GFP mouse kidney (in PBS and 1% bovine serum albumin) by isolating GFP+ cells using FACSAria cell sorting.
Flow Cytometric Analysis
Single cells (1 × 105) from kidney, peripheral blood mononuclear cells or spleen, were resuspended in FACS buffer9 and incubated with antibodies against CD14, CD11b, CD11c, CD45, MHC class II (I-A/I-E) (PE, 1:200, eBioscience), CD86, CD115, CD34 (PE, 1:200, Pharmingen), CD16/32, F4/80 (APC, eBioscience), CD64 (Alexa Fluor 647, 1:200, BD Biosciences), Ly6-C (FITC, 1:200, BD Biosciences), and 7/4 (Alexa Fluor 647, 1:200, AbD) for 30 minutes, 4°C in the presence of 1% mouse serum. After washing with FACS wash buffer,9 and resuspending in 200 μl FACS buffer, cells were analyzed using BD FACSCalibur flow cytometer.
Reverse Transcription-PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen). Purity of determined by A260 to A280. cDNA was synthesized using oligo(dT) and random primers.10 PCR was performed for mouse Twist using specific primers 5′-gAAAATggACAgTCTAgAgACTCTg-3′ and 5′-gTggCTgATTggCAAgACCTCTTg-3′, annealing at 50°C, 35 cycles; Snai1, 5′-AgCCCAACTATAgCgAgCTg-3′ and 5′-CCAggAgAgAgTCCCAgATg-3′; bone morphogenetic protein-7 (Bmp7), 5′-TACgTCAgCTTCCgAgACCT-3′ and 5′-gCTCAggAgAggTTggTCTg-3′, 50°C, 32 cycles; Id1, 5′-CATgAACggCTgCTACTCAC-3′ and 5′-gTggTCCCgACTTCAgACTC-3′, 50°C, 29 cycles; GAPDH 5′-ACTCCACTCACggCAAATTC-3′ and 5′-CACATTgggggTAggAACAC-3′, annealing at 50°C, 23 cycles. Quantitative PCR was determined using methods previously described10 and the following additional primer pairs: Twist 5′-CCCCACTTTTTgACgAAgAA-3′ and 5′-AAAATggAgCCAgTCCACAg-3′, Bmp7 5′-gTACgTCAgCTTCCgAgACC-3′ and 5′-ggTggCgTTCATgTAggA gT-3′. Purity of the pericyte and fibroblast cDNA was confirmed by excluding contamination with transcripts for Kim-1 (epithelial cells), F4/80 antigen, Emr-1 (macrophages), and CD31 endothelial cells.11
Statistical Analysis
Error bars are SE of mean. To calculate Td, curves were fit to plots of cell number against time using Prism software (Graphpad). To mathematically calculate the cell cycle time Tc an equation was derived to analyze the increase in cell number with time during which there was a linear increase in the proportion of pericytes entering cell cycle. The equation was derived and solved using Matlab/Simulink software (Mathworks, Natick, MA).
Results
Pericytes, Perivascular Fibroblasts, and Podocytes Express Coll-GFP in Normal Kidney
Mice expressing EGFP under regulation by the coll1a1 promoter and enhancers was generated as previously described7,8 and herein are referred to as Coll-GFP mice (Figure 1A). In healthy kidney, glomerular podocytes express GFP indicative of coll1a1 transcription. Podocytes co-expressed podocin (Figure 1B) and synaptopodin (Figure 1C). Mesangial cells did not express coll1a1 (Figure 1C). Surrounding arterioles and venules, a population of fine perivascular cells expressed coll1a1 but vascular smooth muscle cells that were labeled with αSMA antibodies did not fluoresce with GFP (Figure 1D). The perivascular _coll1a1-_expressing cells are known as perivascular fibroblasts.12,13 The ratio of perivascular fibroblasts to vascular smooth muscle cells in arterioles was 0.33 ± 0.05. In the interstitium of cortex and medulla, peritubular capillaries were lined intermittently with coll1a1 producing cells. These cells were predominantly subendothelial, in direct apposition with CD31-expressing endothelial cells and exhibited pedicle-like attachment plaques suggestive of the ‘peg and socket processes’ where _coll1a1-_producing cells directly interact with endothelial cells (Figure 1_E_).12,14 These characteristics define peritubular _coll1a1-_producing cells as pericytes. To be sure of their locale, endothelial basement membrane (EBM) was labeled with antibodies against laminin. GFP-expressing pericytes could be seen clearly within the EBM (Figure 1F). In some cases the cell body lay outside the EBM whereas pedicle-like attachment plaques were identified within the EBM. Importantly in normal kidney no GFP-expressing cells labeled for the endothelial marker CD31 or the intermediate filament αSMA (Figure 1G). The ratio of pericytes to endothelial cells in the kidney interstitium was 0.40 ± 0.06.
In Fibrotic Kidney Coll1a1-Producing Cells Correlate Imperfectly with Expression of α-SMA
To explore the contribution of _coll1a1-_producing cells to fibrosis in the kidney we induced UUO, a robust and standardized model for inducing inflammatory fibrosis in the kidney. After 7 days of obstruction in coll-GFP mice many interstitial cells expressed GFP, but GFP was not detected in epithelial cells indicating that epithelial cells were not a source of collagen I (Figure 2A). Podocyte expression of GFP was unaffected and mesangial cells did not express GFP (not shown). Tissue sections were labeled for expression of αSMA (Figure 2A). There was a close but imperfect correlation of αSMA+ cells and GFP+ cells in kidneys 7 days post obstruction (Figure 2_B_). Notably, 1% of _coll1a1-_producing interstitial cells did not express αSMA and 25% of αSMA+ cells in the interstitium did not express detectable levels of GFP by fluorescence microscopy. Thus not all α-SMA cells are generating coll1a1 message in fibrotic kidney. S100A4 (fibroblast specific protein-1) has been reported to be an alternative marker of myofibroblasts in the kidney. Antibody labeling of S100A4 did not colocalize with _coll1a1-_producing cells (Figure 2_C_), indicating S100A4 was a poor marker of _coll1a1-_producing cells in the kidney. However, S100A4+ cells co-expressed markers of monocyte/macrophage leukocytes (Table 1), indicating that in vivo S100A4 labeled leukocytes not fibroblasts. To explore further the molecular markers of _coll1a1-_producing cells we labeled tissue sections from coll-GFP UUO kidney for endothelial cells (CD31) (Figure 2D), myeloid leukocytes (CD11b) (Figure 2E), and leukocyte common antigen CD45. In sections from mice (n = 5/group) at days 3, 5, 7, 10, and 14, there was no overlap of GFP with CD31, or CD11b but small numbers of leukocytes labeling with CD45 expressed GFP that was quantified as 0.09% ± 0.04% of CD45+ cells co-expressing GFP. To be certain we had detected leukocytes that generated coll1a1 transcript we generated single cells from fibrotic kidney, and identified single cells clearly co-expressing GFP and CD45 (Figure 2F) suggesting that some _coll1a1-_producing cells were derived from leukocytes.
Figure 2.

_Coll1a1-_GFP+ interstitial cells from d7 ureteral obstruction correlate imperfectly with αSMA+ interstitial cells, and a minority co-express the leukocyte marker CD45. A: Photomicrograph of d7 UUO kidney from coll-GFP mice (left panel) co-immunolabeled with antibodies against αSMA (right panel). Note the absence of epithelial tubule expression of GFP and the heterogeneity of αSMA and GFP co-expression (magnification = original × 200). B: Graphs of number of cells per HPF (left panel), and proportion (right panel) of GFP+ interstitial cells that co-express αSMA and the proportion of αSMA+ cells that co-express _coll1a1-_GFP in normal and d7 UUO kidney. In UUO kidney 25% of αSMA+ cells do not express _coll1a1-_GFP but 100% of coll1a1-GFP cells express dSMA. C–E: Fluorescent micrograph of d7UUO kidney from coll-GFP mice co-immunolabeled with antibodies against (C) S100A4 (red), magnification = original ×1000; (D) CD31 (red), magnification = original ×1000; or (E) CD11b (red), magnification = original ×200. F: Single cell preparation form d7 UUO kidney showing a single cell co-expressing CD45 (red) and _coll1a1-_GFP with the nucleus shown (blue), magnification = original ×1000.
Table 1.
The Proportion of S100A4+ Interstitial Cells Co-Expressing Other Cell Markers in Fibrotic Kidney
| Col1-GFP+ | αSMA+ | CD11b+ | CD68+ | F4/80+ | |
|---|---|---|---|---|---|
| % | 0 (0) | 6.5 (3.9) | 93.1 (3.2) | 74.6 (6.5) | 28.4 (4.0) |
Fibrocytes Make a Minor Contribution to the Population of Coll1a1-Producing Cells
To explore further the contribution of bone marrow-derived _coll1a1_-producing cells to fibrosis in the kidney we generated bone marrow (BM) chimeric mice transplanting coll-GFP mouse BM into lethally irradiated recipient wild type mice, then after confirming chimerism, performed UUO surgery to induce kidney fibrosis and kidneys were examined at 0, 2, 3, 5, 7, 10, and 14 days (Figure 3). Small numbers of BM-derived _coll1a1_-GFP cells were detected exclusively in perivascular areas, with a predilection for venules, in fibrotic kidneys but not in normal kidneys or contralateral unobstructed kidneys, consistent with the description of fibrocytes (Figure 3, A, H). However in sagittal kidney sections there were on average only 10 BM-derived _coll1a1_-GFP cells amounting to fewer than 0.1% of all myofibroblasts, and in keeping with our observations in the wild-type mice. The pattern of recruitment of BM _coll1a1_-GFP cells to the kidney was not consistent with the development of fibrosis: whereas myofibroblasts increased exponentially with time and plateau (see Figure 4), fibrocytes appeared late in kidney disease and decrease in number as fibrosis progresses (Figure 3H). The BM-derived _coll1a1_-GFP cells in kidney co-expressed CD34 (58.1 ± 10.2%) and CD45 (100%) and some markers of myeloid lineage cells in tissue sections consistent with previous descriptions of fibrocytes,15 but did not express αSMA (0%) or S100A4 (0%) (Figure 3, C–F; Table 2). Single cells were purified from fibrotic kidneys and BM-derived _coll1a1_-GFP cells assessed by flow cytometry for leukocyte markers (Table 2) confirming CD34 and CD45 co-expression, but also high levels of MHC class II and B7–2, suggesting a potential role in antigen presentation. Lung, heart, small, large intestine, liver, and pancreas contained no _coll1a1_-producing cells in the BM chimeras. Blood leukocytes did not express _coll1a1_-GFP by flow cytometry. However, BM and spleen (red pulp) contained small numbers of fibrocytes that were induced within 48 hours of UUO surgery (Figure 3G). By flow cytometry, splenic fibrocytes had similar markers to kidney fibrocytes, notably lacking CD11b and CD11c but expressing other myeloid lineage dendritic cell markers (Table 2). We extended these studies to a unilateral kidney ischemia-reperfusion model that develops interstitial fibrosis 2 weeks after injury (5.8 ± 0.6% area of fibrosis day 15 post ischemia reperfusion compared with 0.2 ± 0.05% in sham surgery). In these studies, chimerism was confirmed and although many fibrocytes appeared in spleen, early after injury, none were seen in kidney 2 days following injury but 3.0 ± 1.2 and 0.6 ± 0.2 fibrocytes per sagittal kidney section were identified 7 days, and 15 days, respectively, following ischemia reperfusion injury, further supporting our data that fibrocytes play little role in kidney fibrosis. We went on to determine the role of fibrocytes in skin wounding. Full thickness skin wounds were examined in BM coll-GFP chimeric mice. Although many wound fibroblasts were detected, no fibrocytes were identified (Figure 3H) lending support to our observations that fibrocytes play no direct role in the progression of fibrosis.
Figure 3.

Fibrocyte recruitment to kidney and spleen in response to ureteral obstruction. Male coll-GFP bone marrow was transplanted into female WT lethally irradiated mice. Chimerism was confirmed at 6 weeks by assessing leukocytes for Y chromosome. A: Image of perivascular GFP+ cells in d7 UUO kidney from coll-GFP ⇒WT bone marrow chimeric mice (magnification = original ×400). B: Confirmation of extensive fibrosis in this model by picrosirius red staining of d14 UUO kidney from chimeric mice. Note prominent perivascular fibrosis (arrows) as well as interstitial fibrosis. C–D: Image of d7 in UUO kidney from coll-GFP ⇒WT BM chimeric mice showing GFP+ cells co-expressing CD34 and CD45, confirming these cells to be fibrocytes (magnification = original ×400). E–F: Image of d7 UUO kidney showing BM-derived _coll1a1_-GFP+ cells lacking αSMA or S100A4 expression (magnification = original ×400). G: Red pulp of spleen from coll-GFP ⇒WT BM chimeric mice, 48 hours after UUO surgery, note the presence of GFP+ cells. H: Time course of fibrocyte recruitment to fibrotic kidney, spleen and full thickness skin wound. Number of cells is per sagittal section for kidney, transverse section of spleen, and transverse section of skin wound.
Figure 4.

Pericyte migration, proliferation, and differentiation accounts for the appearance of the majority of myofibroblasts in the adult kidney following ureteral obstruction. A: Photomicrographs of pericytes and their relationship to endothelial cells (labeled for CD31 expression) at 0 hours, 9 hours, and 24 hours after UUO surgery. At 0 hours, pericytes are in apposition to endothelial cells and GFP expression is low. At 9 hours GFP expression is increased, the area occupied by pericytes has increased and some have detached from endothelial cells (arrow). At 24 hours many pericytes have migrated away from endothelial cells (arrows) and show evidence of cell enlargement or spreading (magnification = original ×400). B: Time course of GFP area assessed by morphometry in kidney interstitium. By 12 hours there is increased GFP+ area in the kidney compared with sham, reflecting pericyte enlargement or spreading. C: Time course of number of interstitial _coll1a1_-GFP+ cells/HPF following UUO surgery. From day 0 through day 4 there is an exponential increase in _coll1a1_-GFP+ cells. The exponential curve was fit to the data giving a Td of 2.2 days. D: Time course of % of Ki-67+ interstitial _coll1a1_-GFP+ cells. E: Time course of NG2 co-expression with _coll1a1_-GFP+ interstitial cells. Note induction of NG2 within 6 hours of UUO disease. F: Time course of αSMA co-expression with _coll1a1_-GFP+ interstitial cells. Induction of αSMA occurs later than NG2. G–H: Photomicrographs of NG2 and PDGFRβ expression by _coll1a1_-GFP+ interstitial cells 48 hours following induction of UUO. Note right hand panels show red and blue channels only for clarity.
Table 2.
Fibrocyte Cell Surface Markers in Spleen and Kidney of Coll-GFP Bone Marrow Chimeric Mice after Kidney Fibrosis
| CD45 | CD14 | CD115 | CD16/32 | CD64 | 7/4 | Ly6C | CD34 | |
|---|---|---|---|---|---|---|---|---|
| Kidney IHC | + | + | ND | + | +/− | ND | ND | +/− |
| Kidney FACS | ++ | + | + | + | +/− | + | + | +/− |
| Spleen FACS | ++ | + | + | + | +/− | + | + | +/− |
| I-A/I-E | B7–2 | CD11b | F4/80 | CD11c | αSMA | S100A4 | Desmin | |
|---|---|---|---|---|---|---|---|---|
| Kidney IHC | ND | ND | − | − | − | − | − | − |
| Kidney FACS | ++ | + | − | − | − | ND | ND | ND |
| Spleen FACS | ++ | + | − | − | − | ND | ND | ND |
The Expansion of the Interstitial Coll1a1-Producing Cell Population in Progressive Fibrosis Can Be Explained by Proliferation of the Initial Population of Pericytes
Our previous and current studies indicate myofibroblasts do not derive from kidney epithelial cells or circulating leukocytes (Humphreys et al, manuscript submitted for publication). We were intrigued by the possibility that pericytes might be the source of myofibroblasts because they transcribe the coll1a1 gene basally, and are distributed in along peritubular capillaries and _coll1a1_-GFP perivascular fibroblasts are distributed along small blood vessels, similarly to myofibroblasts. To study this further we assessed early time-points in the development of UUO to trace pericytes and the appearance of myofibroblasts (Figure 4A). Initially all pericytes can be seen adherent to capillaries, but within 9 hours of UUO, pericytes increased expression of GFP indicating induction of coll1a1 transcript, and increased spreading as detected by increased GFP area (Figure 4, A and B). Within 24 hours, pericytes detached and moved away from capillaries and by 48 hours there was a notable increase in the population of pericytes (Figure 4, A and C). Because EGFP will remain in a cell for more than 24 hours after GFP transcription is curtailed, it is very unlikely that the events observed were due to de novo appearance of a new cell type and the disappearance of pericytes. To study the increase in the population of _coll1a1_-GFP cells further we labeled cells with antibodies against Ki-67, a cell cycle marker detecting cells in all phases of the cell cycle (Figure 4D). We quantified the number of interstitial _coll1a1_+ cells with time and compared this with the proportion of _coll1a1_+ cells in cell cycle (Figure 4, C and D). There was an exponential increase in _coll1a1_+ cells in the interstitium over the first 4 days of the development of kidney fibrosis with an estimated doubling time (Td) of 2.2 days (Figure 4C). This increase was compared with the proportion of _coll1a1_+ cells in cell cycle (Figure 4D). By 24 hours, 43.7% of _coll1a1_+ interstitial cells were in cycle and this increased linearly to >75% at 48 hours. There was no evidence of apoptotic pericytes by terminal deoxynucleotidyl transferase dUTP nick-end labeling staining at these time points. In other studies, the cell cycle time for mouse lymphocytes or malignant cells is reported to be between 12 hours and 45 hours.16,17 It is not possible to accurately quantify cell cycle time from studying tissue sections.18 Nevertheless in the 2 hours preceding the 24-hour time-point, 9% of _coll1a1_+ interstitial cells incorporated BrdU into new DNA (traversed G1 through S phase of cell cycle) suggesting that cell cycle time in _coll1a1_-GFP cells would be within the confines of published studies on mouse cell proliferation. We used a mathematical model to study the expansion of the population of interstitial _coll1a1_+ cells:
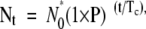
where Nt = number at time t, N0 = number at time 0, P = proportion in cell cycle, and Tc = cell cycle time. Using Td to derive Nt and t, and assuming a linear increase in P over the first 2 days of disease (derived from Figure 4D) we calculated that with a doubling time of 2.2 days the cell cycle time would be 45 hours.
A cell cycle time of 45 hours represents a high cell cycle time compared with those documented for malignant cells or bone marrow cells and means that our studies are therefore consistent with the hypothesis that the majority of the increase in _coll1a1_+ cells in the kidney interstitium is from proliferation of existing pericytes that migrate from the subendothelial space.
There is no unequivocal marker for pericytes, however in other studies, particularly of retinal pericytes, αSMA, desmin, NG2, and antibodies against the 3G5 epitope have been used.12,19,20,21,22,23 We labeled tissues for αSMA and NG2 (Figure 4, E–G). In normal kidney, mesangial cells and vascular smooth muscle cells expressed NG2 (not shown), but only 2.8% of pericytes labeled with the marker NG2. Only 6 hours after disease induction, before an increase in _coll1a1_+ cell number there was increased expression of NG2 by pericytes/_coll1a1_+ cells (Figure 4E), consistent with the hypothesis that myofibroblasts derive from pericytes. After 48 hours, more than half (53.2%) of _coll1a1_+ cells expressed NG2 (Figure 4, E, G). By 96 hours, all (100%) of the _coll1a1_+ cells expressed NG2. In contrast, in normal kidney no pericytes expressed αSMA. Six hours after disease induction pericytes remained negative for αSMA and after 24 hours of disease only 6.4% of _coll1a1_+ cells expressed αSMA. However over the subsequent 24 hours the expression in αSMA in this population of cells increased markedly (Figure 4F). After 48 hours of disease, more than half of interstitial _coll1a1_+ cells expressed both NG2 and αSMA (Figure 4, E–G), and were therefore by definition all myofibroblasts. By 7 days, 98% of the _coll1a1_+ interstitial cells expressed αSMA (Figure 2B). PDGFRβ has been shown in other studies to be expressed by pericytes, as well as fibroblasts and to be an important receptor for recruitment of pericytes by endothelial cells and important in successful paracrine signaling by endothelial cells to pericytes.20,24,27 In normal adult kidney all pericytes labeled with antibodies against PDGFRβ, and following the induction of disease all _coll1a1_+ cells continued to express PDGFRβ (Figure 4H), consistent with pericytes being myofibroblast precursors. In normal adult mice, 11.0 ± 1.2% of all PDGFRβ expressing cells did not express _coll1a1_-GFP. At no time-point did _coll1a1_+ cells co-express CD31, a marker of endothelial cells.
In Neonatal Mice, Pericytes Express NG2 and αSMA, and as Pericytes Differentiate into Myofibroblasts these Markers Are Retained
It was surprising to us that pericytes in the adult kidney did not express markers others have reported to label pericytes.19,20,21,22 However, many studies of pericytes have focused on the neonatal eye. Pericytes are reported to lose markers with maturity.12 We hypothesized that kidney pericytes of young mice might retain pericyte markers and co-express _coll1a1_-GFP, then progressively lose pericyte markers after development. Therefore following UUO surgery in young mice the fate of those pericytes could be followed using established pericyte markers. Kidneys from P12 (neonates 12 days after birth) coll-GFP mice were analyzed, because at this time-point the kidney is fully developed.28 P12 kidneys had many interstitial _coll1a1_+ cells in the subendothelial area (Figure 5A), that were in apposition with CD31+ endothelial cells. 98.4% of _coll1a1_+ cells expressed NG2 and all of them expressed αSMA and PDGFRβ (Figure 5, A–C). Thus in fully developed neonatal kidney pericytes co-expressed other pericyte markers, consistent with the definition of myofibroblasts. However, by P16, 4 days later, in normal mice only 74.0% of pericytes expressed NG2 and 94.5% expressed αSMA, confirming that as pericytes mature in the kidney they lose some pericyte markers (Figure 5, B and C). We induced UUO in P12 mice observing proliferation of pericytes and expansion in _colla1_+ interstitial cells (Figure 5, D and E). Whereas in healthy mice, pericytes lost NG2 and αSMA markers with time, in diseased mice, pericytes/_colla1_+ interstitial cells did not lose markers, rather the population of colla1+, NG2+, αSMA+, PDGFRβ+ cells expanded to account for all of the cells we would define as myofibroblasts (Figure 5, B–E). These studies therefore support the data in adult mice indicating that pericytes are the source of myofibroblasts in the kidney. The Td for neonatal pericyte cell expansion was calculated to be 3.2 days, somewhat longer than in adult mice.
Figure 5.

Normal neonatal mouse kidney _coll1a1-_GFP pericytes express pericyte markers and differentiate into myofibroblasts following ureteral obstruction. A: Low power (magnification = original ×200) view of _coll1a1_-GFP+ pericytes in P12 normal kidney. Higher power views (magnification = original ×400) of pericytes in P12 normal kidneys showing apposition to CD31+ endothelial cells (upper center panel), co-expression of PDGFRβ (upper right), NG2 (left lower and center panels), and αSMA (lower right). Note, in addition to positive staining of NG2 in _coll1a1_-GFP+ pericytes, NG2 is cleaved to a soluble form of the proteoglycan that binds to tubule basement membrane. Isotype control antibody labeling for NG2 showed no staining of pericytes or tubule membrane (not shown). B: Time course showing percentage of _coll1a1_-GFP+ interstitial cells that co-express NG2 in normal aging kidney (P12–P16) or following UUO in P12 mice. Note normal kidney pericytes lose NG2 with aging, but this is prevented by UUO. C: Time course showing percentage of _coll1a1_-GFP+ interstitial cells that co-express αSMA in normal aging of the kidney (P12 to P16) or following UUO in P12 mice. Note normal kidney pericyte lose αSMA with aging, but this is prevented by UUO. D: Time course of number of _coll1a1_-GFP+ interstitial cells/HPF following UUO in P12 mice and also in P16 healthy mice. Note a linear increase in _coll1a1_-GFP+ interstitial cells following UUO, and that the number of _coll1a1_-GFP+ interstitial cells in healthy mice decreases with aging. E: Time course showing percentage of Ki-67+ interstitial _coll1a1_-GFP+ cells.
Snai1 and Id1, Transcription Factors Implicated in Fibrosis, Are Induced in Coll1a1-Producing Cells Following Ureteral Obstruction
Our studies have identified pericytes as the major source of myofibroblasts in the kidney. To understand potential molecular mechanisms by which pericytes differentiate into myofibroblasts we selected candidate transcription factors or signaling molecules implicated in the appearance of myofibroblasts. Some have been reported to promote EMT in epithelial cells in culture, but our recent studies do not support EMT as a source of fibroblasts in vivo (Humpheys et al, manuscript submitted for publication). The transcription factor Twist has been implicated in tumorigenesis and EMT in vitro.29 In young mice some pericytes and other interstitial cells (but not epithelial cells) express Twist with low-level nuclear expression (Figure 6A). In diseased kidney, Twist immunolabeling could not be interpreted due to consistently high background levels (not shown). To study induction of Twist further, pericytes were purified from normal kidneys and myofibroblasts purified from UUO kidney by flow cytometric sorting, relying on GFP fluorescence to separate these cells from others. Purity was confirmed by lack of epithelial, endothelial, and leukocyte markers (not shown). Transcripts were amplified by reverse transcription-PCR and compared with transcripts from adult whole kidney (UUO or control kidney) by both conventional PCR and quantitative PCR. Although Twist transcripts increased in UUO kidney compared with control kidney, Twist transcript levels were not affected by differentiation of pericytes to fibroblasts (Figure 6, B and C). The transcription factors Snai1 and Id1 have also been implicated in the development of fibrosis.30,31 Snai1 transcripts increased in fibrotic kidney compared with control kidney and were expressed by pericytes but increased in myofibroblasts compared with pericytes (Figure 6, B and C). Id1 transcripts were down-regulated in diseased whole kidney compared with healthy kidney, but Id1 transcripts were increased in myofibroblasts compared with pericytes. Furthermore, transcripts for Bmp7, a cytokine of the transforming growth factor superfamily, believed to play a role in inhibiting fibrosis in part through the regulation of Id1 expression, were expressed by pericytes and were down-regulated in myofibroblasts (Figure 6, B and C).
Figure 6.

The transcription factors Snai1 and Id1 but not Twist are up-regulated by pericytes when they differentiate into myofibroblasts in vivo. A: P12 neonatal kidney showing Twist expression in cytoplasm and nucleus of many cortical interstitial cells that lack _coll1a1_-GFP expression, and also a minority of pericytes (arrow), but not epithelial cells (magnification = original ×400). (B) PCR and (C) representative quantitative PCR showing Twist, Snai1, Id1, and Bmp-7 transcripts in adult healthy kidney, UUO kidney (day 7), purified pericytes, and purified _coll1a1_-GFP+ myofibroblasts d7 UUO. Snail transcripts and Id1 transcripts are up-regulated in myofibroblasts as compared with pericytes.
Discussion
These studies have identified two sources of _colla1-_producing cells in the ureteric obstruction model of kidney fibrosis: fibrocytes and pericytes. The former makes a minor contribution <0.1% of collagen producing cells while the latter intrinsic cell population accounts for the majority of myofibroblasts in the kidney. Using genetic fate mapping techniques we have recently excluded a significant role for epithelial transdifferentiation in vivo in the appearance of myofibroblasts in the ureteral obstruction model of kidney fibrosis (Humphreys et al in submission). While our current studies do not exclude endothelial transdifferentiation we did not find evidence of endothelial cells co-expressing _coll1a1-_GFP, suggesting that endothelial cells in vivo are not a significant source of myofibroblasts.
Our studies do not use genetic fate mapping techniques to follow the differentiation of pericytes in the kidney into myofibroblasts. Nevertheless, kinetic analysis in combination with the use of coll-GFP as a pericyte marker in adult mice strongly indicates that the majority, if not all, myofibroblasts derive from the _coll1a1_+ pericytes in the interstitium and perivascular fibroblasts surrounding arterioles. The studies of _coll1a1_+ cells in the neonatal kidney also provide evidence that pericytes are the major precursor cell of myofibroblasts. As presented, we cannot exclude in the adult kidney a second population of kidney cells that contributes to myofibroblasts that in the normal kidney does not initially express _coll1a1-_GFP, but subsequently induces GFP expression following injury. It is possible that small numbers of coll1a1 negative pericytes exist in the normal adult kidney. Small numbers of PDGFRβ+, Coll1a1 negative cells were noted in the kidney interstitium in normal adult kidney but not seen in neonatal kidney. The identity of these cells has not been established but PFGFRβ is not specific for pericytes/myofibroblasts. Although we cannot exclude transdifferentiation of endothelial cells into myofibroblasts, we did not record any examples of CD31+ endothelial cells inducing GFP expression in coll-GFP mice. Thus we have accounted for all of the major cell types (epithelial cells and leukocytes) that could contribute to the myofibroblast population in the diseased kidney.
Precisely a pericyte lies within the EBM and forms peg and socket points of contact with the apposing endothelial cell. This definition has derived in large part from studies of the neonatal retina where the ratio of pericytes to endothelial cells is 1.0. In the adult kidney we found that that ratio of pericytes to endothelial cells was significantly lower, at 0.4, and that not all of the pericytes were completely within the EBM. However, nearly all showed evidence of processes extending through the CBM and forming pedicle-like attachments directly to the underlying endothelial cell. In electron microscopic studies of rat kidney, similar cells were named cortical interstitial fibroblasts.13 In this context it is worth noting that several recent studies have also implicated an endogenous ‘fibroblast’ in the normal kidney as a precursor to the myofibroblast, though none has used kinetic modeling or genetic fate mapping.32,33,34
The fact that pericytes are a major source of myofibroblasts serves to readjust the focus of fibrosis research to the vasculature. Rather than epithelial injury serving as the driving force for fibrosis, our studies lead one to speculate that in fact vascular injury or vascular factors are the most likely triggers for pericyte migration and differentiation into myofibroblasts. Indeed, UUO of the kidney is known to result in down-regulation of renal blood flow and glomerular filtration rate, suggesting that circulating factors may be important triggers of pericyte migration. Understanding the signals between injured or ischemic endothelium and neighboring pericytes requires further research. Pericytes have been reported to serve as paracrine cells supporting vascular integrity, and providing important angiogenic factors including Angiopoietin-1 and -2.12,23,35 A lack of pericytes has been associated with aneurysm formation and spontaneous hemorrhage.27 PDGF signaling from endothelial cells to pericytes is necessary for pericyte recruitment along developing capillaries and vascular stabilization. Recent studies in the eye suggest that pericytes and macrophages may cooperate to delete endothelial cells during scheduled developmental vascular regression.23 In these studies pericyte release of angiopoietin-2 is necessary for withdrawal of a survival signal to the endothelial cell. Fibrotic injury has been associated with loss of capillaries in various settings following in some cases by aberrant revascularization. This is notable because in the kidney, fibrosis is associated with a reduction in peritubular capillaries.36 Furthermore, in diabetic vascular disease of the retina, pericyte loss has been found to be an important initiating factor in the development of the vascular abnormalities that constitute diabetic retinopathy.26 Thus one central question is whether the development of fibrosis results in effective pericyte loss from peritubular capillaries in the kidney, and this effective pericyte loss is a cause for peritubular capillary loss, and the consequent chronic ischemia observed in many chronic kidney diseases.
PDGFRβ has been used as a marker for pericytes and has been implicated as an important receptor for recruitment and survival of pericytes by paracrine secretion of PDGFB by endothelial cells. All pericytes in the healthy adult kidney express PDGFRβ, and all myofibroblasts continue to express PDGFRβ. PDGF secretion has been implicated as an autocrine growth factor for myofibroblasts in vitro, but its role in vivo in the initiation and progression of fibrosis in the kidney is less clear,37 although in skin wound-healing, PDGFRβ blockade has been reported to delay wound closure.38 Macrophages are necessary for proliferation of kidney fibroblasts and induction of matrix deposition.39 Future studies should determine whether macrophage delivered PDGF is important in the initiation of fibrosis and pericyte differentiation. NG2 has been reported to be a marker of pericytes in the eye and brain, but reports indicate NG2 is expressed only by active pericytes, reports that are consistent with our findings in the kidney.12 Our studies show that quiescent pericytes down-regulate and lose pericyte markers, only acquiring them after injury-induced activation.
The transcription factor S100A4 has been strongly implicated in the development of fibrosis in the kidney.40 However our studies identify S100A4 in macrophages and not in collagen producing cells. We have previously demonstrated an important role for macrophages in the development of fibrosis in the kidney and other organs.10,39 The mechanism requires macrophage signaling to myofibroblasts because macrophages do not generate collagen matrix themselves. One explanation for the previously reported role of S100A4 in fibrosis is that it is an important transcription factor in the development of profibrotic macrophages. Further studies will be required to elucidate whether that is the mechanism of action of S100A4.
Many of the genes that are postulated to regulate the development of fibrosis have been studied in epithelial cells undergoing EMT in vitro,41,42,43 and some substantiated in vivo. To identify potential regulators of pericyte differentiation into fibroblasts we selected candidate transcription factors. Twist transcripts were not regulated by differentiation, but although pericytes had transcripts for Snai1 and Id1, they were consistently up-regulated in myofibroblasts compared with pericytes, and Bmp-7 was down-regulated in myofibroblasts compared with pericytes, consistent with their reported roles in the development of fibrosis. Further studies will be required to determine the precise role of these transcription factors and cell signaling molecules in pericyte differentiation and migration.
Although kidney fibrosis research has focused on epithelial cells as the cause of fibrosis, our findings that pericytes are the source of myofibroblasts may not be surprising. There are reports that pericytes can differentiate into vascular smooth muscle cells, fibroblasts, osteoblasts, adipocytes, and chondrocytes.12,44 In disease states pericyte to osteoblast differentiation has been suggested to contribute to ectopic calcification. In fibrotic skin diseases, pericytes have been identified as the primary source of fibroblasts,45 and in the liver and pancreas, Stellate cells lie in apposition to endothelial cells and may be a specialized pericyte.46 It is interesting that podocytes express coll1a1-GFP, suggesting they may have the capacity to play a more predominant role in glomerular scarring than previously thought. In murine nephrotoxic nephritis in the coll-GFP mouse, both mesangial cells and podocytes generate coll1a1 transcripts (manuscript in preparation). The glomerular basement membrane in health, does not contain fibrillary collagen, although may contain minor amounts of non-fibrillary collagen-type I, α chain,47,48 most likely synthesized by podocytes. The fact that podocytes share coll1a1-GFP expression with pericytes points to a pericyte-like role for podocytes in endothelial health. It is interesting therefore that genetic deletion of vascular endothelial growth factor in podocytes only, is sufficient to trigger endothelial death and severe thrombotic glomerular injury.49
Our studies confirmed the existence of a cell defined as fibrocyte, which did not express αSMA. Although we detected fibrocytes in injured kidney, spleen, and BM they were not detected in the circulation, suggesting that fibrocytes differentiate locally into _coll1a1_+ cells from either pre-existing cells or from circulating leukocytes such as a monocyte. Further, the pattern of recruitment in injury to spleen and BM and lack of sustained recruitment to kidney and no recruitment to skin wound suggest that they are acute phase respondent cells, not fibroblasts. Rather than lay down fibrotic matrix their presence in the spleen combined with highly expressed antigen presentation molecules suggests they function more like dendritic cells than fibroblasts. Nevertheless, fibrocytes appear to constitute a novel class of myeloid dendritic cells because they lack CD11b, and express CD34.
In conclusion, our studies identify a population of _coll1a1_+ pericytes in normal kidney that are the major source of myofibroblasts occurring in the ureteral obstruction model of kidney fibrosis.
Acknowledgments
Thanks to Dr. Sushrut Waikar (Harvard Medical School) for assistance with mathematical modeling, Deneen Kozoriz (HMS) for assistance with FACS sorting, Dr. William Stallcup (Burnham Inst.) for anti-NG2 and anti-PDGFRβ antibodies, Dr. Inna Gitelman (Ben Gurion Univ., Israel), Dr. Jing Yang (UCSD) for anti-Twist antibodies, and Dr. Martin Pollak (HMS) for anti-podocin and anti-synaptopodin antibodies.
Footnotes
Address reprint requests to Jeremy S. Duffield, Laboratory of Inflammation Research, Harvard Institutes of Medicine, 5th Floor, 4 Blackfan Circle, Boston, MA, 02115. E-mail: jduffield@rics.bwh.harvard.edu or Shuei-Liong Lin, E mail: linsl@ntumc.org.
Supported by NIH grant DK73299 (J.S.D.), and grants from American Society of Nephrology (Gottschalk Award), Genzyme Renal Innovations Program, Promedior Inc. (J.S.D.), and an award from the National Taiwan Science Council NSC-095-SAF-I-564-601 (S.L.L.).
These studies were presented in part at the Annual Meeting of the American Society of Nephrology, San Francisco, 2007.
References
- Grimm PC, Nickerson P, Jeffery J, Savani RC, Gough J, McKenna RM, Stern E, Rush DN. Neointimal and tubulointerstitial infiltration by recipient mesenchymal cells in chronic renal-allograft rejection. N Engl J Med. 2001;345:93–97. doi: 10.1056/NEJM200107123450203. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Bonventre JV. Kidney tubular epithelium is restored without replacement with bone marrow-derived cells during repair after ischemic injury. Kidney Int. 2005;68:1956–1961. doi: 10.1111/j.1523-1755.2005.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest. 2005;115:1756–1764. doi: 10.1172/JCI23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roufosse C, Bou-Gharios G, Prodromidi E, Alexakis C, Jeffery R, Khan S, Otto WR, Alter J, Poulsom R, Cook HT. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol. 2006;17:775–782. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med. 2004;82:175–181. doi: 10.1007/s00109-003-0517-9. [DOI] [PubMed] [Google Scholar]
- Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, Brenner DA, Rippe RA. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology. 2003;37:267–276. doi: 10.1053/jhep.2003.50067. [DOI] [PubMed] [Google Scholar]
- Krempen K, Grotkopp D, Hall K, Bache A, Gillan A, Rippe RA, Brenner DA, Breindl M. Far upstream regulatory elements enhance position-independent and uterus-specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr. 1999;8:151–163. [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, Bonventre JV. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest. 2005;115:1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–1668. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- Kaissling B, Le Hir M. Characterization and distribution of interstitial cell types in the renal cortex of rats. Kidney Int. 1994;45:709–720. doi: 10.1038/ki.1994.95. [DOI] [PubMed] [Google Scholar]
- Kaissling B, Hegyi I, Loffing J, Le Hir M. Morphology of interstitial cells in the healthy kidney. Anat Embryol (Berl) 1996;193:303–318. doi: 10.1007/BF00186688. [DOI] [PubMed] [Google Scholar]
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Oostendorp RA, Audet J, Eaves CJ. High-resolution tracking of cell division suggests similar cell cycle kinetics of hematopoietic stem cells stimulated in vitro and in vivo. Blood. 2000;95:855–862. [PubMed] [Google Scholar]
- Spang-Thomsen M, Vindelov LL. Proliferation kinetics of a human malignant melanoma serially grown in nude mice. Cell Tissue Kinet. 1984;17:401–410. doi: 10.1111/j.1365-2184.1984.tb00599.x. [DOI] [PubMed] [Google Scholar]
- Brons PP, Raemaekers JM, Bogman MJ, van Erp PE, Boezeman JB, Pennings AH, Wessels HM, Haanen C. Cell cycle kinetics in malignant lymphoma studied with in vivo iododeoxyuridine administration, nuclear Ki-67 staining, and flow cytometry. Blood. 1992;80:2336–2343. [PubMed] [Google Scholar]
- Chan-Ling T, Page MP, Gardiner T, Baxter L, Rosinova E, Hughes S. Desmin ensheathment ratio as an indicator of vessel stability: evidence in normal development and in retinopathy of prematurity. Am J Pathol. 2004;165:1301–1313. doi: 10.1016/S0002-9440(10)63389-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmbold P, Nayak RC, Marsch WC, Herman IM. Isolation and in vitro characterization of human dermal microvascular pericytes. Microvasc Res. 2001;61:160–165. doi: 10.1006/mvre.2000.2292. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Monosov E, Stallcup WB. NG2 proteoglycan expression by pericytes in pathological microvasculature. Microvasc Res. 2002;63:129–134. doi: 10.1006/mvre.2001.2376. [DOI] [PubMed] [Google Scholar]
- Rao S, Lobov IB, Vallance JE, Tsujikawa K, Shiojima I, Akunuru S, Walsh K, Benjamin LE, Lang RA. Obligatory participation of macrophages in an angiopoietin 2-mediated cell death switch. Development. 2007;134:4449–4458. doi: 10.1242/dev.012187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fassler R, Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–1857. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Lin J, Renner O, Shani M, Lundqvist A, Betsholtz C, Brownlee M, Deutsch U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes. 2002;51:3107–3112. doi: 10.2337/diabetes.51.10.3107. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Park JS, Valerius MT, McMahon AP. Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development. 2007;134:2533–2539. doi: 10.1242/dev.006155. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25:5603–5613. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R. Endoglin differentially modulates antagonistic transforming growth factor-beta1 and BMP-7 signaling. J Biol Chem. 2007;282:13934–13943. doi: 10.1074/jbc.M611062200. [DOI] [PubMed] [Google Scholar]
- Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130:141–155. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol. 2005;123:335–346. doi: 10.1007/s00418-005-0788-z. [DOI] [PubMed] [Google Scholar]
- Fujigaki Y, Muranaka Y, Sun D, Goto T, Zhou H, Sakakima M, Fukasawa H, Yonemura K, Yamamoto T, Hishida A. Transient myofibroblast differentiation of interstitial fibroblastic cells relevant to tubular dilatation in uranyl acetate-induced acute renal failure in rats. Virchows Arch. 2005;446:164–176. doi: 10.1007/s00428-004-1155-5. [DOI] [PubMed] [Google Scholar]
- Uemura A, Ogawa M, Hirashima M, Fujiwara T, Koyama S, Takagi H, Honda Y, Wiegand SJ, Yancopoulos GD, Nishikawa S. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J Clin Invest. 2002;110:1619–1628. doi: 10.1172/JCI15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13:1–7. doi: 10.1097/00041552-200401000-00001. [DOI] [PubMed] [Google Scholar]
- Taneda S, Hudkins KL, Topouzis S, Gilbertson DG, Ophascharoensuk V, Truong L, Johnson RJ, Alpers CE. Obstructive uropathy in mice and humans: potential role for PDGF-D in the progression of tubulointerstitial injury. J Am Soc Nephrol. 2003;14:2544–2555. doi: 10.1097/01.asn.0000089828.73014.c8. [DOI] [PubMed] [Google Scholar]
- Rajkumar VS, Shiwen X, Bostrom M, Leoni P, Muddle J, Ivarsson M, Gerdin B, Denton CP, Bou-Gharios G, Black CM, Abraham DJ. Platelet-derived growth factor-beta receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am J Pathol. 2006;169:2254–2265. doi: 10.2353/ajpath.2006.060196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, Neilson EG. Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol Ther. 2001;3:149–159. doi: 10.1006/mthe.2000.0251. [DOI] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Monkawa T, Tsuji M, Inukai M, Itoh H, Hayashi M. Snail1 is involved in the renal epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2007;362:63–68. doi: 10.1016/j.bbrc.2007.07.146. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang J, Luo JH, Dedhar S, Liu Y. Tubular epithelial cell dedifferentiation is driven by the helix-loop-helix transcriptional inhibitor Id1. J Am Soc Nephrol. 2007;18:449–460. doi: 10.1681/ASN.2006030236. [DOI] [PubMed] [Google Scholar]
- Collett GD, Canfield AE. Angiogenesis and pericytes in the initiation of ectopic calcification. Circ Res. 2005;96:930–938. doi: 10.1161/01.RES.0000163634.51301.0d. [DOI] [PubMed] [Google Scholar]
- Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther. 2005;7:R1113–1123. doi: 10.1186/ar1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomhoff R, Wake K. Perisinusoidal stellate cells of the liver: important roles in retinol metabolism and fibrosis. FASEB J. 1991;5:271–277. doi: 10.1096/fasebj.5.3.2001786. [DOI] [PubMed] [Google Scholar]
- Phillips CL, Pfeiffer BJ, Luger AM, Franklin CL. Novel collagen glomerulopathy in a homotrimeric type I collagen mouse (oim). Kidney Int. 2002;62:383–391. doi: 10.1046/j.1523-1755.2002.00451.x. [DOI] [PubMed] [Google Scholar]
- Dean DC, Peczon BD, Noelken ME, Hudson BG. Bovine glomerular basement membrane. Characterization of an alpha-size collagenous polypeptide. J Biol Chem. 1981;256:7543–7548. [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]