Estimating Odds Ratios in Genome Scans: An Approximate Conditional Likelihood Approach (original) (raw)
Abstract
In modern whole-genome scans, the use of stringent thresholds to control the genome-wide testing error distorts the estimation process, producing estimated effect sizes that may be on average far greater in magnitude than the true effect sizes. We introduce a method, based on the estimate of genetic effect and its standard error as reported by standard statistical software, to correct for this bias in case-control association studies. Our approach is widely applicable, is far easier to implement than competing approaches, and may often be applied to published studies without access to the original data. We evaluate the performance of our approach via extensive simulations for a range of genetic models, minor allele frequencies, and genetic effect sizes. Compared to the naive estimation procedure, our approach reduces the bias and the mean squared error, especially for modest effect sizes. We also develop a principled method to construct confidence intervals for the genetic effect that acknowledges the conditioning on statistical significance. Our approach is described in the specific context of odds ratios and logistic modeling but is more widely applicable. Application to recently published data sets demonstrates the relevance of our approach to modern genome scans.
Introduction
In genetic studies, it is widely recognized that the control of genome-wide error requires the use of stringent thresholds for significance testing. For genome-wide linkage scans, standard LOD significance thresholds in the range 3.0 to 4.0 correspond to point-wise p values in the range 10−4 to 10−5, depending on the model and study design.1 For modern genome-wide association scans (GWASs), 100,000 to 1 million SNP markers may be genotyped, and control of family-wise error or false discovery rates typically requires point-wise significance thresholds in the range 10−7 to 10−8.2–4 The use of such stringent thresholds is offset somewhat by the belief that GWAS offer greater power than linkage studies for detecting complex disease genes.5 Nonetheless, the application of stringent thresholds distorts the inferential process, producing estimates of disease risk effect sizes that may be, on average, far greater in magnitude than the true effect.1,2,6–16 This phenomenon has been described as a form of “winner's curse” by Zöllner and Pritchard16 and others, or as a form of regression to the mean,15 and has profound importance for genome scans. Although the problem has been described as primarily an issue of bias, we demonstrate below that the variance of risk estimates can also be greatly inflated by the selection procedure. Moreover, standard confidence intervals for risk estimates will have very poor coverage properties, although this issue seems to have received less attention.
Consider a genome association scan for a complex disease in which ten genomic regions contain disease genes, and each region has a 20% chance of meeting genome-wide significance. Assuming independence of regions, the genome scan has respectable power 1 − (1 − 0.2)10 = 0.89 to achieve significance in at least one region. However, a repeated genome scan of equal size will have power of only 0.2 for any one region and thus probably not result in “replication” of the first study. A follow-up study might focus on a single significant region, with fewer markers and paying a lower penalty for multiple comparisons. But if the results of the initial genome scan are used as a guide, the follow-up study is likely to be underpowered, by relying on an inflated estimate of locus disease risk.
As a statistical phenomenon, the winner's curse should not be confused with additional sources of bias, including variations due to genotyping technologies, or heterogeneity of patient populations from which samples are drawn.12,17,18 The winner's curse is investigated in detailed simulations elsewhere,8,9,13–16 including a recent paper by Garner,8 who clarified that the bias can be understood predominantly through the behavior of Wald statistics for log-odds ratios.
Although the bias is simple to understand and to document, reducing or eliminating it may be nontrivial. Zöllner and Pritchard16 have described a likelihood approach that requires maximization over numerous parameters, including genotype frequencies and penetrance parameters, along with conditioning on declared statistical significance. Their procedure reduces the bias in risk estimation but cannot be performed with standard statistical software. Yu et al.15 have recently applied bootstrapping to correct for significance bias. Both of these bias correction approaches are technically feasible for genome scans, but they would be highly computationally intensive in that setting.
We describe our alternative approach for estimating genetic effects in terms of odds ratios, which have numerous advantages that have made them standard for analysis of case-control designs.19 A crucial advantage for case-control studies is that the odds ratio (OR) may be estimated consistently, whether the study design is prospective or retrospective,20 and the OR has an interpretation distinct from nuisance parameters such as genotype frequencies. Moreover, in logistic models, the OR retains interpretability in the presence of covariates, and such a retention is increasingly important for complex disease investigations.
In this paper, we introduce a method to correct for significance bias in disease association studies, by using an approximate conditional likelihood. The approach is directly based on the log(OR) estimate and its standard error as reported by standard statistical software and applies to dominant, recessive, or additive genetic models. No modification is necessary when covariates such as population-stratification variables have also been fit in the model. The approach may even be applied to published results without access to the original data. In addition, we develop a method to construct accurate confidence intervals for the OR.
We illustrate the performance of our approach via extensive simulations of a disease SNP analyzed by logistic regression. The simulations cover a range of models, disease allele frequencies, and OR values. Compared to naive OR estimation, our approach provides greatly reduced bias and mean squared error, particularly for the modest effect sizes likely to be encountered in complex diseases. In addition, our confidence-interval procedure provides coverage that is accurate or slightly conservative. Performing simulations for OR values near the null presents a challenge because significant results are very rare when applying genome-wide thresholds. We thus employ a screening approach in which a deterministic trend statistic is used to identify data sets potentially significant in logistic regression.
Material and Methods
We assume a genetic model with one parameter for the effect of disease genotype, which includes recessive, dominant, and additive models. We use β=log(OR) to denote the true loge odds ratio for disease risk conferred by a referent genotype, or for the contribution of each allele in an additive model. A single locus test statistic for disease association can be expressed as an estimate for β divided by an estimate for its standard error,
which is compared to the asymptotic null distribution N(0,1). We will refer to β^ and SE^(β^) as naive estimators because they are obtained from standard statistical procedures without acknowledging selection based on significance. For our problem, we wish to estimate β only when the SNP is significant in two-sided testing, i.e., |z|>c for a value c corresponding to genome-wide significance. By explicitly considering this selection, below we obtain three new estimators and a confidence interval procedure. Our approach offers marked improvements over β^ and standard confidence intervals. Our exposition includes mathematical and motivational details that we believe will considerably demystify the problem, which has until now appeared more obscure and complex than necessary. The performance of our new estimators is described in subsection Simulations.
Significance Bias
When logistic regression is used to test for genetic association, the Wald statistic for genetic effect assumes the specific form of (1), with numerator and denominator obtained from maximum likelihood and the information matrix.20–22 However, the essence of our approach applies to a wide variety of testing procedures, for which the key requirements typically hold: (1) asymptotically normality of β^ and (2) consistency of the standard error estimate, so that SE^(β^)/SE(β^)→1. Expressing the test statistic in the form of Equation (1) provides a straightforward illustration of significance bias and points the way toward corrected estimation procedures. Related test statistics that are based on maximum likelihood ratios or efficient scores, or that are directly based on contingency tables, are all asymptotically equivalent to (1) for local departures from the null hypothesis H0:β=0,23 although this asymptotic equivalence is not necessary to apply our approach. The remainder of this subsection is similar to Garner,8 but our explicit and expanded treatment provides the grounds for later development.
For large samples, SE^(β^) does not vary markedly in repeated data realizations. Thus, the estimate β^ and its statistical significance are highly correlated,8 and the problem can be restated as single-parameter estimation for a truncated normal distribution. To see this, we define μ=β/SE^(β^), with Z∼˙N(μ,1). Our use of this approximation follows from the standard result Z−μ=(β^−β)/SE^(β^)→DN(0,1) for increasing sample size.24 The statistical procedures to follow are developed entirely within this “μ version” of the problem, which has been greatly simplified by the variance standardization.
Our naive estimate of μ is μ^=z, and the expectation can be shown analytically to be
| Eμ(Z||Z|>c)=μ+ϕ(c−μ)−ϕ(c+μ)Φ(−c+μ)+Φ(−c−μ), | (2) | | ---------------------------------------------- | --- |
where ϕ and Φ are the density and cumulative distribution function of a standard normal, respectively (see Appendix A). This is the two-sided rejection version of a result given by Garner.8 As we detail in the Results, the bias can be substantial in realistic settings. In the special case of the null hypothesis μ=0, it is clear from Equation (2) that the naive estimate z is unbiased because the two-sided testing procedure is equally likely to falsely declare positive or negative risk (i.e., a protective effect of the referent genotype). It is not clear that the lack of bias for naive estimation under the null has been fully appreciated (e.g., Figure 2 in Zöllner and Pritchard16 does not display the exact null value). However, this lack of bias requires averaging over rejections for both positive and negative z. In any significant data set, μ^ must be less than −c or greater than c and so will be far from the truth under the null. In other words, the lack of bias under the null is offset by very large variance.
An Approximate Conditional Likelihood
The approximating distribution of Z suggests a correspondingly approximate likelihood for μ,
The likelihood applies generally to a wide variety of testing procedures, eliminating any nuisance parameters that have been included in the modeling, including stratification variables, clinical covariates, or the effects of other SNP genotypes. It is easy to show that the maximum-likelihood estimate (MLE) is μ^=z. A standard approach to likelihood testing for H0:μ=0(Ref. 24) involves comparing the maximum log-likelihood ratio LLR=2log(L(μ^)/L(0)) to a χ12 density. It is also simple to show that here LLR=z2, so in terms of both estimation and testing, the likelihood simply recapitulates the initial Equation (1). The advantage to Equation (3), however, is that it provides a simple and transparent approach to handle the conditioning. Acknowledging the event that the SNP is declared statistically significant, we have the conditional likelihood
| Lc(μ)=pμ(z||Z|>c)=pμ(z)Pμ(|Z|>c)=ϕ(z−μ)Φ(−c+μ)+Φ(−c−μ). | (4) | | ------------------------------------------------------------- | --- |
Under Equation (4), the relationship between numerator and denominator is such that, for a given z, it is quite possible that the most likely value for μ is in the interval [−c, _c_], even though z itself is conditioned to be outside that range.
By using this conditional approximate likelihood, we now derive improved estimators of μ. For any proposed value of μ, we can convert back to the desired log-odds ratio by using β=μSE^(β^), where SE^(β^) is obtained from standard approaches (i.e., does not consider the significance selection). One remarkable feature of our approach is that we can apply it to published summary results. To do so, we require only the significance threshold c, β^, and SE^(β^). The standard error, if not provided directly, can be inferred from c, β^, and any one of the following: z, the p value, or an unconditional OR confidence interval.
The Conditional MLE
With the conditional likelihood, the maximum likelihood principle suggests the MLE estimator,
which can be obtained with numerical maximization for any z and c (hereafter “∼” will signify estimates based on the conditional likelihood). Note that in this setting, the conditional maximum-likelihood estimate provides no guarantee of unbiasedness or efficiency, a fact that does not appear to have been considered by other investigators. We have already applied large-sample assumptions in constructing the conditional likelihood (4), but as we show below, other estimators can provide reduced bias or mean-squared error for certain ranges of μ, and therefore β.
Motivated by bias reduction, one might attempt to directly correct the bias in μ^ by solving for μ in the equation Eμ(Z||Z|>c)=z. Such an estimator has intuitive appeal, representing the value of μ for which, after conditioning on significance, we would have expected to observe z. Perhaps surprisingly, this “bias-correction” estimator in fact turns out to be μ˜1. To see this, we take the derivative of the conditional likelihood with respect to μ, for which the identity Lc′(μ˜1)=0 implies
| z=μ˜1+ϕ(c−μ˜1)−ϕ(c+μ˜1)Φ(−c+μ˜1)+Φ(−c−μ˜1). | (5) |
|---|
Comparing Equation (2) to (5) implies that the bias-correction estimator and μ˜1 are the same. Similar estimators have been examined in the context of sequential clinical trials, in which effect parameters are estimated only after a stopping boundary has been reached.25 Despite its secondary motivation as a bias-correction estimator, the conditional MLE μ˜1 is not in fact unbiased because of nonlinearity in the bias of the naive estimator μ^. Moreover, in this setting the conditional MLE has no special optimality properties, and other estimators may be reasonable. Nonetheless, we will show that μ˜1 is markedly improved over the naive estimator, both in terms of bias and mean squared error.
The Mean of the Normalized Conditional Likelihood
The motivation to reduce mean squared error (MSE) suggests another, perhaps less obvious estimator,
| μ˜2=∫−∞∞μLc(μ)dμ/∫−∞∞Lc(μ)dμ, | (6) |
|---|
which is easily calculated numerically. μ˜2 is the mean of the random variable following the distribution Lc(μ), normalized to be a proper density. μ˜2 has favorable MSE properties when averaged across a wide range of μ. This fact follows from an interpretation of μ˜2 as a posterior mean in a Bayesian treatment of the problem with a flat prior on μ.26 However, μ˜2 is considered here as an entirely frequentist estimate, with bias and error examined at each value of μ and judged accordingly. For |z| near the boundary c, μ˜2 typically represents a less aggressive shrinkage toward 0 compared to μ˜1.
A Compromise Estimator
In the treatment below, we will see that the conditional likelihood is typically skewed, and so μ˜1 and μ˜2 can differ appreciably for certain values of z. μ˜2 can show greater MSE than μ˜1 for μ near zero but is more favorable for μ away from zero. Thus, as a practical compromise, we also examine the estimator
which balances the strengths of μ˜1 versus μ˜2.
Illustrations of the Conditional Likelihood
Figure 1 illustrates the conditional and unconditional likelihoods assuming an illustrative constant threshold c = 5.0. Figures 1A–1C correspond to z = 5.2, 5.33, and 6.0, respectively. For each panel, the unconditional likelihood is centered and maximized at z (indicated by a dot on each plot). For Figure 1A, in which z is only slightly above the threshold, the conditional likelihood is in contrast shifted aggressively toward zero (μ˜1=0.66,μ˜2=2.53, and μ˜3=1.60). When z is well above the threshold (z = 6.0; Figure 1C), this shift is much smaller (μ˜1=5.48,μ˜2=4.94, and μ˜3=5.21). For an intermediate z (Figure 1B), the shift is intermediate. Note that our estimates are obtained here for the μ version of the problem, and the conversion β=μSE^(β^) must be performed before the results are interpreted on the log-odds scale.
Figure 1.
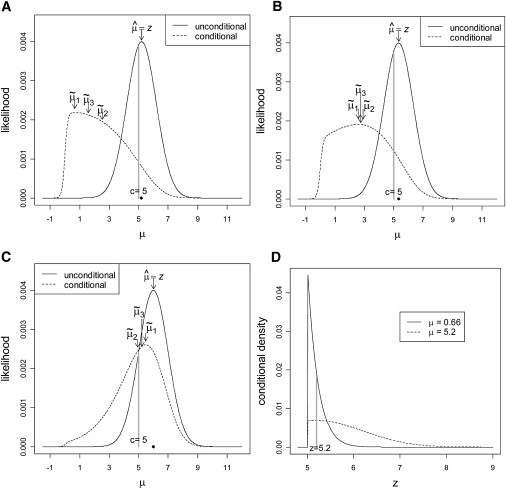
Behavior of the Unconditional and Conditional Likelihoods for μ
Unconditional and conditional likelihoods of μ are presented for (A) z = 5.2, (B) z = 5.33, and (C) z = 6. The location of the observed z is indicated by a black dot on each plot. The conditional likelihood changes considerably for small changes in z near c. For larger z, the conditional likelihood approaches the unconditional likelihood. Likelihoods for μ<−c are negligible and not shown. (D) shows conditional densities of z for μ = 0.66 and μ=5.2, with the relative likelihoods highlighted for a fixed value z = 5.2.
As desired, the conditional likelihood shows a clear shift toward zero. But why is the shift so extreme, e.g., when z = 5.2? Such a z value (which is equivalent to μ^) has already met genome-wide multiple-testing correction for statistical significance, but a shrinkage from μ^=5.2 to μ˜1=0.66 (for example) will effect a corresponding proportional reduction in the log-odds ratio. Thus, it seems our proposed estimation procedures can often adjust the estimated effect size to be practically insignificant. To see why the result is reasonable, consider that the conditional likelihood, as a frequentist construction, makes no judgment about the prior plausibility of various values of μ. When presented with a value z for each μ, it considers only the chance that z would have arisen, given that |z|>c. Figure 1D presents the (truncated normal) conditional densities for z under μ = 0.66 and μ = 5.2. These μ values were chosen because they represent the conditional and unconditional MLEs when z = 5.2. Note that these curves are conditional densities for z, not likelihoods. However, for a fixed value of z, the relative heights of the two curves reflect the conditional likelihoods for the two competing values of μ. From the curves, we can see the value z = 5.2 is 2.77× more likely to arise when μ = 0.66 than when μ = 5.2. Expressed in another way, when μ values are truly of large magnitude, then z tends to overshoot the threshold c by a greater amount than was observed here for z = 5.2. Thus, in this instance, we would conclude that μ is not likely to be of large magnitude.
Our three proposed estimators can be easily computed numerically, and simple R and Excel programs to do so are available at our website. By using the threshold c = 5.0 for illustration, we have calculated the conditional expectations and MSEs for the three estimators, shown in Figures 2A and 2B. The three corrected estimators provide dramatically reduced bias compared to the naive estimator for much of the range of μ. For μ=0, by symmetry all estimators are unbiased. For |μ| considerably larger than c, all methods will give estimators near z and will be nearly unbiased. The corrected estimators tend to undercorrect for small μ and overcorrect for large μ. The conditional MLE μ˜1 can be viewed as a first-order attempt to correct the bias, whereas z occupies the same range whether μ is small or large. In a sense, the corrected estimate splits the difference between the two extremes, leading to the observed pattern.
Figure 2.
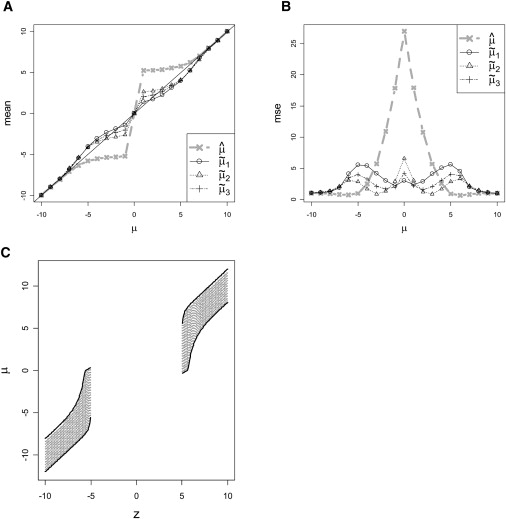
Estimators and Confidence Intervals for μ with Significance Threshold c= 5
(A) The expectation of naive estimator μ^ shows substantial bias and (B) very large mean squared error for much of the range of μ, whereas the corrected estimators have lower bias and MSE (C) shows upper and lower confidence bounds for μ as a function of the observed statistic z.
The MSE for μ^=z is extremely large for μ near zero, as predicted. MSEs for the corrected estimators are considerably smaller in the range of small to moderate μ. As described above, these estimators are easily converted to the corresponding improved log(OR) estimators β˜1, β˜2, and β˜3. Moreover, for large samples, the bias and MSE properties for μ will predominantly carry over to real data, essentially with a rescaling of the axes to convert μ to β.
Conditional Confidence Intervals
Proper interpretation of the corrected μ estimates requires an understanding of estimation error, conditioned on statistical significance. Standard confidence interval (CI) procedures fail in this setting. For example, after conditioning on significance, a standard 95% CI for μ cannot contain 0, for otherwise it would not have been significant. Thus, when μ=0 the standard CI procedure has zero conditional coverage probability. Zöllner and Pritchard16 addressed this issue by using a standard maximum likelihood ratio approach applied to the conditional likelihood. In our setting, a 1−η CI created in this manner would consist of all μ values such that 2log(Lc(μ˜1)/Lc(μ))≤q1−η, where q1−η is the 1−η quantile of a χ12 density. However, we have shown via numerical integration that in the μ version of the problem, the true coverage probability of this CI procedure can exhibit markedly conservative or anticonservative departures from 1−η, depending on the true μ. Approaches that use the second derivative at lnLc(μ˜1) to estimate the error variance also fail. The difficulty arises because the conditional MLE is not normally distributed nor is the shape of Lc(μ) approximately normal for a realized data set.
To create confidence intervals with correct conditional coverage, we return to the original Neymanian concept of a confidence region,23,27 a concept that can always be applied when the distribution of a test statistic is known for each value of the unknown parameter. Let A(μ,1−η) be an acceptance region depending on μ such that Pμ(Z∈A(μ,1−η)||Z|>c)=1−η. Given an observed z, the confidence region consists of all values μ such that z∈A(μ,1−η). It is straightforward to show that this approach gives exact coverage probability 1−η for any μ. Among possible acceptance regions, we choose A(μ,1−η) as the interval between the η/2 and 1−η/2 quantiles of the conditional density pμ(z||Z|>c). Note that, although we have presented three competing point estimates for μ, our procedure yields only a single CI. Figure 2C shows the upper and lower confidence limits for our CI procedure for each z. Note that for |z| near c, the confidence interval can contain μ=0. This does not contradict the statistical significance—the intent of the procedure is to obtain correct coverage for any μ (including μ=0) after conditioning on significance. The conversion of the confidence limits to the β scale is (μlowerSE^(β^),μupperSE^(β^)). Although our procedure is guaranteed correct conditional coverage in the idealized μ setting, our CI for β relies on large-sample normality assumptions for β^. Thus, we investigate empirical coverage of our procedure in the Results.
Simulations
To describe our simulations, we begin with basic notation for disease association studies. We let y denote the disease status (0 = control, 1 = case) for an individual and x denote the SNP genotype predictor value. For a biallelic SNP with major allele A and minor allele a, x is defined as follows for genetic models with respect to a:
recessiveadditivedominantx={0,AA0,Aa1,aax={0,AA1,Aa2,aax={0,AA1,Aa1,aa.
We assume the logistic model for a randomly sampled individual in the population
log(P(Y=1|x)/(1−P(Y=1|x)))=α+βx,
for some α, and β is the log-odds ratio for a unit increase in x. Rather than specify α directly, it is more interpretable to solve for α for a specified allele frequency and disease prevalence π. The marginal frequency of x is denoted p(x) and is easily calculated from Hardy-Weinberg assumptions. With fixed disease prevalence, the identity π=∑xexp(α+βx)1+exp(α+βx)p(x) was used for calculating α. Finally, solving for the genotype probabilities conditioned on case-control status yields
P(X=x|Y=1)=p(x)πexp(α+βx)1+exp(α+βx),P(X=x|Y=0)=p(x)1−π11+exp(α+βx).
A standard result is that logistic modeling for β applies even when the data are sampled retrospectively.20
Each data set was simulated and analyzed in R v.2.5.1. We will denote the total sample size n=ncases+ncontrols and ncases=ncontrols throughout. Most simulations consisted of n=1000. This sample size is relatively small for a genome scan and was intentionally chosen to emphasize any departures from normality or difficulties in estimating SE(β^). We also examined larger sample sizes for several of the setups to examine the effect of sample size on bias, MSE, and confidence coverage. We assumed a disease prevalence of 0.01 throughout—the retrospective sampling is not very sensitive to this specification. We examined β ranging from −0.7 (OR≈0.5) to 0.7 (OR≈2.0). This range corresponds to biological plausibility for complex disease11 and ensures that simulations span the range from low power to high power. For simplicity, we used c = 5.0, corresponding to a single p value of 5.7 × 10−7, near the genome-wide threshold considered by others.2–4
For recessive models, we considered MAF values of 0.25 and 0.5—lower values created small expected cell counts that were problematic for sample sizes of 500 in each group. For the additive and dominant models, we considered minor allele frequency (MAF) values of 0.05, 0.1, 0.25, and 0.5. A single setup consisted of the genetic model, MAF, and β, and sufficient simulations were performed for each setup so that 1000 significant data sets were obtained. Setups with β=0 required on the order of 109 to 1011 simulations for this rarified threshold. We sped up the analysis by first applying a chi-square test (Cochran-Armitage trend test for the additive model) to the data sets, which can be obtained without iterative maximization. The chi-square statistic was determined to have a close correspondence to z2 obtained from the more computationally intensive logistic regression, and a chi-square statistic ≥24 was determined to capture essentially all data sets with z2≥c2=25. Data sets meeting the chi-square criterion were analyzed via logistic regression in R glm. For data sets achieving final significance as determined by logistic regression, β^ and SE^(β^) were used for obtaining β˜1,β˜2,β˜3, and conditional confidence intervals.
Results
In all scenarios described here, expectations and mean-squared errors are calculated conditional on significance, i.e., |z|>c.
Bias
The top row of Figure 3 plots the means for each of the naive and corrected estimators versus β (with corresponding OR values) for all models, with MAF = 0.25. The naive estimator shows very large bias, especially for moderate β. All of the corrected estimators show dramatically reduced bias across most of the range examined. For each model, the corrected estimates tend to undercorrect for small (magnitude) β while overcorrecting for large β. All of the methods become nearly unbiased for large β, as they must, because the conditional and unconditional likelihoods are nearly identical when |z| is well beyond c. In terms of bias, β˜1 performs best among the corrected estimates for small β. However, the overcorrection of the conditional MLE can be substantial for moderate to large β, especially for the recessive model. β˜2 shrinks the estimates toward zero less dramatically, resulting in undercorrection for a larger part of the range of β. β˜3 strikes a balance between the other two corrected estimates and has much improved bias for moderate β under the recessive model. All estimators are effectively unbiased for β=0. A subtle asymmetry in the plots for positive and negative log(OR), most evident in the recessive model, occurs because MAF < 0.5 and, for a fixed prevalence, the logistic intercept α depends on β.
Figure 3.
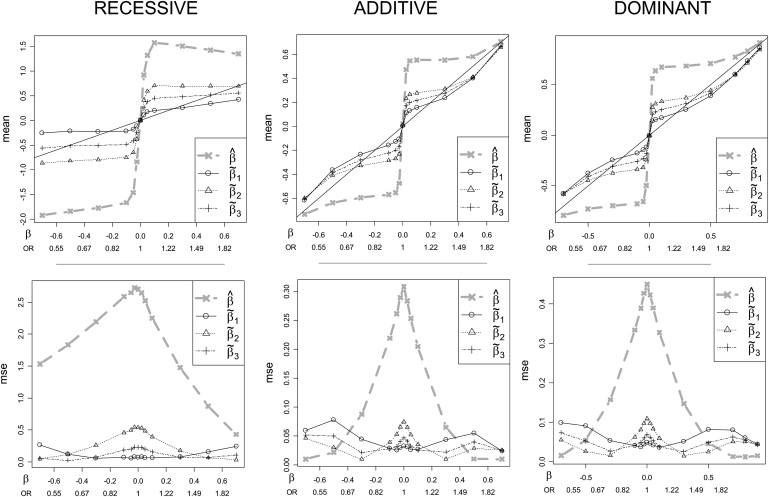
Expectations and Mean Squared Errors for the Three Genetic Models under MAF = 0.25
The corrected estimators show greatly improved performance for much of the range of β. The left panels correspond to the recessive model, the middle panels correspond to the additive model, and the right panels correspond to the dominant model. The top row shows expected values for the naive and conditional likelihood estimators versus β. The bottom row shows mean squared errors for the estimators. The y axes are rescaled to highlight details—the MSE is considerably larger for the recessive model because of scarcity of the risk homozygotes.
Mean Squared Error
The corresponding MSE values for the estimators are shown in the bottom row of Figure 3. The naive estimator β^ exhibits extremely large MSE for most β values examined. For β=0, this is due to high variance, whereas for moderate β, the naive estimator has low variance but high bias. The corrected estimators show dramatically improved MSE for β in the interval [−0.3, 0.3] (OR ranging from 0.74 to 1.35) that encompasses the bulk of significant associations thus far for complex diseases.3,4 The MSEs of β˜1 and β˜2 are predominantly complementary. At β=0, the MSE (β˜1) is fairly low, whereas MSE (β˜2) peaks. For larger magnitude β, the roles reverse. As expected, β˜3 exhibits a more even MSE across the range, and represents a reasonable choice for stable error characteristics. For the additive and dominant models, β^ exhibits very low MSE for large β. This phenomenon is not as attractive as it appears, essentially resulting from a boundary effect in which β^ is nearly constant because z is just barely significant. In particular, for β outside of the plotted range, MSE (β^) rises again to the var(β^) value encountered in the unconditional setting.
The empirical bias and MSE observed in our simulations essentially follow the results from the μ version of the estimation problem, with a rescaling of the axes to convert μ to β. Our empirical results for the remaining MAF values are plotted in Figure S1 available online and predominantly follow the results described for MAF = 0.25. Figure 4 shows a portion of these results for the additive model, in which the MSE is shown to drop for all estimators as the MAF increases. This occurs because for small MAF, the MSE is largely driven by the heterozygote genotype counts, which increase with the MAF. The key point of Figure 4 is that the relative advantages of the corrected estimators are preserved across a wide range of MAF values.
Figure 4.
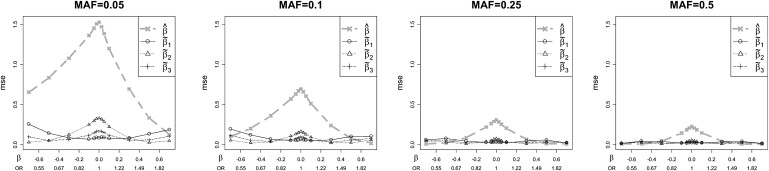
Mean Square Errors of the Estimators versus β for MAF Values Ranging from 0.05 to 0.5
The additive model is assumed, with n = 1000. The MSEs drop for larger MAF, but the relative performance of the estimators is maintained.
Confidence Coverage
Figure 5 presents the estimated coverage probabilities of 95% and 90% CIs with MAF = 0.25 for the three models. The top row shows the results for n = 1000. The coverage is close to the nominal level for almost all the setups, except for conservativeness near β=0 for the recessive model. The coverage of the naive confidence intervals is also depicted in the figure, dropping dramatically out of the axis range to zero coverage for β of small magnitude. For n = 2000, the coverage of the proposed procedure improves further, with a region of modest overcoverage for recessive models. Results for other MAF values are similar, and are presented in Figure S2.
Figure 5.
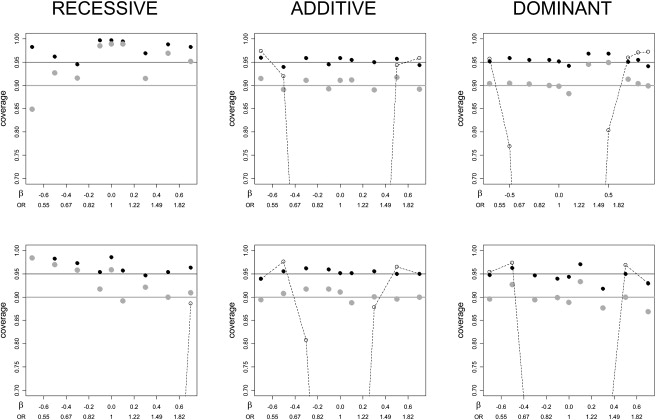
Estimates of the CI Coverage Probability Plotted against β for the Three Genetic Models, MAF = 0.25
Black dots correspond to 95% CIs; gray dots correspond to 90% CIs. The dashed curves represent coverage of standard 95% CIs that do not acknowledge the significance selection. The top row shows n = 1000 (500 cases and 500 controls). The bottom row shows n = 2000 (1000 cases and 1000 controls). Coverage is close to nominal, except for regions of overcoverage in the recessive model because of small cell counts (note that the y axis range begins at 0.7). For all models, the coverage will approach the nominal value as the sample size increases further.
Sample Sizes, Thresholds, and Covariates
Our setup conditions represent a wide range of realistic scenarios but cannot represent all situations and complicating factors. Fortunately, the large-sample behavior of the constructed approximate likelihood provides considerable robustness for our conclusions. Figure S3 shows the results of increasing sample size for several realistic β values for the additive model when MAF = 0.25. The bias and MSE for all the estimators are reduced as the sample size increases. For each sample size, the corrected estimators show superior bias and MSE compared to the naive estimator.
In maximum-likelihood settings, the distribution of the Wald test statistic is predominantly driven by β/SE(β^). This is also true for our conditional likelihood, because β/SE(β^) determines the noncentrality of the z statistic. For a fixed ratio ncases:ncontrols, the standard error is proportional to 1/n. Thus, for the setups in Figure 3 and Figure S1, a doubling of the sample size to n = 2000 (for example, and assuming cases and controls remain in the same ratio) would produce qualitatively similar results, with perhaps a slight improvement for the corrected estimates as the normality approximation improves. Moreover, we can make the results quantitatively comparable by appropriate rescaling. For example, for any value β for n = 1000, the comparable results for n = 2000 should correspond to β′=β2. Figure S4A demonstrates an empirical example of this effective rescaling equivalence for the additive model, MAF = 0.25. Thus, the conclusions from our simulations extend to larger sample sizes.
Similarly, variations on the threshold c do not have much impact. A value of c = 5.5 would be considered quite conservative for genome scans, corresponding to Bonferroni control of family-wise error at 0.05 for 1.3 million SNPs. Empirical investigation requires many more simulations to achieve significance, but we find that the qualitative behavior of the estimators is unchanged (Figure S4B).
Finally, we simulated an example in which the additive model is fit (MAF = 0.25), and the logistic regression includes an additional continuous covariate [distributed N(0,1), one fitted regression coefficient] and a discrete covariate [distributed Binomial(2,0.05), two fitted coefficients]. The covariates were independent of case-control status and the test-locus genotype. The Wald statistic is relatively insensitive to inclusion of these extra parameters, and the relative change in degrees of freedom quite minimal. Accordingly, the results for our corrected estimators are virtually unchanged compared to the model without covariates (Figure S4C—only β˜1 is shown). Covariate considerations are increasingly important in genome scans, for example to control for confounding population stratification.
Analyses of Published Data Sets
Table 1 illustrates our reanalysis of an association study with a modest number of SNPs, as well as two GWASs, all of which had been analyzed with additive models. We begin with a brief description of the three studies, followed by our reanalysis. Yu et al.15 examined the lymphoma results described in Wang et al.,18 with 48 SNPs and a p value threshold 0.1/48 ≈ 0.002. The standard OR results were compared to bootstrap bias-corrected15 results, as well as the results from a larger pooled analysis involving seven studies.28 The SNPs rs1800629 and rs909253 were found to be significant, with ORs 1.54 and 1.40, respectively. Todd et al.3 list four significant SNPs resulting from two type 1 diabetes (T1D [MIM 222100]) GWAS studies, highlighting for especial consideration those SNPs with p value less than 5 × 10−7. In addition, the authors conducted a larger case-control follow-up study to confirm these results. Scott et al.4 performed numerous analyses of several type 2 diabetes (T2D [MIM 125853]) data sets (FUSION, DGI, and WTCCC/UKT2D). We consider here only the SNPs reported by the T2D authors using the declared genome-wide significance threshold (p < 5 × 10−8) for the combined analysis of all studies.
Table 1.
Original versus Corrected Odds-Ratio Estimates for Three Published Genetic Association Studies
| Study | SNP | Minor Allele Frequency | p Value | Reported OR,a (95% CI) | Bootstrapb Estimates | Bias-Corrected Estimatesc | Biasc-Corrected (95% CI) | Follow-Upd OR, (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| OR˜1 | OR˜2 | OR˜3 | |||||||
| Association Study of Lymphoma, Wang et al.18 (318 Cases and 766 Controls) | |||||||||
| rs1800629 | 0.217 | 5.7 × 10−4 | 1.54 | 1.29 | 1.08 | 1.25 | 1.16 | (0.94,1.85) | 1.29 |
| rs909253 | 0.358 | 7.4 × 10−4 | 1.4 | 1.18 | 1.06 | 1.18 | 1.12 | (0.95,1.56) | 1.16 |
| GWAS of T1D, Todd et al.3 (2000 Cases and 3000 Controls) | |||||||||
| rs17696736 | 0.423 | 7.27 × 10−14 | 1.37 (1.27,1.49) | - | 1.37 | 1.36 | 1.37 | (1.25,1.49) | 1.16 (1.09,1.23) |
| rs2292239 | 0.34 | 1.49 × 10−9 | 1.3 (1.20,1.42) | - | 1.26 | 1.23 | 1.25 | (1.08,1.42) | 1.28 (1.20,1.36) |
| rs12708716 | 0.322 | 1.28 × 10−8 | 0.77 (0.70,0.84) | - | 0.82 | 0.84 | 0.83 | (0.71,1.00) | 0.83 (0.78,0.89) |
| rs2542151 | 0.163 | 8.4 × 10−8 | 1.33 (1.20,1.49) | - | 1.04 | 1.15 | 1.09 | (0.99,1.44) | 1.29 (1.19,1.40) |
| GWAS of T2D, Scott et al.4 (9521 Cases and 12183 Controls) | |||||||||
| rs7903146 | 0.18 | 1.0 × 10−48 | 1.37 (1.31,1.43) | - | 1.37 | 1.37 | 1.37 | (1.31,1.43) | |
| rs4402960 | 0.30 | 8.9 × 10−16 | 1.14 (1.11,1.18) | - | 1.14 | 1.14 | 1.14 | (1.10,1.18) | |
| rs10811661 | 0.85 | 7.8 × 10−15 | 1.2 (1.14,1.25) | - | 1.2 | 1.2 | 1.2 | (1.14,1.26) | |
| rs8050136 | 0.38 | 1.3 × 10−12 | 1.17 (1.12,1.22) | - | 1.17 | 1.16 | 1.16 | (1.10,1.22) | |
| rs5219 | 0.46 | 6.7 × 10−11 | 1.14 (1.10,1.19) | - | 1.13 | 1.11 | 1.12 | (1.05,1.19) | |
| rs7754840 | 0.36 | 4.1 × 10−11 | 1.12 (1.08,1.16) | - | 1.11 | 1.1 | 1.11 | (1.05,1.16) | |
| rs1111875 | 0.52 | 5.7 × 10−10 | 1.13 (1.09,1.17) | - | 1.1 | 1.09 | 1.1 | (1.00,1.17) |
With only the published odds ratios, p values, and stated significance thresholds, we produced bias-corrected odds ratios for all of these studies. Our corrected β estimates are exponentiated so that odds ratios were obtained: For example, OR˜1=exp(β˜1). For the two lymphoma SNPs (Table 1, top section), the p values are slightly above the threshold, and our bias-corrected estimates shrink the naive OR estimates markedly. Our estimated values match well with the bootstrap-corrected values obtained by Yu et al.,15 as well as the pooled analysis results from Rothman et al.28
For the four T1D SNPs (Table 1, middle section), our analysis results in noticeably less extreme OR estimates (Table 1, middle section) than those reported by Todd et al.3 The corrected ORs and CIs for the most extreme SNP, rs17696736, are only slightly changed from the published estimated of 1.37 because the result is so extreme (p = 7.27 × 10−14). However, the follow-up study obtained a considerably lower value (OR = 1.16), with the 95% CI not overlapping the earlier estimates, thereby suggesting possible heterogeneity in population sampling. For the two least significant T1D SNPs among those considered, the corrected ORs show a more substantial change. It is worth noting that the OR estimate corresponding to the SNP rs12708716 was shrunk from 0.77 to ∼0.82 by our methods, whereas the estimated OR from the follow-up study was 0.83. We also note that for the four significant T1D SNPs, as well as an additional three SNPs approaching significance (Table 1 of Todd et al.3), the follow-up study always gave a less extreme OR estimate than the initial studies. This result is strong empirical evidence for significance bias and showed that corrected OR approaches are needed.
The bottom portion of Table 1 gives the results for the combined T2D studies. All of the p values are considerably beyond the significance threshold, and so the corrected estimates are nearly unchanged from the original estimates. This phenomenon is hopeful, in the sense that with very large studies, OR estimates can be attained that will not be shrunk to irrelevance by corrected OR estimates.
Discussion
We have presented an approach that greatly reduces significance bias for odds ratios in genome association scans and that is much simpler than competing approaches. We favor the use of β˜3 as a general-purpose estimator with fairly uniform MSE as a function of β. However, all of the three corrected estimators have greatly superior performance compared to the naive estimator. Although developed for case-control applications, our methodology is an effective blueprint to perform inference whenever a Wald-like statistic has been used to declare significance. Thus, the general approach can be used in numerous other settings, including regression-based quantitative-trait association analyses. Our results are qualitatively similar to those of other investigators15,16 (e.g., see bias curves similar to ours in Figure 2 of Zöllner and Pritchard16). Additional comparisons to these approaches should be performed in future work, although comparison is complicated by differing genetic models. To our knowledge, our approach is the only method that can perform bias correction based only on published summary tables.
The widespread application of conditional likelihood estimators in genome scans will no doubt be discouraging to genetic investigators, who may expend considerable time and expense only to find that a significant SNP is estimated to have a very weak effect. Nonetheless, we view this process as healthy and necessary for the genetics community, in particular to tamp down expectations that significant findings will be easily replicated. The use of our estimators may also have an additional benefit of discouraging excessive massaging of data and trying various test procedures to achieve genome-wide significance. If a SNP suddenly becomes significant after numerous data manipulation procedures have been applied, its z statistic is likely to be only slightly above the threshold c. Thus, as we observed in the μ version of the problem, the conditional-likelihood estimator will be dramatically shrunk toward the null. Thus, the estimated SNP effect size will be very modest, as is appropriate here for a likely spurious finding.
Our current approach does not explicitly consider multistage or other sequential designs, in which SNPs meeting a loose standard of significance are used for further testing in a follow-up sample. However, for multistage designs in which almost all SNPs that will eventually be declared significant are carried forward to later stages, the approach may be used directly. Also, our results technically hold for a SNP randomly selected from those achieving the significance threshold, and thus an additional bias may be anticipated for the most highly significant SNPs among a collection of significant SNPs. Although we believe this second source of bias is much less than that produced by significance selection, it is the subject of continuing investigation.
Our rejection-sampling scheme was feasible, but it required a massive number of simulations to provide accurate results. Future work in this area may benefit from the practical development of importance sampling or related computational techniques to provide flexible and accurate simulations conditioned on significance.
Appendix A
Derivation of the Conditional Mean for the Naive Estimator
E(Z||Z|>c)=K−1[∫−∞−czϕ(z−μ)dz+∫c∞zϕ(z−μ)dz],whereK=Φ(−c+μ)+Φ(−c−μ)=μ+K−1[∫−∞−c−μxϕ(x)dx+∫c−μ∞xϕ(x)dx],x=z−μ=μ+K−1[(2π)−12∫∞12(c+μ)2e−ydy+(2π)−12∫12(c−μ)2∞e−ydy],y=12x2=μ+K−1[(2π)−12e−12(c−μ)2−(2π)−12e−12(c+μ)2]=μ+ϕ(c−μ)−ϕ(c+μ)K
Supplemental Data
Four figures are available at http://www.ajhg.org/.
Supplemental Data
Document S1. Four Figures
Web Resources
The URLs for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
- R code and a simple Excel calculator, www.bios.unc.edu/∼fwright/genomebias
Note Added in Proof
While this paper was in press, a paper by Zhong, H. and Prentice, R.L. proposed a similar methodology, with some differences in approaches to point and interval estimation: Zhong, H. and Prentice, R.L. (2008). Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics, in press.
Acknowledgments
This paper was supported in part by the Carolina Environmental Bioinformatics Center (EPA RD-83272001) and NIH grants R01 GM074175 and R01 HL068890. We thank the anonymous reviewers, who provided numerous suggestions that led to improvements in the manuscript.
References
- 1.Lander E., Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 2.Zondervan K.T., Cardon L.R. Designing candidate gene and genome-wide case-control association studies. Nat. Protocols. 2007;2:2492–2501. doi: 10.1038/nprot.2007.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 6.Allison D.B., Fernandez J.R., Heo M., Zhu S., Etzel C., Beasley T.M., Amos C.I. Bias in estimates of quantitative-trait-locus effect in genome scans: Demonstration of the phenomenon and a method-of-moments procedure for reducing bias. Am. J. Hum. Genet. 2002;70:575–585. doi: 10.1086/339273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chanock S.J., Manolio T., Boehnke M., Boerwinkle E., Hunter D.J., Thomas G., Hirschhorn J.N., Abecasis G., Altshuler D., NCI-NHGRI Working Group on Replication in Association Studies Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 8.Garner C. Upward bias in odds ratio estimates from genome-wide association studies. Genet. Epidemiol. 2007;31:288–295. doi: 10.1002/gepi.20209. [DOI] [PubMed] [Google Scholar]
- 9.Goring H.H., Terwilliger J.D., Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 2001;69:1357–1369. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirschhorn J.N., Lohmueller K., Byrne E., Hirschhorn K. A comprehensive review of genetic association studies. Genet. Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Ioannidis J.P., Ntzani E.E., Trikalinos T.A., Contopoulos-Ioannidis D.G. Replication validity of genetic association studies. Nat. Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 12.Lohmueller K.E., Pearce C.L., Pike M., Lander E.S., Hirschhorn J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 13.Siegmund D. Upward bias in estimation of genetic effects. Am. J. Hum. Genet. 2002;71:1183–1188. doi: 10.1086/343819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L., Bull S.B. Reduction of selection bias in genomewide studies by resampling. Genet. Epidemiol. 2005;28:352–367. doi: 10.1002/gepi.20068. [DOI] [PubMed] [Google Scholar]
- 15.Yu K., Chatterjee N., Wheeler W., Li Q., Wang S., Rothman N., Wacholder S. Flexible design for following up positive findings. Am. J. Hum. Genet. 2007;81:540–551. doi: 10.1086/520678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zöllner S., Pritchard J.K. Overcoming the winner's curse: Estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 2007;80:605–615. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balding D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 18.Wang W.Y., Barratt B.J., Clayton D.G., Todd J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 19.Aschengrau A., Seage G.R. Jones & Bartlett Publishers; Sudbury, MA: 2003. Essentials of Epidemiology in Public Health. [Google Scholar]
- 20.McCullagh P., Nelder J.A. Chapman & Hall/CRC; Boca Raton, FL: 1989. Generalized Linear Models. [Google Scholar]
- 21.Agresti A. Wiley-Interscience; Hoboken, NJ: 2002. Categorical Data Analysis. [Google Scholar]
- 22.Cox D.R., Snell E.J. Chapman & Hall/CRC; Boca Raton, FL: 1989. Analysis of Binary Data. [Google Scholar]
- 23.Rao C.R. Wiley-Interscience; Hoboken, NJ: 2002. Linear Statistical Inference and Its Application. [Google Scholar]
- 24.Wald A. Tests of statistical hypotheses concerning several parameters when the number of observations is large. Trans. Am. Math. Soc. 1943;54:426–482. [Google Scholar]
- 25.Liu A.Y., Troendle J.F., Yu K.F., Yuan V.W. Conditional maximum likelihood estimation following a group sequential test. Biometrical Journal. 2004;46:760–768. [Google Scholar]
- 26.Leonard T., Hsu J.S.J. Cambridge University Press; Cambridge: 2001. Bayesian Methods: An Analysis for Statisticians and Interdisciplinary Researchers. [Google Scholar]
- 27.Lehmann E.L., Casella G. Springer; New York: 1998. Theory of Point Estimation. [Google Scholar]
- 28.Rothman N., Skibola C.F., Wang S.S., Morgan G., Lan Q., Smith M.T., Spinelli J.J., Willett E., De Sanjose S., Cocco P. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: A report from the InterLymph Consortium. Lancet Oncol. 2006;7:27–38. doi: 10.1016/S1470-2045(05)70434-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Four Figures