Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome (original) (raw)
Abstract
22q11.2 Deletion Syndrome (22q11.2DS) is a common microdeletion syndrome with congenital and late-onset features. Testing for the genomic content of copy number variations (CNVs) may help elucidate the 22q11.2 deletion mechanism and the variable clinical expression of the syndrome including the high (25%) risk for schizophrenia. We used genome-wide microarrays to assess CNV content and the parental origin of 22q11.2 deletions in a cohort of 100 adults with 22q11.2DS (44 with schizophrenia) and controls. 22q11.2DS subjects with schizophrenia failed to exhibit de novo CNVs or any excess of novel inherited CNVs outside the 22q11.2 region. There were no significant effects of parental origin of the 22q11.2 deletion, deletion length, parental age or family history on expression of schizophrenia. There was no evidence for a general increase of de novo CNVs in 22q11.2DS. A novel finding was the relative paucity of males with de novo 22q11.2 deletions of paternal origin (P = 0.019). The Y chromosome may play a mediating role in the mechanism of 22q11.2 deletion events during spermatogenesis, resulting in the previously observed excess of maternal de novo 22q11.2 deletions. Hemizygosity of the 22q11.2 region appears to be the major CNV-related risk factor for schizophrenia in 22q11.2DS. The results reinforce the need for further efforts to identify specific molecular mechanisms underlying this expression and to identify the 1% of patients with schizophrenia who carry 22q11.2 deletions.
INTRODUCTION
22q11.2 deletion syndrome (22q11.2DS), also known as velocardiofacial syndrome or DiGeorge syndrome (MIM nos 188400, 192430), is the most common microdeletion syndrome, occurring in approximately one in every 4000 live births (1). De novo 22q11.2 deletions (approximately 90%) are more common than inherited deletions (approximately 10%) in newly diagnosed patients. The high recurrence rate of these mutations is related to the presence of low-copy repeat sequences, which predispose the 22q11.2 region to structural rearrangement (2,3). A slight excess of de novo 22q11.2 deletions arising on the maternally inherited chromosome is unexplained (4,5).
The 22q11.2DS phenotype is highly variable with features including velopharyngeal insufficiency, congenital cardiac defects and learning difficulties. Most 22q11.2DS cases, however, share similar 3 Mb hemizygous microdeletions, suggesting that factors other than deletion length might contribute to variable clinical expression (6,7).
Adults with 22q11.2DS have a rate of schizophrenia approaching 25%, whereas in the general population approximately 1% of schizophrenia is associated with 22q11.2 deletions (8,9). It is likely that there are elevated rates of spontaneous mutations in schizophrenia in the general population (10). In addition to recurrent 22q11.2 deletions, recent reports (11–14) provide some evidence for elevated rates of rare novel CNVs and/or recurrent CNVs in schizophrenia. These studies suggest that CNVs at loci other than 22q11.2 may contribute to the expression of schizophrenia in 22q11.2DS. To test this hypothesis and to examine characteristics of the 22q11.2 deletion itself, we scanned the genomes of a cohort of 100 Canadian adults diagnosed with 22q11.2DS.
RESULTS
Genome-wide copy number variation
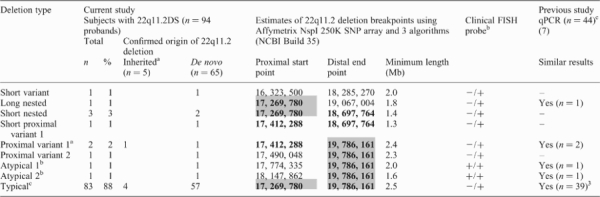
Table 1 summarizes the 22q11.2DS-related deletions observed in this study. Table 2 summarizes CNVs detected elsewhere in the genome. We found no evidence for an excess of CNVs or any specific targeted CNV regions in subjects with 22q11.2DS and schizophrenia, or in 22q11.2DS overall compared with controls. We were also able to compare the CNV content between 65 unaffected parents in whom de novo 22q11.2 deletions were confirmed not to have arisen and 53 unaffected parents on whose chromosome 22 the 22q11.2 deletions were confirmed to have arisen as de novo events. These analyses showed no significant differences between these two groups of parents in number, size and proportion of losses of genome-wide CNVs (Supplementary Material, Table S1).
Table 1.
22q11.2 deletions in 94 adult probands with 22q11.2 deletion syndrome (22q11.2 DS)
 |
|---|
Table 2.
Genome-wide copy number variants (CNV) in 22q11.2 deletion syndrome (22q11.2 DS) with and without schizophrenia and in unaffected parents
| 250K Affymetrix array 22q11.2DS study | 500K Affymetrix array (13) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 22q11.2DS patientsa (n = 95) | Unaffected parentsa (n = 122) | Popgen controls (n = 500) | |||||||||
| 22q11.2DS Schizophrenia versus non-psychoticb | All 22q11.2DS patients versus unaffected parentsb | ||||||||||
| All (n = 95) | Schizophrenia (n = 42) | Non-psychotic (n = 53) | Statistic | df | P | Statistic | df | P | |||
| Total number of CNV | 494 | 209 | 285 | 657 | 3695 | ||||||
| Stringentb | 216 | 91 | 125 | 286 | 1558 | ||||||
| Average CNV per genome | 5.2 | 5 | 5.4 | 5.4 | 7.4 | ||||||
| Stringentb | 2.3 | 2.2 | 2.4 | z = −1.20 | – | 0.2 | 2.3 | z = −0.43 | – | 0.7 | 3.1 |
| Median size in kb (range) | 177 (3–1987) | 177 (3–1987) | 181 (3–1987) | 194 (2–2731) | 151 | ||||||
| Stringentb | 212 (14.8–1987) | 211 (24.7–1987) | 215 (14.8–1987) | z = −0.36 | – | 0.7 | 206 (3–2731) | z = −0.51 | – | 0.6 | 224 |
| Number >1 Mb CNV (% of total) | 45 (9.1%) | 19 (9%) | 26 (9.1%) | 72 (10.9%) | 343 (9%) | ||||||
| Stringentb | 28 (13%) | 13 (14.3%) | 15 (12%) | χ2 = 0.24 | 1 | 0.6 | 46 (16.1%) | χ2 = 0.95 | 1 | 0.3 | 250 (16%) |
| Losses, % of total | 36% | 35.4% | 36.5% | 37.6% | 41% | ||||||
| Stringentb | 40.3% | 36.3% | 43.2% | χ2 = 1.05 | 1 | 0.3 | 40.6% | χ2 = 0.004 | 1 | 1.0 | 30% |
| Number of novel CNV (% of total) | 100 (20.2%) | 42 (20.1%) | 58 (20.4%) | 117 (18.1%) | 690 (19%) | ||||||
| Stringentb [number of different loci involved] | 39 (18.1%) [39] | 18 (19.8%) [18] | 21 (16.8%) [21] | χ2=0.32 | 1 | 0.6 | 44 (15.4%) [43] | χ2 = 0.64 | 1 | 0.4 | 237 (15.2%) [219] |
There was only one confirmed de novo CNV outside the 22q11.2 region (a 636 kb gain at 4q35.1) that occurred in a non-psychotic 22q11.2DS proband. This is within the expected rates, given a de novo mutation rate of approximately 1% at this resolution of analysis in control populations (15). Inspection of approximately 100 inherited CNVs in subjects with 22q11.2DS revealed on average fewer regions of interest in this study than in our recent study of autism spectrum disorders (16). There was a novel recurrent/overlapping CNV at 18p11.31 in four subjects with 22q11.2DS and schizophrenia, three with 8.5–10 kb losses that were not validated with quantitative polymerase chain reaction (qPCR) and one with a confirmed 338 kb gain inherited from an unaffected parent. Although a possible region of interest for psychotic disorders (17,18), the CNV region contained no genes. Supplementary Material, Tables S2 and S3 show all CNVs, besides the major 22q11.2 deletion, in subjects with 22q11.2DS and their unaffected parents, respectively.
22q11.2 deletions and related copy number variations
The majority (83/94; 88.3%) of probands with 22q11.2DS had the common 3 Mb 22q11.2 deletion. The other 11 probands carried one of eight variants of the 22q11.2 deletion (Table 1). Single nucleotide polymorphism (SNP) array results broadly agreed with qPCR results previously published for 44 of the subjects (7) (Table 1). Subjects with schizophrenia carried the typical (39/44; 88.6%) or one of the three variant 22q11.2 deletion breakpoints.
We also examined the 22q11.2 deletion region for CNVs that could predispose to deletion formation. As expected in this region, there were polymorphic CNVs in some parents, including two on whose chromosome the deletion had arisen. A rare 142 508 bp loss (17 269 780–17 412 288), involving the typical 22q11.2 deletion's proximal breakpoint, was found with one algorithm in a proband and unaffected father on whose chromosome arose the 22q11.2 deletion with an atypical proximal breakpoint about 360 kb distal at 17 774 335 (Atypical 1, Table 1). The rarity of this CNV in control populations (0.2–0.8%) (16,19) makes it possible that it was a factor in the origin of this atypical deletion which is estimated to comprise <1% of all 22q11.2 deletions identified (6). However, there was no evidence that this or any other CNV in the 22q11.2 region predisposed to other 22q11.2 deletion events.
Parental origin of 22q11.2 deletions and expression of schizophrenia
Expression of schizophrenia was not related to inherited or de novo status of 22q11.2 deletions. Of the probands with inherited 22q11.2 deletions (Fig. 1), three of seven (42.9%; one confirmed, two probable) had schizophrenia. Of the 65 probands with confirmed de novo deletions (Table 1), in whom parental origin of deletion could be resolved for 60 (Fig. 1), there were no significant differences in parental origin of de novo 22q11.2 deletion between the schizophrenia (14/26, 53.9% maternal) and non-psychotic groups (22/34, 64.7% maternal; _χ_2 = 0.72, df = 1, P = 0.39), or in mean intellectual level (IQ) between probands with maternal or paternal origin of de novo 22q11.2 deletion (70.8, SD 7.9 versus 71.9, SD 9.5, respectively; t = −0.48, df = 22, P = 0.63). IQ results were similarly non-significant within the schizophrenia sub-group (data not shown).
Figure 1.

22q11.2DS, copy number variants (CNVs) and parental origin of deletion. Summary of samples used for genome-wide CNV analyses and of results for analyses of parental origin of 22q11.2 deletions. Confirmed results for parental origin of deletion are based on CNV and fluorescence in situ hybridization (FISH) data for 46 trios and 19 duos. In six cases, FISH results allowed inference of a de novo (n = 5) or inherited (n = 1) 22q11.2 deletion when parental DNA was unavailable and, in five cases CNV results for siblings helped confirm parental origin. Probable results are based on CNV and FISH data for 11 duos, supplemented with clinical data for the parent whose DNA was unavailable for study.
Other factors examined also showed no evidence that they significantly affected risk for schizophrenia in 22q11.2DS. In first-degree relatives, with fluorescence in situ hybridization (FISH) data confirming no 22q11.2 deletion, there was a family history of psychotic illness in two of 44 (4.5%) subjects with schizophrenia and one of 56 (1.8%) in the non-psychotic group. There were no significant differences between these groups in maternal or paternal age (data not shown).
Sex distribution and parental origin of 22q11.2 deletions
Table 3 shows the sex distribution of probands with confirmed de novo 22q11.2 deletions and known parental origin. As previously observed in 22q11.2DS, we found a non-significant excess of de novo maternal deletions (60%; P = 0.12). Further analyses showed that this was associated with sex of the offspring, with males significantly less likely than females to have a de novo 22q11.2 deletion of paternal origin [relative risk (RR) = 0.44, 95% CI = 0.20–0.94; P = 0.019]. This appeared to be due to an unexpected paucity of males with deletions arising on paternally inherited chromosomes (Table 3). Results were similar for an expanded sample (Fig. 1) including nine subjects with ‘probable’ de novo origin of 22q11.2 deletion (RR = 0.47, 95% CI = 0.25–0.91; P = 0.014). Similar rates of subjects with schizophrenia were present in all four of the possible groups (data not shown) and there was no significant difference in sex distribution between the schizophrenia (20 male, 24 female) and non-psychotic (24 male, 32 female) 22q11.2DS groups (_χ_2 = 0.07, df = 1, P = 0.80).
Table 3.
Confirmed parental origin of de novo 22q11.2 deletions and sex distribution in 22q11.2 deletion syndrome (22q11.2 DS)
| Probands with 22q11.2DS | Parental origin of de novo 22q11.2 deletion | Analyses | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal | Paternal | Chi-square of equal proportions of parental origin | Relative risk (RR) for maternal origin of deletion in males | Overall chi-square of parental origin by sex | ||||||||
| n | (%) | n | (%) | χ2 | df | P | RR | 95% CI | χ2 | df | P | |
| Males | 20 | (33.3) | 6 | (10.0) | 7.54 | 1 | 0.006 | 1.63 | 1.08–2.47 | 5.48 | 1 | 0.019 |
| Females | 16 | (26.7) | 18 | (30.0) | 0.12 | 1 | 0.73 | |||||
| Total (n = 60) | 36 | (60.0) | 24 | (40.0) | 2.40 | 1 | 0.12 |
We expanded our examination of skewed sex distribution to include unaffected full sibling live births. Tests of equal proportions showed no significant differences in the six families with male probands of paternal de novo origin 22q11.2 deletions (three brothers, five sisters), 18 families with female probands of paternal origin (18 brothers, 21 sisters), 16 with female probands of maternal origin (14 brothers, 16 sisters) and 20 families of male probands of maternal origin (18 brothers, 14 sisters). There were also no significant differences in maternal or paternal age between subjects with maternal or paternal de novo origin of deletion overall or when male and female probands were examined separately (data not shown).
DISCUSSION
Our results suggest that the expression of schizophrenia in individuals with 22q11.2DS primarily arises from effects related to the 22q11.2 deletion itself. There was no indication that other CNVs elsewhere in the genome or that the parent of origin of de novo 22q11.2 deletions influenced the schizophrenia phenotype. An interesting observation was that the previously observed maternal excess of 22q11.2 deletions appears to be related to a relative paucity of 22q11.2 deletions of paternal origin in males, that is, in those deletions arising during spermatogenesis and segregated with a Y chromosome in the resulting sperm.
Expression of schizophrenia in 22q11.2DS
In contrast to the recent reports for schizophrenia in the general population (11–14), we found no evidence for an overall increase in novel genome-wide CNVs in 22q11.2DS. There was also no indication in our sample that specific recurrent CNVs, other than 22q11.2 deletions, were associated with expression of schizophrenia. While some rarer CNVs may be involved in increasing risk for schizophrenia in individual cases in the general population (11,13,14,20), these do not appear to be a necessary additional mechanism in the expression of schizophrenia in individuals with 22q11.2 deletions. We found no significant effects of parental origin of the 22q11.2 deletion, deletion length, paternal age or family history of psychotic illness on developing schizophrenia in 22q11.2DS. Our results are consistent with those showing no relationship of parental origin of de novo 22q11.2 deletions and expression of attention deficit hyperactivity disorder or obsessive–compulsive disorder in 22q11.2DS (21). Previous results using small samples that have suggested a parental origin of deletion effect on neuropsychiatric expression (22) are likely to be spurious. Collectively, these results provide no support for imprinting effects on neuropsychiatric expression in 22q11.2DS (23). Hemizygosity of the 22q11.2 region appears to be the main factor in neuropsychiatric expression in the syndrome (9).
Notwithstanding our negative statistical results, we compared individual CNVs in our 22q11.2DS sample to those reported in recent studies of schizophrenia from the general population that used various ascertainment strategies but similar CNV detection methods and diagnostic criteria (DSM-IV) (11–14) (Supplementary Material, Table S4). We were particularly interested in determining any overlap between individual non-22q11.2 deletion CNVs in our 22q11.2DS subjects with schizophrenia and those reported in these studies. Five subjects with schizophrenia from our sample had CNVs that shared some similarities in position, though not necessarily loss/gain status or length, to three CNVs reported in these studies. One 22q11.2DS subject with schizophrenia had a 1.48 Mb loss at 15q13.2-15q13.3 (chr 15:28,748,100-30,231,500) that was inherited from an unaffected parent. This CNV, in a region flanked by segmental duplications, has previously been reported in controls (chr 15:28,393,128-30,740,356; variation_8795) (24) as has a large inversion (chr 15:28,524,307-30,602,466) in this region (25). The CNV we observed was similar in position but approximately 100 kb smaller than the 1.54–2.47 Mb losses reported in 15 patients with schizophrenia and eight controls in two multi-site studies with slightly overlapping samples (13,14). Interpretation of these findings will be aided by further data on ethnicity of samples and the phenotype of controls and inheritance of this CNV in schizophrenia. Three subjects with 22q11.2DS and schizophrenia had large (>1.6 Mb) losses at 15q11.2, one of which was inherited from an unaffected parent. We found similar CNVs in 20 (16.4%) unaffected parents who did not transmit this CNV to the offspring with 22q11.2DS and in one non-psychotic proband with 22q11.2DS. These 15q11.2 CNVs show partial overlap with the proximal end of a 471 kb loss observed in 26 cases with schizophrenia and 79 controls in one study (13). Both the large >1.6 Mb and more distal 471 kb CNVs have previously been reported in controls (24). Finally, one subject with 22q11.2DS and schizophrenia had inherited from an unaffected parent a novel 141 kb gain in a 2q31.3 region with no genes but contained within the extent of a 2.5 Mb de novo loss reported in a patient with childhood-onset schizophrenia (11). These comparisons with the literature do not seem sufficiently compelling to modify our overall conclusions with respect to CNVs, other than the 22q11.2 deletion, and expression of schizophrenia in 22q11.2DS.
What then underlies the variable expression of schizophrenia in 22q11.2DS? Altered expression of several genes within the 22q11.2 region may be necessary to increase susceptibility of schizophrenia in 22q11.2DS (26–28). There is some evidence that SNPs in certain genes within the 22q11.2 deletion region on the intact homologous chromosome may be involved. For example, we have found significant association with schizophrenia and the PIK4CA gene in 22q11.2DS (29) using the same SNPs as those found to be associated in a general population sample of schizophrenia (30). This is in contrast to the negative findings of the COMT functional allele and schizophrenia in 22q11.2DS (31). Other small genetic variants in candidate genes for schizophrenia elsewhere in the genome may also modify the large effects of the hemizygous 22q11.2 deletion, as shown for other phenotypic features of 22q11.2DS (32,33). There also remains the possibility that various individual CNVs increase the likelihood of expressing schizophrenia in 22q11.2DS.
Origin of de novo 22q11.2 deletions
We found that males with 22q11.2DS who had a maternal origin of a de novo 22q11.2 deletion were represented in a similar proportion to females with either maternal or paternal origin of deletion, but that males had a significantly lower chance of having a paternal origin of the deletion. This is consistent with the meta-analyses of previous studies (4,5) and our own results showing an excess of de novo deletions of maternal origin in 22q11.2DS. Previous studies have not consistently documented the sex distribution of patients however, hampering further comparisons. Larger samples will be needed to replicate the paucity of paternal origin of de novo 22q11.2 deletions in males which, although significant, could be due to chance in a relatively small sample.
Explaining this finding is difficult. The mechanism underlying deletions in 22q11.2DS mainly involves non-allelic homologous recombination (NAHR) between misaligned low copy repeats during gametogenesis (4,34). It is possible that there is a female bias in NAHR causing de novo 22q11.2 deletions (5). Our results suggest however that de novo 22q11.2 deletions may occur with approximately equal frequency in oogenesis and during spermatogenesis, but that somehow the Y chromosome affects the likelihood of observing a 22q11.2 deletion. There is a sequence homology between the Yq12 region and the low copy repeats, LCR22-2 and LCR22-4, that are most commonly involved in the 22q11.2 interchromosomal exchanges and NAHR that give rise to 22q11.2 deletions. These regions share a 2.4 kb human satellite sequence subclass involving AT-rich tripartite repeats (35). Babcock et al. (35) have posited that these repeat elements may play a structural role in mediating NAHR between LCR22-2 and LCR22-4 if the presence of the Yq12 repeat prevents recombination between the LCRs, thereby enhancing meiotic stability of the 22q11.2 region. Such a mechanism could be related to physical proximity of these heterochromatic regions in the nuclear periphery (35,36). The report of a subject with 22q11.2DS resulting from a de novo Y;22 translocation (37) may be consistent with interchromosomal interaction between these chromosomes. To explain the possibility that, in sperm with a 22q11.2 deletion chromosome, there may be an elevated ratio of those with an X to those with a Y chromosome, however, would appear to implicate the segregation process, as a spermatocyte with a de novo 22q11.2 mutation would have both sex chromosomes present. One could speculate that proximity of the Y chromosome to the deletion chromosome 22 could interfere with normal segregation and/or error correction, resulting in an unbalanced, non-viable sperm (38), e.g. with two deletion 22q11.2 chromosomes.
Studies of sperm could help investigate possible mechanisms. Further studies of NAHR and allelic homologous recombination (AHR, i.e. normal meiotic recombination) in 3-generation families are also needed to determine whether polymorphic size of LCRs in the 22q11.2 region (39) or clustering of recombination events within certain families and high rates of AHR in the 22q11.2 region in female meiosis (5) may also play a role in the observed sex distribution of de novo 22q11.2 deletions.
Advantages and limitations
This is the largest sample of adults with 22q11.2DS, and one of the largest ever investigated with respect to the parent of origin of de novo 22q11.2 deletions (5). Nevertheless, there were only six men with confirmed paternal de novo origin of the deletion. There were fewer fathers than mothers available in parent–offspring pairs but a systematic ascertainment bias appears unlikely to explain the results. Although including the largest number of patients with 22q11.2DS-schizophrenia yet studied, our sample size was relatively limited. However, if there were a three-fold excess of genome-wide novel or rare CNVs as proposed in a recent study (11), our sample would have had sufficient power to show this. Few subjects (four of 56, 7.1%) in the non-psychotic group were younger than the median age at onset of psychosis (20.5 years) and there were no trends for differences between the schizophrenia and non-psychotic subgroups on any CNV-related parameter (Table 2), suggesting that misclassifying some subjects as non-psychotic would not have materially affected the results.
CONCLUSIONS
In summary, hemizygosity of the 22q11.2 region seems to confer the major CNV risk for the expression of schizophrenia in 22q11.2DS. While further investigations are required to determine molecular risk factors that mediate de novo 22q11.2 deletion events during gametogenesis, the results reinforce the need for genetic counseling of individuals with 22q11.2DS about the risk for schizophrenia and further efforts to identify the 1% of patients with schizophrenia who carry 22q11.2 deletions.
MATERIALS AND METHODS
Subjects with 22q11.2DS
We diagnosed 100 Canadian adults (44 male, 56 female) with 22q11.2DS originating from 94 families; six families had two affected subjects (five parent–offspring pairs and one half-sibling pair). All subjects met clinical screening criteria for 22q11.2DS in adults (40) and had confirmed chromosome 22q11.2 microdeletions using FISH and probes from the 22q11.2 region (41) (Table 1). Observed ethnicity of probands was 85/94 (90.4%) European, three of 94 (3.2%) African or mixed African, three of 94 (3.2%) Asian and three of 94 (3.2%) other origins, consistent with the genotype data. Probands (median age 31.4; range 17.6–56.3) were ascertained as previously described (8), many through psychiatric sources or referrals, ensuring a high rate of schizophrenia. Informed consent was obtained and the study was approved by the Research Ethics Boards of the University of Toronto, Centre for Addiction and Mental Health and University Health Network.
All 22q11.2DS subjects had direct assessments, review of medical records and information from relatives (8,31,42). Lifetime psychiatric diagnoses were obtained by research psychiatrists using standard methods and DSM-IV criteria (31). Subjects with schizophrenia (n = 37), schizoaffective disorder (n = 6) and psychotic disorder, not otherwise specified (n = 1), met the criteria for major psychotic disorders [mean age at onset 20.9 (SD 5.4) years], collectively termed ‘schizophrenia’ for this study. The 56 other subjects comprised the non-psychotic subgroup. Intellectual level was assessed using standard methods to obtain Full Scale IQ (n = 86) or IQ estimates (n = 8) (43). As expected (43), subjects with schizophrenia were significantly older (38.3, SD 9.8 versus 30.6, SD 10.6 years; t = 3.74, P < 0.001) and had lower mean IQ (68.6, SD 9.7 versus 73.0, SD 10.2; t = −2.16, P = 0.03) than those in the non-psychotic group.
Parents
We included data for as many of the biological parents of this adult sample as possible; no parental data existed for four probands who were adopted. Mean paternal and maternal ages at birth for 96 subjects with 22q11.2DS were 31.3 (SD 5.8) and 27.9 (SD 4.7) years, respectively. FISH data confirmed the absence of a 22q11.2 deletion in 73 mothers and 64 fathers. Extensive family history data from multiple sources provided information on the demographics and health status of first-degree relatives, including common phenotypic features of 22q11.2 DS.
Copy number variation detection
We genotyped each available DNA sample [99 subjects with 22q11.2DS; 122 unaffected parents (69 mothers, 53 fathers); five unaffected siblings] for approximately 250 000 SNPs with the Affymetrix GeneChip® Human Mapping 250K NspI Array according to established protocols (16,39,44). Duos (proband and one parent) and trios (proband and both parents) comprised the sample sets used to investigate parental origin of de novo 22q11.2 deletions and inheritance of genome-wide CNVs (Fig. 1).
NspI array scans were analyzed for CNV content with three algorithms: DNA Chip Analyzer (dChip) (45,46), Copy Number Analysis for GeneChip (CNAG) (47) and Genotyping Microarray based CNV Analysis (GEMCA) (16,48). To minimize potential batch effects, analysis with dChip was performed in groups of approximately 50 probands/subjects according to when the arrays were hybridized. For CNAG version 2.0, we set the reference pool to include all samples and performed an automatic batch pairwise analysis with sex-matched controls. For GEMCA, we used two designated DNA samples (NA10851 and NA15510) as references for pairwise comparison with all proband experiments. We further filtered these results by including only those CNVs that were common to both pairwise experiments. In all instances, CNVs were merged if they were detected in the same individual by more than one algorithm with the outside probe boundaries (16). In general, CNAG Merged (5.6/genome) and GEMCA (5.5/genome) algorithms show more CNVs per genome than dChip Merged (3.0/genome) (16). We report all data but in analyses we used a ‘stringent’ data set, i.e. CNVs called by more than one algorithm, because these data have a high rate of validation (16).
Controls
Comparison control data comprised CNVs observed in 500 Europeans from the German PopGen project (16,49) and entries in the Database of Genomic Variants (containing 8006 CNVs at 3933 loci) (24). A CNV was considered to be ‘novel’ in subjects if it was ≥10 kb, contained at least three probes, and at least 20% of its total length was unique when compared with controls. We also compared the results to CNVs found in a cohort of 1152 non-disease controls of European origin from the Ontario population that had been analyzed using the same methods (16,19).
Parental origin of 22q11.2 deletions and other copy number variations
We used SNP data to infer the parental origin of 22q11.2 deletions for trios and a combination of SNP and FISH data for duos. Where parent of origin could not be resolved using SNP data, microsatellite marker analyses were performed using 11 markers (sequence available upon request) across the 22q11.2 region (16). For some duos, parent of origin could be determined but that parent was unavailable for genotypic study. In those cases, we estimated whether 22q11.2 deletions were probably inherited or de novo in origin using the available clinical data. We assigned a ‘probable inherited’ 22q11.2 deletion status if the unavailable parent had two or more major features of 22q11.2DS (40) and a ‘probable _de novo_’ 22q11.2 deletion status if the unavailable parent had none of these features and other offspring did not meet the clinical criteria for 22q11.2DS (40).
Copy number variation validation of genome-wide copy number variations
We validated all de novo and other potentially interesting novel CNVs (16). Position coordinates were determined using NCBI Build 35, and results were confirmed using inheritance patterns and qPCR.
Statistical analysis
Chi-square or two-sided Fisher's exact tests and estimates of relative risk with 95% CI were used to compare categorical variables. Wilcoxon rank sum tests were used to compare ordinal data, and two-tailed Student's _t_-tests for continuous variables. Main comparisons were between schizophrenia and non-psychotic groups on genome-wide CNVs and parent of origin of de novo 22q11.2 deletions; we also examined parental ages at birth and family history of psychotic illness in first-degree relatives. All analyses were performed using SAS 9.1.3 (SAS Institute, Cary, NC, USA). _P_-values <0.05 were considered significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported by grants from the Canadian Institutes of Health Research (MOP-79518), W. Garfield Weston Foundation, a Distinguished Investigator award from the National Alliance for Research on Schizophrenia and Depression (A.S.B.), and Canada Research Chair in Schizophrenia Genetics (A.S.B.). C.R.M. is supported by the National Alliance for Research on Schizophrenia and Depression (NARSAD). S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genetics and Genomics at the University of Toronto and the Hospital for Sick Children.
Supplementary Material
[Supplementary Data]
ACKNOWLEDGEMENTS
The authors thank the patients and their family members for their participation, staff and students at our Clinical Genetics Research Program, staff at The Centre for Applied Genomics (supported by Genome Canada/Ontario Genomics Institute) and colleagues at the Toronto Congenital Cardiac Centre for Adults, Hospital for Sick Children and Centre for Addiction and Mental Health and many others for referring patients. Funding to pay the open access charges was provided by Canadian Institutes of Health Research grant MOP-79518.
Conflict of Interest statement. None of the authors have financial interests that might present a conflict of interest.
REFERENCES
- 1.Oskarsdottir S., Vujic M., Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch. Dis. Child. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edelmann L., Pandita R.K., Morrow B.E. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet. 1999;64:1076–1086. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaikh T.H., Kurahashi H., Emanuel B.S. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet. Med. 2001;3:6–13. doi: 10.1097/00125817-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Thomas N.S., Durkie M., Potts G., Sandford R., Van Zyl B., Youings S., Dennis N.R., Jacobs P.A. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11.23, 15q11-q13 and 22q11. Eur. J. Hum. Genet. 2006;14:831–837. doi: 10.1038/sj.ejhg.5201617. [DOI] [PubMed] [Google Scholar]
- 5.Torres-Juan L., Rosell J., Sanchez-de-la-Torre M., Fibla J., Heine-Suner D. Analysis of meiotic recombination in 22q11.2, a region that frequently undergoes deletions and duplications. BMC Med. Genet. 2007;8:14. doi: 10.1186/1471-2350-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saitta S.C., Harris S.E., Gaeth A.P., Driscoll D.A., McDonald-McGinn D.M., Maisenbacher M.K., Yersak J.M., Chakraborty P.K., Hacker A.M., Zackai E.H., et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum. Mol. Genet. 2004;13:417–428. doi: 10.1093/hmg/ddh041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weksberg R., Stachon A.C., Squire J.A., Moldovan L., Bayani J., Meyn S., Chow E.W.C., Bassett A.S. Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum. Genet. 2007;120:837–845. doi: 10.1007/s00439-006-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassett A.S., Chow E.W.C., Husted J., Weksberg R., Caluseriu O., Webb G.D., Gatzoulis M.A. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am. J. Med. Genet. A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassett A.S., Chow E.W.C. Schizophrenia and 22q11.2 Deletion Syndrome. Curr. Psychiatry Rep. 2008;10:148–157. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bassett A.S., Bury A., Hodgkinson K.A., Honer W.G. Reproductive fitness in familial schizophrenia. Schizophr. Res. 1996;21:151–160. doi: 10.1016/0920-9964(96)00018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A., et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 12.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 13.Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupski J.R. Genomic rearrangements and sporadic disease. Nat. Genet. (Supplement) 2007;30:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 16.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y., et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee O., Meera P., Ghosh S., Kubendran S., Kiran K., Manjunath K.R., Subhash M.N., Benegal V., Brahmachari S.K., Majumder P.P., et al. Evidence of linkage and association on 18p11.2 for psychosis. Am. J. Med. Genet. B. 2006;141:868–873. doi: 10.1002/ajmg.b.30363. [DOI] [PubMed] [Google Scholar]
- 18.Faraone S.V., Skol A.D., Tsuang D.W., Young K.A., Haverstock S.L., Prabhudesai S., Mena F., Menon A.S., Leong L., Sautter F., et al. Genome scan of schizophrenia families in a large Veterans Affairs Cooperative Study sample: evidence for linkage to 18p11.32 and for racial heterogeneity on chromosomes 6 and 14. Am. J. Med. Genet. B. 2005;139:91–100. doi: 10.1002/ajmg.b.30213. [DOI] [PubMed] [Google Scholar]
- 19.Zogopoulos G., Ha K.C., Naqib F., Moore S., Kim H., Montpetit A., Robidoux F., Laflamme P., Cotterchio M., Greenwood C., et al. Germ-line DNA copy number variation frequencies in a large North American population. Hum. Genet. 2007;122:345–353. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]
- 20.Kirov G., Gumus D., Chen W., Norton N., Georgieva L., Sari M., O'Donovan M.C., Erdogan F., Owen M.J., Ropers H.H., et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 2007;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 21.Michaelovsky E., Gothelf D., Korostishevsky M., Frisch A., Burg M., Carmel M., Steinberg T., Inbar D., Apter A., Weizman A. Association between a common haplotype in the COMT gene region and psychiatric disorders in individuals with 22q11.2DS. Int. J. Neuropsychopharmacol. 2008;11:351–363. doi: 10.1017/S1461145707008085. [DOI] [PubMed] [Google Scholar]
- 22.Eliez S., Antonarakis S.E., Morris M.A., Dahoun S.P., Reiss A.L. Parental origin of the deletion 22q11.2 and brain development in velocardiofacial syndrome. Arch. Gen. Psychiatry. 2001;58:64–68. doi: 10.1001/archpsyc.58.1.64. [DOI] [PubMed] [Google Scholar]
- 23.Maynard T.M., Meechan D.W., Heindel C.C., Peters A.Z., Hamer R.M., Lieberman J.A., LaMantia A.S. No evidence for parental imprinting of mouse 22q11 gene orthologs. Mamm. Genome. 2006;17:822–832. doi: 10.1007/s00335-006-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:941–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 25.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F., et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meechan D.W., Maynard T.M., Gopalakrishna D., Wu Y., LaMantia A.S. When half is not enough: gene expression and dosage in the 22q11 deletion syndrome. Gene Expr. 2007;13:299–310. doi: 10.3727/000000006781510697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sivagnanasundaram S., Fletcher D., Hubank M., Illingworth E., Skuse D., Scambler P. Differential gene expression in the hippocampus of the Df1/+ mice: a model for 22q11.2 deletion syndrome and schizophrenia. Brain Res. 2007;1139:48–59. doi: 10.1016/j.brainres.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z., Baldini A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum. Mol. Genet. 2007;17:150–157. doi: 10.1093/hmg/ddm291. [DOI] [PubMed] [Google Scholar]
- 29.Vorstman J.A.S., Chow E.W.C., Ophoff R.A., Engeland H., Beemer F.A., Kahn R.S., Sinke R.J., Bassett A.S. Association of the PIK4CA schizophrenia-susceptibility gene in adults with the 22q11.2 deletion syndrome. Am. J. Med. Genet. B. 2008 doi: 10.1002/ajmg.b.30827. Published online July 21, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jungerius B.J., Hoogendoorn M.L., Bakker S.C., Van't Slot R., Bardoel A.F., Ophoff R.A., Wijmenga C., Kahn R.S., Sinke R.J. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol. Psychiatry. 2007;12:1–9. doi: 10.1038/sj.mp.4002080. [DOI] [PubMed] [Google Scholar]
- 31.Bassett A.S., Caluseriu O., Weksberg R., Young D.A., Chow E.W.C. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol. Psychiatry. 2007;61:1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guris D.L., Duester G., Papaioannou V.E., Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Amati F., Biancolella M., Farcomeni A., Giallonardi S., Bueno S., Minella D., Vecchione L., Chillemi G., Desideri A., Novelli G. Dynamic changes in gene expression profiles of 22q11 and related orthologous genes during mouse development. Gene. 2007;391:91–102. doi: 10.1016/j.gene.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 34.Shaw C.J., Lupski J.R. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum. Mol. Genet. 2004;13:57–64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- 35.Babcock M., Yatsenko S., Stankiewicz P., Lupski J.R., Morrow B.E. AT-rich repeats associated with chromosome 22q11.2 rearrangement disorders shape human genome architecture on Yq12. Genome Res. 2007;17:451–460. doi: 10.1101/gr.5651507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carvalho C., Pereira H.M., Ferreira J., Pina C., Mendonca D., Rosa A.C., Carmo-Fonseca M. Chromosomal G-dark bands determine the spatial organization of centromeric heterochromatin in the nucleus. Mol. Biol. Cell. 2001;12:3563–3572. doi: 10.1091/mbc.12.11.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lupski J.R., Langston C., Friedman R., Ledbetter D.H., Greenberg F. Di George anomaly associated with a de novo Y;22 translocation resulting in monosomy del(22)(q11.2) Am. J. Med. Genet. 1991;40:196–198. doi: 10.1002/ajmg.1320400214. [DOI] [PubMed] [Google Scholar]
- 38.Nicklas R.B. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- 39.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassett A.S., Chow E.W.C. 22q11 Deletion syndrome: a genetic subtype of schizophrenia. Biol. Psychiatry. 1999;46:882–891. doi: 10.1016/s0006-3223(99)00114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Driscoll D.A., Salvin J., Sellinger B., Budarf M.L., McDonald-McGinn D.M., Zackai E.H., Emanuel B.S. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J. Med. Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassett A.S., Chow E.W.C., AbdelMalik P., Gheorghiu M., Husted J., Weksberg R. The schizophrenia phenotype in 22q11 Deletion Syndrome. Am. J. Psychiatry. 2003;160:1580–1586. doi: 10.1176/appi.ajp.160.9.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chow E.W.C., Watson M., Young D.A., Bassett A.S. Neurocognitive profile in 22q11 Deletion Syndrome and schizophrenia. Schizophr. Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moessner R., Marshall C.R., Sutcliffe J., Skaug J., Pinto D., Vincent J., Zwaigenbaum L., Frenandez B., Roberts W., Szatmari P., et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Consortium T.A.G.P. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao X., Li C., Paez J.G., Chin K., Janne P.A., Chen T.H., Girard L., Minna J., Christiani D., Leo C., et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 47.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C., et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 48.Komura D., Shen F., Ishikawa S., Fitch K.R., Chen W., Zhang J., Liu G., Ihara S., Nakamura H., Hurles M.E., et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006;16:1575–1584. doi: 10.1101/gr.5629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinto D., Marshall C., Feuk L., Scherer S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007;16:168–173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Data]