High-resolution mapping and isolation of a yeast artificial chromosome contig containing fw2.2: A major fruit weight quantitative trait locus in tomato (original) (raw)
Abstract
A high-resolution physical and genetic map of a major fruit weight quantitative trait locus (QTL), fw2.2, has been constructed for a region of tomato chromosome 2. Using an F2 nearly isogenic line mapping population (3472 individuals) derived from Lycopersicon esculentum (domesticated tomato) × Lycopersicon pennellii (wild tomato), fw2.2 has been placed near TG91 and TG167, which have an interval distance of 0.13 ± 0.03 centimorgan. The physical distance between TG91 and TG167 was estimated to be ≤ 150 kb by pulsed-field gel electrophoresis of tomato DNA. A physical contig composed of six yeast artificial chromosomes (YACs) and encompassing fw2.2 was isolated. No rearrangements or chimerisms were detected within the YAC contig based on restriction fragment length polymorphism analysis using YAC-end sequences and anchored molecular markers from the high-resolution map. Based on genetic recombination events, fw2.2 could be narrowed down to a region less than 150 kb between molecular markers TG91 and HSF24 and included within two YACs: YAC264 (210 kb) and YAC355 (300 kb). This marks the first time, to our knowledge, that a QTL has been mapped with such precision and delimited to a segment of cloned DNA. The fact that the phenotypic effect of the fw2.2 QTL can be mapped to a small interval suggests that the action of this QTL is likely due to a single gene. The development of the high-resolution genetic map, in combination with the physical YAC contig, suggests that the gene responsible for this QTL and other QTLs in plants can be isolated using a positional cloning strategy. The cloning of_fw2.2_ will likely lead to a better understanding of the molecular biology of fruit development and to the genetic engineering of fruit size characteristics.
Keywords: Lycopersicon esculentum, mapping, fruit development, domestication
In nature there are numerous examples of quantitative traits that display continuous variation due to the interaction of polygenes, or quantitative trait loci (QTLs), with the environment. Early genetic experiments with beans (1), wheat (2), and tobacco (3) suggested that continuous variation in phenotype could be accounted for by a large number of polygenes, each contributing a small effect. More recently, through the use of molecular linkage maps, a systematic search of entire genomes for QTLs has revealed that in many cases a large proportion of the total phenotypic variance is attributable to a few major QTLs (4–12). Such QTL marker studies have changed the original hypothesis that polygenes each have an equally small effect on the phenotype. Instead, it appears that for traits displaying continuous variation, the phenotype is the result of the action of major QTLs together with the environment and QTLs of lesser effects.
Tomato fruit weight is a classic example of a quantitative trait displaying continuous variation (13). Previous studies indicated that between 5 (14) and 20 (15) genes are involved in the inheritance of tomato fruit weight. Recently, molecular marker studies conducted by Paterson et al. (6) found 11 QTLs affecting tomato fruit mass in a cross between the domesticated tomato (Lycopersicon esculentum Mill.) and the related wild tomato species,Lycopersicon cheesmanii. In all cases, the wild species allele caused a reduction in fruit mass. The phenotypic variance explained by these 11 fruit mass QTLs ranged from 4.7% to 42.0%, with the majority (55%) of individual QTL accounting for greater than 10% of the phenotypic variance.
Recently we identified a major fruit weight QTL, fw2.2, on chromosome 2 that is common to both green and red fruited tomato species (16). fw2.2 accounts for 5–30% of the phenotypic variance in segregating populations (6, 16), or up to 47% in an F2 nearly isogenic line (NIL) population (16). Cultivated and wild tomatoes are apparently differentiated by a major allelic substitution at fw2.2 (16).
In this paper, we report the development of a high-resolution physical and genetic map of the fw2.2 locus and the development of a tomato yeast artificial chromosome (YAC) contig spanning the region. Because the phenotypic effect of the fw2.2 QTL can be mapped to a small interval, it is likely that the action of this QTL is due to a single gene. The development of a high-resolution map of_fw2.2_ may lead to the molecular cloning of this key locus controlling fruit weight, which may open the door to understanding the molecular biology of fruit development and potentially to the genetic engineering of fruit size characteristics.
MATERIALS AND METHODS
Plant Material.
Seed for the F2 Lycopersicon pennellii NIL population was generated as described previously in a cross between the domesticated tomato (L. esculentum) cv M82-1-8 and the wild tomato species, L. pennellii (16, 17). In the present study, a total of 3472 F2 plants, nearly isogenic for the region spanning the fw2.2 locus, were subjected to restriction fragment length polymorphism (RFLP) analysis (see below). Based on this analysis, 55 F2 NIL plants were identified that contained recombination events within the_fw2.2_ region spanning the RFLP markers CD66 to TG361 (see Fig. 1). These 55 F2 NIL recombinants were used to develop a high-resolution map using molecular markers in the region of_fw2.2_. Of these 55 recombinant plants, 51 were used for phenotypic analysis of fruit weight. The remaining four recombinants failed to produce an apical meristem. A single homozygous recombinant NIL, 939–2, containing the wild species (L. pennellii) DNA in the region spanning CD66 and TG361, was selected by RFLP analysis and used along with the recurrent parent, M82-1-8, as controls with which to compare the recombinants. Five replications of recombinant plants and controls were separately transplanted to the field in Ithaca, NY, and Davis, CA, for phenotypic evaluations.
Figure 1.
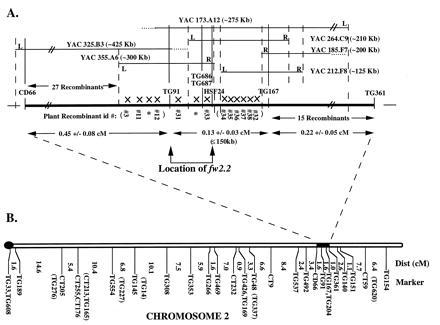
High-resolution physical and molecular map of_fw2.2_ on tomato chromosome 2. (A) The high-resolution map of fw2.2 is delineated by CD66 and TG361. The map position (dashed vertical lines) of YAC iPCR “right”-end (R) and “left”-end (L) sequences was determined by genetic mapping using the recombinants (X), while the physical placement (solid vertical lines) of YACs was determined by RFLP analysis using genomic, cDNA, and RAPD molecular markers. Ends of YACs that could not be genetically mapped are shown with dotted horizontal lines. The physical size, in kilobases (kb), of the YACs and the distance spanning TG91 and TG167 (≤150 kb) was determined by PFGE and DNA hybridization. The location of fw2.2 (bracketed by arrows) was determined by the phenotypic data associated with recombinants #31 and #33 (Fig. 2). The map distances with standard deviations, in centimorgans (cM), were calculated based on the number of recombinants obtained from the F2 population divided by the total number of meioses (6944) times 100. Plants that died (∗) were included in the number of recombinants for mapping purposes. The map order of the recombinants in parentheses has not been determined. (B) Tomato chromosome 2 high-density linkage map (L. esculentum × L. pennellii; ref. 20). The Kosambi mapping function was used to convert recombination frequencies to map distances in centimorgans (cM) (33). Markers with tick marks were ordered with LOD > 3. Markers enclosed in parentheses were located to corresponding intervals with LOD < 3. The black knob indicates the location of the centromere. The black box indicates the region corresponding (expanding dotted line) to the high-resolution map of fw2.2 in A.
Phenotypic Analysis.
Five replications of individual recombinant plants and the controls, NIL 939–2 and tomato cultivar M82-1-8, were evaluated for fruit weight by weighing 10 representative fruits from each plant.
DNA Extraction and RFLP Analysis.
DNA from leaf tissue was extracted for polymerase chain reactions (PCR), random amplified polymorphic DNA (RAPD) analysis, and DNA blots using a microprep procedure (18). Microprep DNA was digested with either _Bst_NI or _Eco_RV and further subjected to DNA blot analysis as described by Bernatzky and Tanksley (19). Molecular markers (cDNA and genomic DNA clones) from the tomato high-density linkage map (20), RAPDs, and YAC end sequences were used to survey the fw2.2 region spanning CD66 and TG361 (see Fig. 1). Probes were labeled with [32P]dCTP by primer extension (21).
RAPD Analysis, Purification, and Sequence Information.
Six hundred decanucleotide primers were obtained from Operon Technologies (Alameda, CA), and were used to amplify 50 ng of DNA from the NIL, 939–2, and the tomato cultivar, M82-1-8, as described by Martin_et al._ (22). DNA was amplified using an MJR Thermocycler (MJ Research, Cambridge, MA), further resolved on 2% agarose electrophoresis gels in 1× TAE buffer (40 mM Tris·acetate/1 mM EDTA, pH 8.0), and detected by staining in 2 mg/ml ethidium bromide. Selected RAPD products were purified to a single band by excising a portion of the polymorphic band, followed by a second round of PCR using the same primers and conditions. Subsequent single band RAPD products were purified using a QIAquick PCR purification kit as described by the manufacturer (Qiagen, Chatsworth, CA). The two RAPD primers discussed in this paper have the following nucleotide sequence: OPW-01, 5′-d[CTCAGTGTCC]-3′; OPG-08, 5′-d[TCACGTCCAC]-3′.
Subcloning and Mapping of RAPDs OPW-01 and OPG-08.
RAPDs OPW-01 and OPG-08 were subcloned into the vector, PCRII, using the TA cloning kit and procedures described by the manufacturer (Invitrogen). OPW-01 and OPG-08 were subsequently mapped to the fw2.2 region, and were given tomato genomic (TG) numbers, TG686 and TG687, respectively.
Conversion of Molecular Markers to Cleaved Amplified Polymorphic Sequences.
The molecular markers TG91, TG686, TG687, TG167, and TG361 (see Fig. 1) were converted to codominant PCR based molecular markers, using the procedures previously described for cleaved amplified polymorphic sequences (23, 24). Amplification reactions were done using 50 ng genomic DNA/200 μM dNTPs/10 μM primer/1× reaction buffer (10 mM Tris·HCl, pH 8.3/50 mM KCl/1.5 mM MgCl/0.1% gelatin)/1 unit of Taq polymerase in 25 μl of reaction mix using an MJR Thermocycler (MJ Research) programmed for 35 cycles with a denaturing temperature of 94°C for 1 min, an annealing temperature of 50°C for 1 min, and an extension temperature of 72°C for 2 min. Following PCR, the products were digested with the appropriate restriction enzymes (Table 1) to yield codominant markers that were resolved on 2% agarose electrophoresis gels in 1× TAE buffer (40 mM Tris·acetate/1 mM EDTA, pH 8.0) and detected by staining in 2 mg/ml ethidium bromide. No polymorphism was detected for marker CD66 using 20 restriction enzymes. The primer sequences for the molecular markers are shown in Table 1.
Table 1.
PCR conditions for cleaved amplified polymorphic sequence markers
| Marker | OP number | Primer sequences | Genomic PCR product size, kb | Enzyme used |
|---|---|---|---|---|
| TG91 | NA | 5′-d[TGCAGAGCTGTAATATTTAGAC]-3′ | 0.4 | _Dra_I |
| 5′-d[CGGTCTCAGTTGCAACTCAA]-3′ | ||||
| TG167 | NA | 5′-d[GCGAGAGCGAGTTGAGTGTATATC]-3′ | 1.3 | _Taq_I |
| 5′-d[CAGAAGAGAGAAGCTGCAAAGCAG]-3′ | ||||
| TG361 | NA | 5′-d[GTACAGGAGTCCTCTGAGATGATC]-3′ | 0.6 | _Apo_I |
| 5′-d[CAACGACAAGCATTCCAGTC]-3′ | ||||
| TG686 | OP1 | 5′-d[GGTTCATGTTGACTTGACGGTAG]-3′ | 1.2 | _Taq_I |
| 5′-d[CTCAGTGTCCACAAGATCAAATG]-3′ | ||||
| TG687 | OP608 | 5′-d[GACTCATGGAGTAAATGCAATCAC]-3′ | 0.6 | _Apo_I |
| 5′-d[TTCACGTCCACTTGAGGTTTGG]-3′ | ||||
| CD66 | NA | 5′-d[CTCAAGATGTCAATGAAGTGACC]-3′ | 0.25 | NP |
| 5′-d[CTCTGCTCGACAGAGCTGAAC]-3′ |
Isolation and Digestion of High Molecular Weight Tomato and YAC DNA, Pulsed-Field Gel Electrophoresis (PFGE), and Blotting.
High molecular weight DNA was isolated from the tomato cultivar M82-1-8 using a nuclei preparation procedure as described by Liu and Whittier (25). The isolation of high molecular weight YAC DNA was as described by Ausubel et al. (26). Digestion of high molecular weight DNA, PFGE, and blotting were performed as described by Ganal and Tanksley (27). The only modification was that gels were blotted to Hybond N+ (Amersham) instead of GeneScreen Plus (NEN). Pulsed-field conditions included a 40-sec pulse at a constant 125 V using 1% agarose (BM) gels electrophoresed in 0.5× TAE buffer (40 mM Tris·acetate/1 mM EDTA, pH 8.0) for 60 hr.
Isolation and Analysis of Tomato YAC Clones, Yeast Strains, and Media.
Tomato YAC clones were obtained by a method using PCR (28) from a previously constructed library (29) using primers and conditions for the molecular markers TG91, TG686, TG687, TG167, and TG361 as described in conversion to cleaved amplified polymorphic sequences above (Table 1).
YAC “right”- and “left”-end termini were isolated by inverse PCR (iPCR), following digestion with _Alu_I as described by Giovannoni et al. (30).
The Saccharomyces cerevisiae strain AB1380 (MATa, y+, ura3, trp1, ade2-1, can1-100, lys2-1, his5) and pYAC4 vector were provided by D. Burke (Washington University, St. Louis). Yeast cells were grown in YPD media as described by Sherman et al. (31).
Statistical Analyses.
Statistical analyses were performed using qgene (version 2.17) for the Macintosh (32). Mean fruit weight values for the NIL recombinants #3, #11, #12, #31, #33, and #34 were contrasted to the controls, NIL 939–2 and M82-1-8, at_P_ < 0.01.
RESULTS AND DISCUSSION
Molecular Maps of the fw2.2 Region of Chromosome 2.
To precisely determine the genetic distance between molecular markers in the region of fw2.2, a large NIL F2 population (3472 plants) derived from L. esculentum cv M82-1-8 × IL2-5 (L. pennellii introgression line; ref. 17) was screened for recombinants. The map derived from this population (Fig. 1A) was compared with the previously published tomato high-density linkage map of chromosome 2 (Fig. 1B; ref. 20). While the linear order of common markers (CD66, TG91, TG167, and TG361) was confirmed to remain constant between the maps, the genetic distances were quite different. For the interval spanning CD66 and TG361, the distance on the tomato high-density map (Fig. 1B) consisted of 4.2 ± 1.7 centimorgans (cM) in contrast to 0.8 ± 0.1 cM on the NIL high-resolution map (Fig.1A). However, this reduced recombination is consistent with the concept that when foreign DNA is progressively introgressed into NILs, the recombination rate between foreign and cultivated DNA decreases (5, 34).
Three additional molecular markers (TG686, TG687, and HSF24; Fig.1A) were placed on the high-resolution map of_fw2.2_. TG686 and TG687 were derived from a screen of 600 RAPDs based on polymorphisms detected between the NIL, 939–2, and the tomato control, M82-1-8. These two markers were subsequently screened on the 55 recombinants used for high-resolution mapping. The third molecular marker, HSF24, is a gene from tomato that encodes a heat stress transcription factor containing a region with a high degree of similarity to the DNA binding domain of a yeast heat shock factor (35). HSF24 was previously mapped to the fw2.2 region (20) and was further positioned between recombinants #33 and the cluster of recombinants including #34 (Fig. 1A).
Isolation of YACs and YAC-End Mapping.
Six YAC clones (Fig.1A), in the region of fw2.2, were obtained by PCR screening of a tomato YAC library (29) using primers designed for the molecular markers CD66, TG91, TG686, TG687, TG167, and TG361 (Table 1). A physical contig (Fig. 1A) was constructed for these six YACs based on the presence or absence of hybridization signals using the same molecular markers as probes onto filters containing _Eco_RI-digested YAC DNA with tomato DNA as control. _Eco_RI was used to construct the YAC library and therefore produced cleavage products that comigrated with tomato genomic DNA control digests. To more precisely align the YAC ends in the contig, iPCR YAC “right”- and “left”-end termini were isolated and used as probes onto the same Eco_RI digested YACs. All of the iPCR products and molecular markers, including RAPDs, in the fw2.2 region were found to be single copy based on RFLP analysis under moderate stringency conditions (0.5× SSC). This is unusual because only 44% of random tomato genomic clones (n = 50) were found to be single copy under similar stringency conditions (1.0× SSC; ref. 36). It is possible that the_fw2.2 region of chromosome 2, which is located in euchromatin, is enriched for single copy sequences including genes. In addition, the YAC ends were mapped genetically by RFLP analysis using the 55 recombinants in the region of fw2.2. The physical and genetic alignments of the six YAC ends were in complete agreement. None of the molecular markers internal to the YAC sequences indicated any rearrangements after RFLP analysis, even though YACs have been shown to be prone to chimerisms and rearrangements (37–39). The lack of YAC rearrangements may be due to the low frequency of repetitive elements within the fw2.2 region of the genome.
Placement of fw2.2 on the High-Resolution Molecular Map.
fw2.2 was previously mapped to a distal region on the long arm of chromosome 2 near the RFLP markers TG91 and TG167 in the wild tomato species, L. pennellii and Lycopersicon pimpinellifolium (refs. 16 and 40; Fig. 1B). To place fw2.2 more precisely on the high-resolution molecular map (Fig. 1A), individual recombinants in the region spanning CD66 and TG361 were subjected to fruit weight analysis by averaging the weight of 10 fruit from each of five plant replications (Fig. 2). Fruit weight analysis of two recombinants, #31 and #33, places fw2.2 between the molecular markers TG91 and HSF24 and included between the “left”-end of YAC355 and the “right”-end of YAC264 (Figs.1A and 2). This is based on the significant (P < 0.01) difference between the average fruit weights for recombinant #31 (NY, 63.0 g) in comparison to the small-fruited NIL control, 939–2 (NY, 49.6 g), and for recombinant #33 (NY, 50.5 g) in comparison to the large-fruited tomato control, M82-1-8 (NY, 71.9 g). Conversely, there was no significant difference (P_ > 0.01) between the fruit weights of the recombinants #31 and #33 in comparison to the large-fruited tomato control, M82-1-8, and the small-fruited NIL control, 939–2, respectively. The additional recombinants including #3, #11, #12, and #34 were consistent with the placement of_fw2.2 based on their average fruit weights as compared with the large- and small-fruited tomato controls (Fig. 2).
Figure 2.
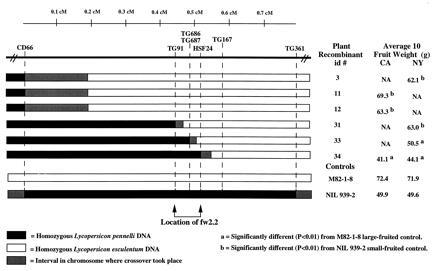
Graphical genotypes of homozygous recombinants in the fw2.2 region of chromosome 2. Five replications of each recombinant plant were grown in California (CA) and New York (NY). The average gram (g) weight of 10 fruit from each recombinant was compared with the large-fruited, M82-1-8, and the small-fruited, NIL 939–2, controls. Recombinants #3, #11, #12, and #31 were significantly larger (b; P < 0.01) for average 10 fruit weight in comparison to the small-fruited control, NIL 939–2, while recombinants #33 and #34 were significantly smaller (a;P < 0.01) for average 10 fruit weight in comparison to the large-fruited control, M82-1-8. Recombinants #31 and #33 delineate the fw2.2 region (bracketed by arrows), based on the smallest region demonstrating statistical significance. Plants for which few or no fruit were harvested due to pest infection were not available (NA) for fruit weight analysis. The black and white boxes indicate the homozygous condition for L. pennellii (NIL 939–2) and L. esculentum (M82-1-8) at the molecular markers, respectively. The gray boxes indicate the approximate position between two molecular markers where the genetic recombination event took place. The genetic distance between molecular markers (separated by dashed lines) is indicated by the scale shown in centimorgans (cM; Fig. 1).
YACs 264 and 355, which contain fw2.2, will be used to screen a fruit-specific cDNA library made from L. pennellii. cDNAs mapping within the region flanked by molecular markers TG91 and HSF24 will aid in the development of a cosmid contig of_fw2.2_ and will permit complementation analysis of candidate cosmid and cDNAs by plant transformation of the large-fruited recurrent parent, M82-1-8.
Relationship Between Physical and Genetic Distances Around the_fw2.2_ Locus.
PFGE blot analysis was used to determine the relationship between the physical and genetic distances for molecular markers anchored on the high-resolution map as well as to determine the size in kilobases of each YAC in the contig. High molecular weight tomato DNA from M82-1-8 was digested with the rare-cutting restriction enzymes _Mlu_I, _Nar_I,_Not_I, _Sal_I, and _Sma_I and probed sequentially with the molecular markers on the high-resolution map (Fig. 1A). TG91 and TG167 hybridized to the same_Mlu_I, _Nar_I, and _Sma_I restriction fragments with the smallest _Mlu_I fragment being ≤150 kb. This represents an upper size estimate since the markers may lie even closer than the restriction fragment indicates. The genetic distance between markers TG91 and TG167 was estimated in the F2 NIL population to be 0.13 ± 0.03 cM (Fig. 1A) so that on average, 1 cM is ≤ 1150 kb (≤150 kb/0.13 cM) for this interval. Based on an estimate of 750 kb/cM (20) for the entire tomato genome, the region spanning markers TG91 and TG167 appears to near the expected kb/cM ratio. Given that the ≤150-kb_Mlu_I restriction fragment is an upper size estimate for the physical distance, this kb/cM ratio may be even less. It therefore appears that fw2.2 is not in a region of suppressed recombination. Lending even more support to this hypothesis is the fact that the genetic distance between the surrounding loci, CD66 and TG91, is 0.45 ± 0.08 cM while the YAC325 spanning this region is 425 kb. Thus, for this interval, 1 cM is ≤950 kb, which is very close to the estimate for the interval TG91 to TG167.
Suppressed recombination can be a major factor limiting one’s ability to use genetic crossovers as landmarks to localize a gene to a small physical region. As an example in tomato, the centromeric region containing the gene _Tm_-2a, which confers resistance to tobacco mosaic virus, has a ratio of approximately 4 megabases/cM (27). This represents a recombination suppression rate of about 4-fold and is thought to be due, at least in part, to a general repression of crossing over around tomato centromeres (20). As a result, it has been extremely difficult to obtain recombinants in the region of _Tm_-2a, which is critical for mapping and thus determining a more precise location of the gene (41). In contrast, the fw2.2 region is located in euchromatin, far away from the centromere of chromosome 2. It appears that recombination suppression is not a major factor and thus should not be a limiting component in our attempt to clone the gene.
Implications for Map-Based Cloning of QTLs in Plants.
Like most traits of biological interest and agricultural importance, tomato fruit weight is a complex quantitative trait controlled by a number of genes (15). In the past, it was impractical to study the effects of individual loci responsible for quantitative traits. Recent advances in genome mapping have made it possible to map and determine the magnitude of the effect of individual loci controlling complex traits (42). However, due to environmental and epistatic influences, current procedures allow the placement of quantitative trait loci with only low resolution (>10 cM intervals of the genome) (42). Such large segments likely contain millions of base pairs of DNA and a multitude of genes, so it is unclear whether individual effects are due to a single gene or a set of linked genes. Low resolution mapping also makes it a daunting task to sort through large segments of DNA to clone the gene(s) responsible for the QTL effects.
Until recently, it was thought that the cloning of a quantitative trait would be impractical due to the lack of the ability to precisely map the gene(s) responsible for the QTL. In the current study, we demonstrate that the effect attributed to a QTL can be mapped down to a sub-centimorgan interval. This was accomplished by using molecular markers to select a subset of recombinant plants containing crossovers in the region of interest and then progeny testing these recombinants for the effect of the quantitative trait. To our knowledge, this marks the first time that a plant QTL has been mapped to such fine precision and isolated on a cloned segment of DNA. These results suggest that the QTL effect of fw2.2 is due to a single gene and provides a foundation toward eventually cloning this gene which will help unravel the basis of fruit development. This may eventually lead to the genetic engineering of novel fruit size and shape characteristics. It also generates hope that other genes responsible for QTL effects in plants can be pinpointed and isolated via a map-based cloning procedure.
Acknowledgments
We thank Drs. E. D. Earle, A. Frary, S. R. McCouch, J. C. Steffens, and J. Xiao for critical reading of the manuscript, and H. Ku for technical assistance. This work was supported in part by grants from the National Research Initiative Cooperative Grants Program, U.S. Department of Agriculture Plant Genome Program and by the Binational Agricultural Research and Development Fund. K.B.A. was supported in part by the Cornell National Science Foundation/Department of Energy/Department of Agriculture Plant Science Center.
Footnotes
Abbreviations: QTL, quantitative trait locus; PFGE, pulsed-field gel electrophoresis; YAC, yeast artificial chromosome; NIL, nearly-isogenic line; cM, centimorgan; RFLP, restriction fragment length polymorphism; RAPD, random amplified polymorphic DNA.
References
- 1.Johanssen W. Elemente der exakten Erblichkeitsllehre. Jena, Germany: Fischer; 1909. [Google Scholar]
- 2.Nilsson-Ehle H. Lunds Univ Arsskr Avd 2. 1909;5:1–122. [Google Scholar]
- 3.East E M. Genetics. 1915;1:164–176. doi: 10.1093/genetics/1.2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paterson A H, Lander E S, Hewitt J D, Peterson S, Lincoln S E, Tanksley S D. Nature (London) 1988;335:721–726. doi: 10.1038/335721a0. [DOI] [PubMed] [Google Scholar]
- 5.Paterson A H, DeVerna J W, Lanini B, Tanksley S D. Genetics. 1990;124:735–742. doi: 10.1093/genetics/124.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paterson A H, Damon S, Hewitt J D, Zamir D, Rabinowitch H D, Lincoln S E, Lander E S, Tanksley S D. Genetics. 1991;127:181–197. doi: 10.1093/genetics/127.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doebley J, Stec A, Wendel J, Edwards M. Proc Natl Acad Sci USA. 1990;87:9888–9892. doi: 10.1073/pnas.87.24.9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doebley J, Stec A. Genetics. 1993;134:559–570. doi: 10.1093/genetics/134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doebley J, Stec A, Gustus C. Genetics. 1995;141:333–346. doi: 10.1093/genetics/141.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorweiler J, Stec A, Kermicle J, Doebley J. Science. 1993;262:233–235. doi: 10.1126/science.262.5131.233. [DOI] [PubMed] [Google Scholar]
- 11.de Vicente M C, Tanksley S D. Genetics. 1993;134:585–596. doi: 10.1093/genetics/134.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradshaw H D, Jr, Wilbert S M, Otto K G, Schemske D W. Nature (London) 1995;376:762–765. [Google Scholar]
- 13.MacArthur J W, Butler L. Genetics. 1938;23:253–268. doi: 10.1093/genetics/23.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powers L. Tech Bull US Dep Agric. 1955;1131:1–64. [Google Scholar]
- 15.Ibaria E A, Lambeth V N. J Am Soc Hortic Sci. 1968;94:498–500. [Google Scholar]
- 16.Alpert K B, Grandillo S, Tanksley S D. Theor Appl Genet. 1995;91:994–1000. doi: 10.1007/BF00223911. [DOI] [PubMed] [Google Scholar]
- 17.Eshed Y, Zamir D. Euphytica. 1994;79:175–179. [Google Scholar]
- 18.Fulton T M, Chunwongse J, Tanksley S D. Plant Mol Biol Rep. 1995;13:207–209. [Google Scholar]
- 19.Bernatzky R B, Tanksley S D. Genetics. 1986;112:887–898. doi: 10.1093/genetics/112.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanksley S D, Ganal M W, Prince J P, de Vicente M C, Bonierbale M W, Broun P, Fulton T M, Giovannoni J J, Grandillo S, Martin G B, Messeguer R, Miller J C, Miller L, Paterson A H, Pineda O, Roder M S, Wing R A, Wu W, Young N D. Genetics. 1992;132:1141–1160. doi: 10.1093/genetics/132.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 22.Martin G B, Williams J K G, Tanksley S D. Proc Natl Acad Sci USA. 1991;88:2336–2340. doi: 10.1073/pnas.88.6.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konieczny A, Ausubel F M. Plant J. 1993;4:403–410. doi: 10.1046/j.1365-313x.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- 24.Chunwongse, J. (1995) Ph.D. dissertation (Cornell Univ., Ithaca, NY).
- 25.Liu Y G, Whittier R F. Nucleic Acids Res. 1994;22:2168–2169. doi: 10.1093/nar/22.11.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Short Protocols in Molecular Bology. 3rd Ed. New York: Wiley; 1995. [Google Scholar]
- 27.Ganal M W, Tanksley S D. Plant Mol Biol Rep. 1989;7:17–27. [Google Scholar]
- 28.Pillen K, Alpert K B, Giovannoni J J, Ganal M W, Tanksley S D. Plant Mol Biol Rep. 1996;14:58–67. [Google Scholar]
- 29.Martin G B, Ganal M W, Tanksley S D. Mol Gen Genet. 1992;233:25–32. doi: 10.1007/BF00587557. [DOI] [PubMed] [Google Scholar]
- 30.Giovannoni J J, Noensie E N, Ruezinsky D M, Lu X, Tracy S L, Ganal M W, Martin G B, Pillen K, Alpert K, Tanksley S D. Mol Gen Genet. 1995;248:195–206. doi: 10.1007/BF02190801. [DOI] [PubMed] [Google Scholar]
- 31.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 32.Nelson, C. (1994) Ph.D. dissertation (Cornell Univ., Ithaca, NY).
- 33.Kosambi D D. Ann Eugen. 1944;12:172–175. [Google Scholar]
- 34.Rick C M. Genetics. 1969;62:753–768. doi: 10.1093/genetics/62.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scharf K S, Rose S, Zott W, Schoffl F, Nover L. EMBO J. 1990;9:4495–4501. doi: 10.1002/j.1460-2075.1990.tb07900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamir D, Tanksley S D. Mol Gen Genet. 1988;213:254–261. [Google Scholar]
- 37.Selleri L, Eubanks J H, Giovannoni M, Hermanson G G, Romo A, Djabali M, Maurer S, Mcelligott D L, Smith M W, Evans G A. Genomics. 1992;14:536–541. doi: 10.1016/s0888-7543(05)80263-0. [DOI] [PubMed] [Google Scholar]
- 38.Haldi M, Perrot V, Saumier M, Desai T, Cohen D, Cherif D, Ward D. Genomics. 1994;24:478–484. doi: 10.1006/geno.1994.1656. [DOI] [PubMed] [Google Scholar]
- 39.Chumakov, I. M., Rigault, P., Le Gall, I., Bellanne-Chantelot, C., Billault, A., et al. (1995)Nature (London) 377, Suppl. 6547, 175–297. [DOI] [PubMed]
- 40.Eshed Y, Zamir D. Genetics. 1995;141:1147–1162. doi: 10.1093/genetics/141.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pillen K, Ganal M W, Tanksley S D. Theor Appl Genet. 1996;93:228–233. doi: 10.1007/BF00225750. [DOI] [PubMed] [Google Scholar]
- 42.Tanksley S D. Annu Rev Genet. 1993;27:205–233. doi: 10.1146/annurev.ge.27.120193.001225. [DOI] [PubMed] [Google Scholar]