Pro-inflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes (original) (raw)
. Author manuscript; available in PMC: 2010 May 1.
Published in final edited form as: Glia. 2009 May;57(7):734–743. doi: 10.1002/glia.20801
Abstract
HIV encephalitis (HIVE), the pathologic correlate of HIV-associated dementia (HAD) is characterized by astrogliosis, cytokine/chemokine dysregulation and neuronal degeneration. Increasing evidence suggests that inflammation is actively involved in the pathogenesis of HAD. In fact, the severity of HAD/HIVE correlates more closely with the presence of activated glial cells than with the presence and amount of HIV-infected cells in the brain. Astrocytes, the most numerous cell type within the brain, provide an important reservoir for the generation of inflammatory mediators, including interferon-γ inducible peptide-10 (CXCL10), a neurotoxin and a chemoattractant, implicated in the pathophysiology of HAD. Additionally, the pro-inflammatory cytokines, IFN-γ and TNF-α, are also markedly increased in CNS tissues during HIV-1 infection. In the present study we hypothesized that the interplay of host cytokines and HIV-1 could lead to enhanced expression of the toxic chemokine, CXCL10. Our findings demonstrate a synergistic induction of CXCL10 mRNA and protein in human astrocytes exposed to HIV-1 and the pro-inflammatory cytokines. Signaling molecules, including JAK, STATs, MAPK (via activation of Erk1/2, AKT, and p38), and NF-κB were identified as instrumental in the synergistic induction of CXCL10. Understanding the mechanisms involved in HIV-1 and cytokine mediated up-regulation of CXCL10 could aid in the development of therapeutic modalities for HAD.
Keywords: Astrocytes, HIV-associated dementia, CXCL10
Introduction
There are more than 40 million people infected with human immunodeficiency virus (HIV)-1 worldwide. Approximately 10-15% of HIV-1 infected individuals suffer from CNS pathologies including HIV-associated encephalitis (HIVE) and HIV-associated dementia (HAD), collectively termed NeuroAIDS (Kaul and Lipton 2006; McArthur et al. 1999; Navia et al. 1986). HIVE, the pathologic correlate of HAD is characterized by increased astrocytosis, microglial activation, enhanced expression of inflammatory mediators and neuronal dysfunction/death (Gonzalez-Scarano and Martin-Garcia 2005; Minagar et al. 2002; Navia et al. 1986). While the exact mechanism by which HIV-1 causes these neuropathologies is not completely understood, increasing evidence suggests neuronal damage results in part from microglial and astroglial mediated inflammation (Deshpande et al. 2005; Gonzalez-Scarano and Martin-Garcia 2005; Minagar et al. 2002). In fact, the severity of HIVE/HAD seems to correlate better with the presence of activated glial cells than with the presence and number of HIV-infected cells in the brain (Glass et al. 1995; Gonzalez-Scarano and Martin-Garcia 2005; Minagar et al. 2002).
Brain tissue derived from patients with HAD reveals increased expression of mRNA for the chemokine, CXCL10, which is both a neurotoxin and a chemoattractant (Sui et al. 2004; van Marle et al. 2004). Astrocytes, the most numerous cell type within the brain, provide an important reservoir for the generation of inflammatory mediators, including CXCL10 (Dong and Benveniste 2001; Minagar et al. 2002; Thompson et al. 2001). Additionally, the pro-inflammatory cytokines, IFN-γ and TNF-α, are markedly increased in CNS tissues during HIV-1 infection in the brain and are implicated in the pathophysiology of HAD (Shapshak et al. 2004; Wesselingh et al. 1997). While both the cellular (IFN-γ and TNF-α) (Majumder et al. 1998a; Majumder et al. 1998b; Ohmori and Hamilton 1993; Ohmori and Hamilton 1995) and viral factors (Tat and gp120) (Asensio et al. 2001; Kutsch et al. 2000) induce CXCL10, it remains unclear how the interplay of host factors and virus modulate chemokine expression.
Chemokines in the brain have been recognized as essential elements in neurodegenerative disease and related neuroinflammation through their regulation of inflammatory responses (Conant et al. 1998; Kelder et al. 1998; Luster and Ravetch 1987) thereby contributing to injury and eventual loss of neurons (Asensio and Campbell 1999; Miller and Meucci 1999). Cerebral expression of various chemokines, including CXCL10 (interferon γ-inducible peptide, or IP-10), and their receptors are increased in HIVE (Kolb et al. 1999; Kolson and Pomerantz 1996; McArthur et al. 1993; Sanders et al. 1998). Increased levels of CXCL10 have been detected in the CSF and plasma of individuals with HIV-1 infection (Kolb et al. 1999) and in the brains of individuals with HAD (Kolson and Pomerantz 1996; McArthur et al. 1993; Sanders et al. 1998). Importantly, CXCL10 levels in the CNS of HIV-1 infected individuals correlated positively with disease progression (28). There is also evidence that CXCL10 participates in the neuropathogenesis of SHIV-infected macaques (Sasseville et al. 1996; Westmoreland et al. 1998) by contributing to the degeneration of neurons possibly through activation of a calcium dependent apoptotic pathway (Sui et al. 2004; Sui et al. 2006). Increased CXCL10 levels were critical for the increased migration of inflammatory cells into the CNS, a hallmark feature of HAD (Nath 1999; Navia et al. 1986).
Additionally, the pro-inflammatory cytokines, IFN-γ and TNF-α, are also markedly increased in CNS tissues during HIV-1 infection and have been implicated in the pathophysiology of HAD (Saha and Pahan 2003; Shapshak et al. 2004; Wesselingh et al. 1997). Furthermore, IFN-γ and TNF-α, interact to synergistically upregulate CXCL10 expression (Majumder et al. 1998a; Nosheny et al. 2007). Besides its induction by host factors, CXCL10 can also be induced by the HIV-1/HIV-1 proteins (Asensio et al. 2001; Kutsch et al. 2000; van Marle et al. 2004). Due to the potentially neurotoxic function of CXCL10 in disease states, it is crucial to analyze how combinatorial interactions of the virus and host factors can lead to an increased pool of this toxic chemokine in the CNS. There is a paucity of information on the combined effects of IFN-γ/TNF-α with HIV-1 on CXCL10 expression. In the current study we hypothesized that IFN-γ and TNF-α not only synergize with each other, but also have the potential to synergize with HIV-1 to induce CXCL10 expression in astrocytes. To our knowledge, this is the first published report characterizing the synergistic enhancement of astroglial CXCL10 expression by IFN-γ/TNF-α and HIV-1. Furthermore, our findings demonstrate that this synergistic increase in CXCL10 expression involves the signaling cascades of the JAK/STAT and MAPK pathways. Together, these insights may be instrumental in the development of therapeutic strategies aimed at treating or preventing HIV-1 neuropathogenesis.
Materials and Methods
Astrocyte cell culture and treatments
Primary human astrocytes (cat# HA1800; ScienCell Research Laboratories, Carlsbad, CA) were plated on poly-L-lysine coated plates (2μg/cm2) at a density of 5,000 cells/cm2 in Astrocyte Medium (cat.# 1801) containing 2% FBS, growth supplement (cat.# 1852), and penicillin/streptomycin solution (cat.# 0503) as described by the supplier. Primary human astrocytes were allowed to grow for two weeks in order to reach 90% confluence. The human astrocytic cell line, A172 (ATCC #CRL-1620; American Type Culture Collection, Manassas, VA), were cultured as described previously (Davis et al. 2002). Since serum contains growth factors that can potentially upregulate constitutive expression of cytokines/chemokines, thereby masking the effects of the treatment agents, we conditioned the cells in the absence of serum 24hrs prior to treatment. The cells (triplicate or quadruplicate wells) were treated for 6-12 hrs (A172) or 24 hours (primary astrocytes), with: 1) a combination of the cytokines IFN-γ (100ng/ml) and TNF-α (30ng/ml), 2) HIV-1 BAL and HIV-1 NL4.3 (both at a multiplicity of infection of 0.01) (Adachi et al. 1986), or 3) HIV-1 BAL/NL4.3 and cytokines. The rationale for using CXCR4 (X4)-tropic NL4.3 in these studies is based on our observation that NL4.3 in combination with the cytokines had a stronger effect on CXCL10 induction than HIV-1 BAL. There are also published reports indicating increased activation of astrocytes by X4 viruses (Bezzi et al. 2001; Kaul et al. 2001).
Control treatments included HIV-1 NL4.3 incubated with neutralizing antibodies against gp120 (25ng) for one hour at room temperature. Gp120 monoclonal antibodies, 1510 and 1511, to HIV-1 V3 were submitted by Dr. Susan Zolla-Pazner and obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Gorny et al. 1991; Gorny et al. 1993). Following the one hour incubation, protein-G sepharose beads (Sigma, St. Louis, MO) were added to the mixture for one hour at room temperature before being spun down. The resulting viral inoculum was added to stimulated/unstimulated A172 cells. For additional virus controls, the NL4.3 virus was subjected to either UV treatment, heat inactivation, or both.
The following specific pharmacological inhibitors were used at the final concentration specified: PI3-K Inhibitor LY294002, PLC inhibitor U73122, MEK inhibitor U0126, JAK inhibitor I, JNK inhibitor II, P38 inhibitor SB203580 (all at 20μM, Calbiochem, Gibbstown, NJ), and NF-κB inhibitor N-_p_-Tosyl-L-phenylalanine chloromethyl ketone (TPCK) (2μM, Sigma, St. Louis, MO) (Arai et al. 2003; Dhillon et al. 2007a; Lee et al. 2000).
CXCL10 mRNA analysis
RNA was extracted from A172 astrocytes that were either untreated or treated with HIV-1 NL4.3 and/or the cytokines IFN-γ and TNF-α was carried out as previously described (Dhillon et al. 2007a). Quantitative analysis of CXCL10 mRNA was done by quantitative Real-Time PCR using the SYBR Green detection method. RT2 PCR primer pair set for CXCL10 was obtained from SuperArray Bioscience and amplification of CXCL10 from first strand cDNA was performed as described earlier (Dhillon et al. 2007b).
CXCL10 protein analysis by ELISA
Supernatants collected from both primary human astrocytes and A172 astrocytes that were either untreated or treated with HIV-1 and/or cytokines, were examined for secreted CXCL10 protein levels using a commercially available ELISA kit (R&D Systems, Minneapolis, MN).
Western Blot Analysis
Treated A172 cells were lysed using the Mammalian Cell Lysis kit (Sigma, St. Louis, MO) and the NE-PER Nuclear and Cytoplasmic Extraction kits (Pierce, Rockford, IL). Samples were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel (12%). The western blots were then probed with antibodies recognizing the phosphorylated forms of Erk1/2, AKT, P38, p706S (Cell Signaling, Danvers, MA 1:200), PI3-K (Santa Cruz Biotechnology, Santa Cruz, CA, 1:100), Stat1-α and Stat-3 (Cell Signaling, 1:500), NF-κB p65 (Cell Signaling, 1:1000), and β-actin (Sigma, St. Louis, MO,1:4000) The secondary antibodies were alkaline phosphatase conjugated to goat anti mouse/rabbit IgG (1:5000). Signals were detected by chemiluminescence (CDP-star; Tropix, Bedford, MA).
Immunocytochemistry
Immunocytochemical analysis for NF-κB activation was performed on A172 astrocytes cultured on coverslips and treated with HIV-NL4.3 and cytokines for 60 minutes. Following treatment cells were fixed and blocked as described previously (Dhillon et al. 2007a). Following blocking, anti-human NF-κB p65 rabbit polyclonal antibody (1: 500, Cell Signaling) was added to each coverslip and incubated for 2 hours at room temperature. The secondary antibody, AlexaFluor 488 goat anti-rabbit IgG, was used at a 1:1000 dilution for 2 hours to view NF-κB activation in cells and DAPI was used to stain the cell nuclei. Fluorescent digital images were obtained as described earlier (Dhillon et al. 2007a). Control coverslips comprised of: 1) cells without any secondary antibody treatment and, 2) cells treated with secondary antibody only.
Results
Synergistic induction of CXCL10 in astrocytes exposed to HIV-1 and pro-inflammatory cytokines
Astrocytes, the most numerous cell type within the brain, provide an important reservoir for the generation of inflammatory mediators in response to HIV-1 infiltration into the brain (Dong and Benveniste 2001; Minagar et al. 2002; Thompson et al. 2001). Several studies have shown that IFN-γ and TNF-α can synergistically induce CXCL10 in multiple cell types, including astrocytes (Croitoru-Lamoury et al. 2003; Oh et al. 1999). Other studies have shown that HIV-1/HIV-1 proteins can also induce CXCL10 expression (Asensio et al. 2001; Kutsch et al. 2000; van Marle et al. 2004). The objective of this study was to determine whether HIV-1 in conjunction with the pro-inflammatory cytokines IFN-γ, and TNF-α can synergistically impact the expression of CXCL10 in astrocytes. Serum-starved A172 cells were treated with CXCR4 tropic HIV-1 NL4.3 and/or the cytokine mix (IFN-γ and TNF-α) for six hours, following which the cells were lysed in Trizol for RNA extraction and Real Time RT-PCR analysis for CXCL10. As shown in Fig. 1 there was a dramatic induction of CXCL10 RNA (about 80,000 fold) in cells treated with both HIV-1 and the cytokine mix compared with astrocytes treated with either the cytokine mix or virus alone.
Figure 1.
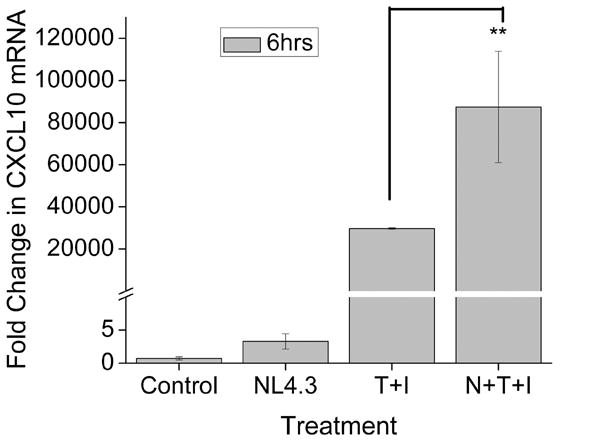
Real Time RT-PCR analysis showing a significant increase in CXCL10 RNA in virus and cytokine treated A172 astrocytes. Cell were stimulated with either HIV-1 NL4.3 (moi of 0.01), the cytokines IFN-γ (100ng/ml) and TNF-α (30ng/ml), or both for 6 hrs followed by total cell lysis and RNA extraction. The graph shows and 80,000 fold induction of CXCL10 RNA in the virus and cytokine stimulated astrocytes over the untreated astrocytes. The data represents the mean ± SD from three independent experiments (**, p < 0.01).
The next step was to determine whether such a dramatic increase in CXCL10 mRNA also translated into a concomitant increase in CXCL10 protein levels. Serum-starved A172 cells were treated with HIV-1 NL4.3 in presence or absence of the cytokine mix for 12 hrs and CXCL10 release in the supernatant was quantified by CXCL10 ELISA. As shown in Figure 2A, virus and cytokine treatment of the astrocyte cell line resulted in a dramatic up-regulation of CXCL10 protein expression. To confirm that the increase in CXCL10 was due to the virus/viral protein interactions with the cell surface and not due to other stimulants in the viral inoculum, two separate anti-gp120 antibodies, 1510 and 1511, were incubated individually with the virus and pulled down with protein-G sepharose beads prior to stimulation of the cells with the virus. The results demonstrated that in the absence of gp120, the viral inoculum was unable co-operate with the cytokine mix to enhance CXCL10 expression, thus underscoring the role of gp120 binding in the synergistic induction of CXCL10.
Figure 2.
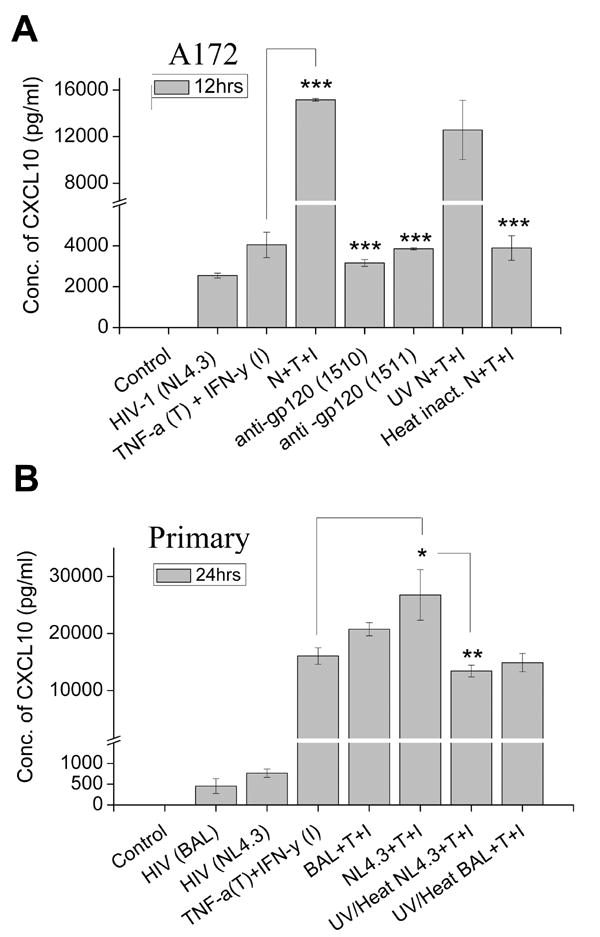
Synergistic induction of CXCL10 protein in astrocytes treated with virus and cytokines. Supernatants from cells treated with either virus or cytokine or both virus and cytokines were collected at A) 12hrs for A172 cells and B) 24hrs for primary human astrocytes and analyzed for CXCL10 protein levels by ELISA. Both A172 and primary human astrocytes showed a significant increase in CXCL10 protein levels in the cells treated with virus in conjunction with the cytokines, then either treatment alone. Treatment of A172 cells with neutralizing gp120 antibodies abrogated the synergistic effect. The data represents the mean ± SD from three independent experiments. Statistical significance from independent experiments was calculated (***, p < 0.001, **, p < 0.01).
Additionally, as negative controls we also used UV and/or heat-inactivated virus. UV-inactivated virus was able to synergize with the cytokines with respect to CXCL10 release, thereby indicating that it is not the viral infection/replication, rather it is the viral protein(s) that could be mediating the effect. Heat treated virus, on the other hand, was unable to synergize the CXCL10 induction. A possible explanation of this could be that heat treatment rendered the denaturation of proteins and thus negatively impact the synergy.
The phenomenon of CXCL10 protein induction by combination of HIV and cytokines was also reproduced in primary normal human astrocytes (Fig.2B). Astrocytes exposed to the cytokines and inactivated virus (UV and heat treated), on the other hand, failed to demonstrate virus-mediated synergy of CXCL10 induction. Conversely, UV-inactivated virus was able to synergize with the cytokines with respect to CXCL10 release (data not shown), again emphasizing that it is not viral infection/replication, rather the viral protein(s) that could be mediating the effect. These data confirmed the importance of HIV-1 in the synergistic enhancement of astroglial CXCL10 expression.
Signaling pathways involved in HIV-1 and cytokine-mediated synergistic induction of CXCL10
Role of the JAK-STAT pathway
Since IFN-γ and TNF-α in conjunction with HIV-1 synergistically induced expression of CXCL10, and because IFN-y is known to mediate its effects primarily via the JAK/STAT1 signaling pathways (Croitoru-Lamoury et al. 2003; Giunta et al. 2006; Oh et al. 1999; Ramana et al. 2002) we chose to assess the role of this signaling pathway in stimulated astrocytes. STAT proteins are a family of transcription factors that are present in many cell types and function as major signal transduction pathway in IFN-γ signaling. Following the binding of ligands to their receptors, JAKs are activated and, in turn, phosphorylate STAT-1α and/or STAT 3 proteins. Phospho-STATs dimerize and translocate into the nucleus, binding to the interferon stimulated response element (ISRE) on the promoter regions of target genes, such as CXCL10 (Darnell et al. 1994). Since IFN-γ is a major inducer of CXCL10 expression and efficiently induces its transcription without intervening protein synthesis, we rationalized that augmented induction of CXCL10 by HIV-1 in co-operation with IFN-γ/TNF-α, must involve increased activation of the JAK/STAT1 signaling system in astrocytes.
Western blot analysis of the nuclear extracts from astrocytes treated with the virus and cytokines showed time-dependent activation of both STAT-1α and STAT-3 (Fig. 3A). As shown in Fig. 3B, pre-treatment of A172 astrocytes with the JAK inhibitor followed by stimulation with HIV-1 and cytokines resulted in a substantial decrease in CXCL10 protein expression to a level lower than that achieved with cytokines alone. These findings suggested a crucial role for JAK/STAT signaling in the synergistic induction of CXCL10 in HIV-1 and IFN-γ/TNF-α treated astrocytes.
Figure 3.
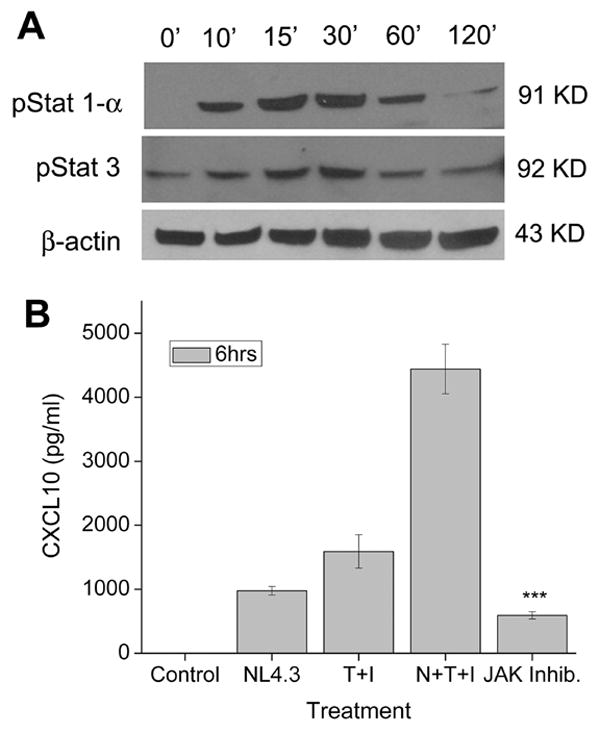
The JAK/STAT pathway is a critical player in the regulation of CXCL10 expression in virus and cytokine stimulated astrocytes. A) Western Blot analysis using phosphorylated antibodies against STAT1α and STAT3 on the nuclear lysates from untreated or HIV-1 NL4.3 (N) and cytokines (T+I) treated A172 cultures. Blots were stripped and reprobed with β-actin for normalization. B) CXCL10 ELISA on supernatants collected from A172 astrocytes treated with JAK inhibitor and HIV-1 (N) and the cytokines (T+I) for 6hrs before analysis. Values are mean ± SD from three independent experiments (***, p < 0.001).
Role of Mitogen activated protein kinase (MAPK) signaling pathways
Mitogen activated protein kinase (MAPK) activation is critical in regulating inflammatory responses, such as cytokine/chemokine secretion in response to multiple stimuli (Means et al. 2000; Park et al. 2001; Popik et al. 1998; Shapiro et al. 1998). Therefore, we next explored the involvement of Erk1/2 and p38 MAP kinases in the regulation of CXCL10 induction by HIV-1 and cytokine mix. As shown in Figure 4A, A172 cells stimulated with HIV-1 and the cytokine mix demonstrated a time-dependent activation of Erk1/2. Confirmation of the specificity of Erk1/2 in the synergistic induction of CXCL10 was further carried out by examining the chemokine expression by ELISA in the supernatants collected from virus and cytokine-stimulated A172 cells in the presence of the pharmacological inhibitor of MEK signaling, U0126. As shown in Figure 4B, levels of CXCL10 were significantly decreased in the presence of the inhibitor, thus confirming the role of Erk1/2 MAPK signaling pathway in the induction of CXCL10.
Figure 4.
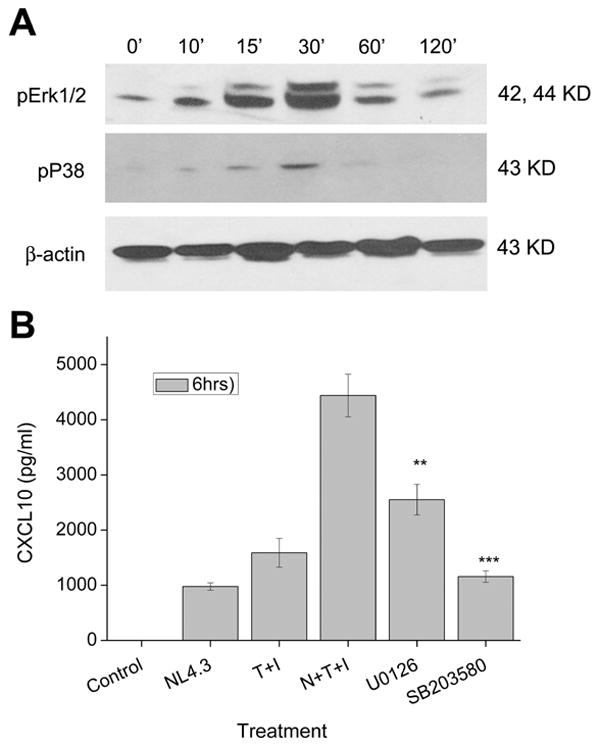
The Erk1/2 and p38 MAPK pathways mediate CXCL10 expression in stimulated astrocytes. A) Western Blot analysis of cytosolic lysates from untreated and virus and cytokine treated A172 astrocytes was conducted at various time points using antibodies against the phosphorylated forms of Erk1/2 and p38. An antibody against β-actin was used to reprobe the blots for normalization. (B) Activation of these pathways were shown to be involved in the synergistic increase in CXCL10 protein expression in these cells through inhibition of the Erk1/2 pathway by U0126 and of the p38 pathway by SB203580. Values are mean ± SD from three independent experiments (***, p < 0.001, **, p < 0.01).
We next assessed the role of another MAPK signaling pathway, p38 in the synergistic induction of CXCL10 in astrocytes. A172 cells stimulated with the virus and cytokines led to the activation of p38 protein as early as 10 min following stimulation with peak activation attained at 30 min (Fig. 4A). To determine the functional role of p38 pathways in CXCL10 regulation, cells were treated with SB203580, a pharmacological inhibitor of p38 MAPK and assessed for CXCL10 release in the supernatant by ELISA (Fig. 4B). In the presence of the inhibitor synergistic increase of CXCL10 with HIV-1 and cytokine mix was significantly decreased thereby indicating the importance of p38 signaling pathway in the induction of the chemokine.
Role of Phospho-inositol 3-kinase (PI3-Kinase) signaling pathway
Phospho-inositol 3-kinase (PI3-K) is a signaling protein that can be activated by various stimuli and is critical in the regulation of extracellular signals along with modulation MAPK activity (Lee et al. 2005). To dissect the role of the PI3-K pathway in HIV-1 and cytokine-mediated synergy of CXCL10 expression, cell lysates from A172 stimulated cells were examined for phosphorylation of PI3-Kinase and its downstream effectors, Akt and p70S6 kinase. As shown in Figure 5A, there was a temporal activation of the signaling proteins of the PI3-Kinase pathway in cells stimulated with the virus and cytokines. PI3-K, which is directly upstream of Akt, was activated as early as 10-15 min, followed by activation of Akt at around 30-120 min and this, in turn, was followed by phosphorylation of p70S6 at 60-120 min (Fig. 5A). To address the functional role of PI3-K in the synergistic induction of CXCL10, A172 cells were pretreated with the PI3-K inhibitor, LY294002, and with the PLC inhibitor U73122 as a negative control, followed by stimulation of the cells with HIV-1 and the cytokines, then subsequently monitoring the supernatants for CXCL10 expression. As shown in Fig. 5B, pretreatment of A71 cells with the PI3-K inhibitor led to a significant decrease in the synergistic induction of CXCL10 expression, while inhibition of PLC did not significantly decrease CXCL10 levels.
Figure 5.
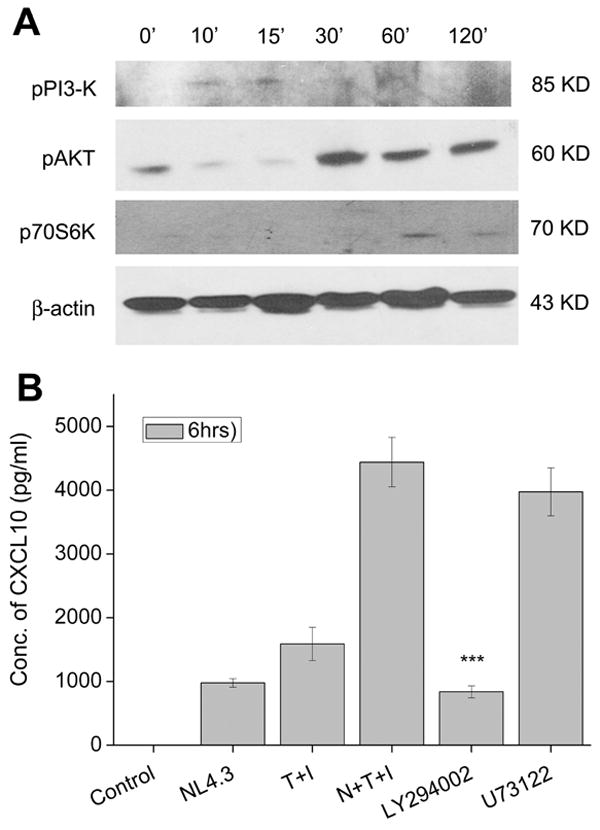
The PI3-Kinase pathway is involved in CXCL10 expression in virus and cytokine treated astrocytes. A) Western Blot analysis of cytosolic lysates from untreated and virus and cytokine treated A172 astrocytes was conducted at various time points using antibodies against the phosphorylated forms of PI3-K, and its downstream targets Akt and p706S-kinase. An antibody against β-actin was used to reprobe the blots for normalization. (B) Confirmation of the involvement of the PI3-K pathway in production of CXCL10 in virus and cytokine treated astrocytes was confirmed by pretreating the cells with an inhibitor of PI3-K, LY294002, and the PLC inhibitor U73122 followed by CXCL10 ELISA. Values are mean ± SD from three independent experiments (***, p < 0.001).
NF-κB Signaling
The transcription factor NF-κB plays a pivotal role in inflammatory and immune responses (Calzado et al. 2007; Giunta et al. 2006). This family of transcription factors is present in the cytosol in an inactive state complexed with the inhibitory IκB proteins. Activation occurs via the phosphorylation of the inhibitory protein and the subsequent release of active p65 subunit of NF-κB (Brasier 2006). Both TNF-α and IFN-γ have been shown to synergistically increase CXCL10 expression through the transcription factor NF-κB (Majumder et al. 1998a; Majumder et al. 1998b; Ohmori and Hamilton 1993; Ohmori and Hamilton 1995). We next examined the role of NF-κB in the synergistic induction of CXCL10 by HIV-1 and cytokines. Nuclear lysates were isolated from A172 cells stimulated with the virus and host factors for different time intervals and probed for the release of p65 subunit of NF-κB in the nuclear fraction by Western blot analysis (Fig. 6A). Stimulation of astrocytes with the virus and cytokines induced time-dependent nuclear translocation of NF-κB p65, which was observed by 10 min and peaked by 60 min as indicated by Western blot analysis (Fig. 6A).
Figure 6.
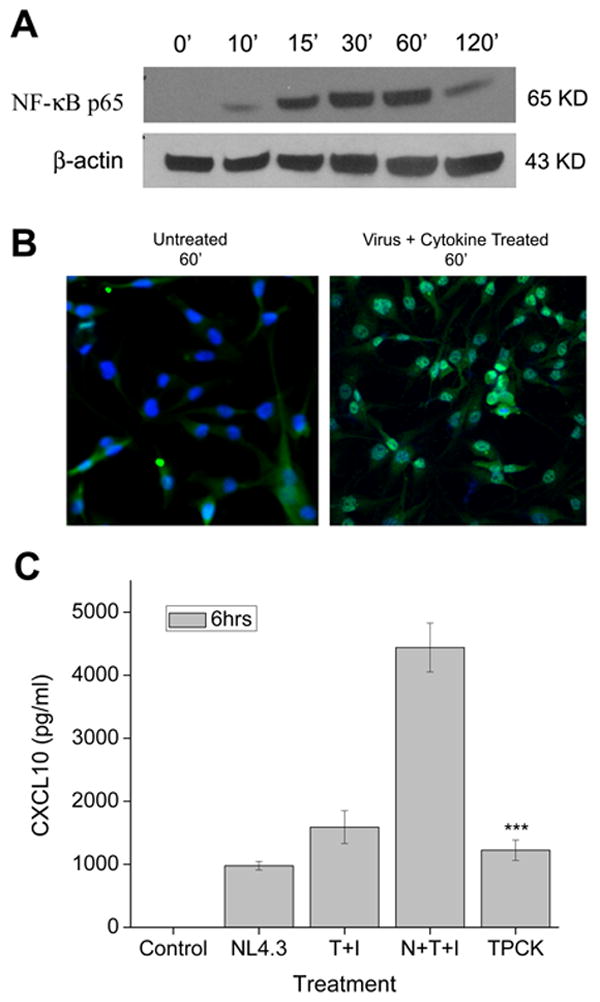
NF-κB plays a role in the synergistic induction of CXCL10 by HIV-1 and cytokines in astrocytes. A) Western Blot analysis of nuclear lysates from untreated and virus plus cytokine treated cells using an antibody against the p65 subunit of NF-κB. B) A172 astrocytes were either untreated or treated with virus plus cytokines for 60 min and stained with a NF-κB p65 antibody followed by treatment with an Alexa Flour 488-conjugated secondary antibody. After 60 min nuclear translocation of NF-κB is clearly evident in the treated cells, as show by the green (NF-κB) and blue (DAPI) stains overlapping (magnification X 250). C) To confirm the role of NF-κB in the synergistic induction of CXCL10 in stimulated astrocytes a CXCL10 ELISA utilizing the NF-κB inhibitor TPCK was conducted. Values are mean ± SD from three independent experiments (***, p < 0.001). cytokine treated astrocytes.
Immunocytochemistry was used to further visualize the nuclear translocation of NF-κB. Figure 6B clearly shows the nuclear translocation of NF-κB p65 in virus and cytokine treated cells versus control untreated cells, where the NF-κB is seen localized to the cytoplasm. To further confirm the role of NF-κB in the synergistic induction of CXCL10 in HIV-1 and cytokine stimulated astrocytes, we pre-treated the cells with TPCK, an inhibitor of NF-κB followed by stimulation of cells. Cell supernatants were then monitored for CXCL10 expression by ELISA. There was a significant decrease in the amount of CXCL10 released from astrocytes stimulated with the virus and cytokine mixture in the presence of TPCK (Fig. 6C) compared with cells not pre-treated with the inhibitor. Clearly, NF-κB plays a pivotal role in the synergistic induction of CXCL10 in astrocytes activated by the pro-inflammatory cytokines, IFN-γ and TNF-α in conjunction with HIV-1.
Discussion
Despite the use of combinational antiretroviral therapy (cART) HIV-associated cognitive impairments afflict 60% of the HIV-1 infected population (Giunta et al. 2006; Oh et al. 1999). HAD, the most severe form of these impairments (Albright et al. 2003; Ramana et al. 2002), is pathologically characterized by astrocytosis, cytokine/chemokine imbalance, and neuronal degeneration (Gonzalez-Scarano and Martin-Garcia 2005; Minagar et al. 2002; Navia et al. 1986). Because of the crucial role astrocytes play in maintaining neuronal homeostasis, astrocyte hyper-activation can severely impact the neuronal environment, resulting in disease pathogenesis (Darlington 2005; Darnell et al. 1994).
During HIV-1 infection there is an imbalance of the pro-inflammatory cytokines, IFN-γ and TNF-α, which have been shown to be markedly increased in CNS tissues (Shapshak et al. 2004; Wesselingh et al. 1997). In addition to cytokine dysregulation, enhanced expression of chemokines, such as CXCL10 and MCP-1, is also known to correlate with the severity of HAD (Lawrence et al. 2006). Functionally, astroglial CXCL10 recruits inflammatory cells into the CNS, and has also been demonstrated to exhibit potent neurotoxic effects (Sui et al. 2004; Sui et al. 2006). Consequently, it is critical to comprehend the regulation of the CXCL10 expression in the inflamed brain. Therefore, the objective of this study was to understand how these dysregulated host cytokines can interact with HIV-1 in astrocytes to amplify CXCL10 expression in the CNS.
Due to the ability of CXCL10 expression to be regulated by diverse stimuli and studies demonstrating the synergistic effects of IFN-γ and TNF-α on CXCL10 in multiple cell types (Cassatella et al. 1997; Hardaker et al. 2004; Hiroi and Ohmori 2003; Means et al. 2000; Park et al. 2001; Popik et al. 1998), we evaluated whether these two cytokines in combination with HIV-1 could further increase CXCL10 expression in astrocytes. Based on our findings, it was evident that HIV-1 in combination with the pro-inflammatory cytokines, synergistically enhanced CXCL10 mRNA and protein expression in both the human A172 astroglia and primary human astrocytes. These findings have implications for increased neuronal toxicity in HIV-1-infected individuals and underscore the importance of host-virus interactions in the pathogenesis of HAD.
Having determined the co-operative interaction of HIV-1 with the pro-inflammatory cytokines, it was of interest to explore the signaling pathways involved in this synergy. IFN-γ activates the JAK/STAT pathway through the α- and β- subunits (Darnell et al. 1994; Lackmann et al. 1998; Lee et al. 2005; Shapiro et al. 1998) leading to activation of JAK1 and JAK2 kinases followed by tyrosine phosphorylation of STAT1 (Brasier 2006; Calzado et al. 2007; Darnell et al. 1994; Muller et al. 1993; Shapiro et al. 1998; Shuai et al. 1993). Herein, we found nuclear translocation and time-dependent phosphorylation of both STAT-1 and STAT-3, with activation as early as 15 min following stimulation of cells. Inhibition of JAK, an upstream effector of STAT1/3, through a specific pharmacological inhibitor drastically reduced the amount of CXCL10 expressed by stimulated astrocytes. These findings suggest that the JAK/STAT pathway plays a crucial role in the cooperative induction of CXCL10 in IFN-γ, TNF-α, and HIV-1 stimulated astrocytes. This data is also in agreement with that reported by Dhillon et al. on the synergistic induction of CXCL10 by IFN-γ and PDGF in macrophages (Cassatella et al. 1997; Dhillon et al. 2007a).
Since HIV-1/viral proteins, TNF-α, and IFN-γ all have the ability to activate MAPK signaling cascades (Asensio et al. 2001; Hardaker et al. 2004; Kutsch et al. 2000; Robichaud and Poulin 2000; Schutze et al. 1995), we next investigated the role of these pathways in the induction of CXCL10 in stimulated astrocytes. In congruence with the findings by Lee et. al. on gp120-stimulated macrophages (Hiroi and Ohmori 2003; Lee et al. 2005), we also found that both the Erk1/2 and p38 MAPK pathways were strongly activated following stimulation of astrocytes with HIV-1 and cytokines. Both of these pathways have also been implicated in the induction of proinflammatory genes (Cassatella et al. 1997; Dhillon et al. 2007a) and such activation is responsible for the transcriptional stabilization of the target proinflammatory genes (Cassatella et al. 1997; Dhillon et al. 2007a).
Furthermore, we also demonstrated temporal activation of the PI3-K pathway, involving sequential phosphorylation of Akt and its downstream p706S-kinase. Confirmation of the role of this pathway in CXCL10 expression was examined by pre-treating the cells with the PI3-K inhibitor, LY294002. A significant decease in the amount of CXCL10 was observed in stimulated cells pre-treated with the inhibitor. Interestingly, a similar pathway has been reported in the enhanced expression of TNF-α in gp120-treated macrophages (Lee et al. 2003; Lee et al. 2005). Furthermore, Western Blot analysis demonstrated clear evidence of Akt activation, a critical survival factor (Chong et al. 2005; Kolson 2002; Zhao et al. 2006) and a downstream target of PI3-K. Taken together, these results indicate a role for the PI3-K pathway in CXCL10 induction in virus and cytokine stimulated astrocytes.
Several studies have shown that astrocytes activated by HIV-1/viral proteins have increased nuclear translocation and activation of the transcription factor NF-κB (Bach et al. 1997; Conant et al. 1996; Lawrence et al. 2006; Schutze et al. 1995; Sheng et al. 2003), which, in turn, can regulate CXCL10 expression. Since activation of the Erk1/2, p38, and PI3-K signaling pathways can converge on a common transcription factor such as NF-κB, and since the CXCL10 promoter has NF-κB binding sites (Lee et al. 2003; Majumder et al. 1998a; Ohmori and Hamilton 1995; Tomura and Narumi 1999), we next examined the activation and translocation of NF-κB in stimulated astrocytes. Our findings showed dramatic and sustained activation of NF-κB in the nucleus of stimulated astrocytes. These findings are consonant with other reports implicating the role of NF-κB in the regulation of various chemokines and cytokines, such as MCP-1 and IL-6, in astrocytes (Bach et al. 1997; Lawrence et al. 2006; Nath et al. 1999; Zhao et al. 2006). The role of NF-κB was further confirmed by pre-treating the cells with TPCK, a NF-κB inhibitor, which resulted in significant decrease in expression of CXCL10. These findings underscore the role of this transcription factor in the synergistic induction of CXCL10.
As mentioned earlier, the CXCL10 promoter region has two NF-κB sites and one ISRE site. The NF-κB site, κB2, in conjunction with the ISRE site are necessary for the synergistic induction of CXCL10 in IFN-γ and TNF-α simulated astrocytes (Majumder et al. 1998b). Both IFN-γ and TNF-α have been shown to activate the JAK/STAT and MAPK/NF-κB pathways. HIV-1/viral proteins also have the ability to activate the MAPK pathways Erk1/2, JNK and p38 (Kutsch et al. 2000) in astrocytes, and viral gp120-mediated induction of CXCL10 is independent of the STAT1 pathway (Asensio et al. 2001; Robichaud and Poulin 2000).
Thus, while activation of MAPK and NF-κB can be attributed to all the three stimuli in astrocytes, STAT 1 activation is unique to IFN-γ and thus IFN-γ plus TNF-α. It has been well documented that activation of the MAPK pathways by HIV-1/viral proteins can lead to the activation and nuclear translocation of NF-κB (Lawrence et al. 2006; Sheng et al. 2003) potentially increasing the amount of NF-κB available to the κB2 site essential for synergistically up-regulating CXCL10. Therefore, while IFN-γ is activating the JAK/STAT pathway providing for the binding of the ISRE site on the CXCL10 promoter, all three stimuli can impact the amount of NF-κB needed for the binding of the two NF-κB binding sites.
In addition, none of the inhibitors tested were able to totally abolish the release of CXCL10 in our study thereby indicating the involvement of more than one pathway in the synergy. This does not however, preclude cross talk between the pathways. In fact, dramatic inhibition of CXCL10 with either the P38 or the Akt inhibitor does indeed imply a cross-talk between the pathways. Additionally, significant down-regulation of CXCL10 was seen in the presence of the either the JAK or the NF-κB inhibitor. This is most likely due to the fact that both STAT and NF-κB transcription factors are involved in the IFN-γ/TNF-α-mediated synergistic modulation of CXCL10. Therefore, inhibiting either of the transcription factors will dramatically abrogate CXCL10 induction in the presence of HIV-1 and the cytokines.
In conclusion, we have shown that HIV-1 NL4.3 in co-operation with the cytokines IFN-γ and TNF-α is capable of synergistically inducing CXCL10 in human astrocytes at both the RNA and protein levels. This induction is likely due to the activation of the JAK/STAT and PI3-K signaling pathways, along with the MAPK pathways, Erk1/2 and p38 (Fig. 7). The significance of the potential synergistic interactions between HIV-1 and soluble host factors and the implications of this type of complex interplay on the progression of HAD is progressively garnering more attention. Our studies demonstrating the signal transduction pathways activated by HIV-1 and the pro-inflammatory cytokines in the enhancement of CXCL10 expression, define a precarious proinflammatory mechanism that exacerbate the pathogenesis of HAD. Due to the neurotoxic potential of CXCL10 these findings lend to important implication in the progression of AIDS-associated dementia. The consequences of CXCL10 over expression may include amplified neuronal dysfunction and death, as well as an enhanced influx of inflammatory cells into the CNS, a combination that creates elevated toxic, pathological responses characteristic of end-stage HAD. These findings have implications in the development of therapeutic strategies aimed at inhibiting glial cell activation to prevent HIV-1 neuropathogenesis.
Figure 7.

Schematic of the signaling pathways involved in the synergistic induction of CXCL10 in astrocytes stimulated with HIV-1 in conjunction with IFN-γ and TNF-α. The major signaling pathways activated include JAK/STAT, MAPK, and PI3-K. The latter two are capable of converging on NF-κB, resulting in the transcription of CXCL10.
Acknowledgments
We acknowledge the gp120 monoclonal antibodies, 1510 and 1511, to HIV-1 V3 were submitted by Dr. Susan Zolla-Pazner and obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Gorny et al. 1991; Gorny et al. 1993).
This work was supported by funding from NIMH MH068212, NIDA DA020392, NIDA DA024442 (SB), NINDS F31NS062665 (RW) and NIH AA014955 (RLD).
Works Cited
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright AV, Soldan SS, Gonzalez-Scarano F. Pathogenesis of human immunodeficiency virus-induced neurological disease. J Neurovirol. 2003;9(2):222–7. doi: 10.1080/13550280390194073. [DOI] [PubMed] [Google Scholar]
- Asensio VC, Campbell IL. Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 1999;22(11):504–12. doi: 10.1016/s0166-2236(99)01453-8. [DOI] [PubMed] [Google Scholar]
- Asensio VC, Maier J, Milner R, Boztug K, Kincaid C, Moulard M, Phillipson C, Lindsley K, Krucker T, Fox HS, et al. Interferon-independent, human immunodeficiency virus type 1 gp120-mediated induction of CXCL10/IP-10 gene expression by astrocytes in vivo and in vitro. J Virol. 2001;75(15):7067–77. doi: 10.1128/JVI.75.15.7067-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4(7):702–10. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. 2006;6(2):111–30. doi: 10.1385/ct:6:2:111. [DOI] [PubMed] [Google Scholar]
- Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem. 2007;14(3):367–76. doi: 10.2174/092986707779941113. [DOI] [PubMed] [Google Scholar]
- Cassatella MA, Gasperini S, Calzetti F, Bertagnin A, Luster AD, McDonald PP. Regulated production of the interferon-gamma-inducible protein-10 (IP-10) chemokine by human neutrophils. Eur J Immunol. 1997;27(1):111–5. doi: 10.1002/eji.1830270117. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95(6):3117–21. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Ma M, Nath A, Major EO. Extracellular human immunodeficiency virus type 1 Tat protein is associated with an increase in both NF-kappa B binding and protein kinase C activity in primary human astrocytes. J Virol. 1996;70(3):1384–9. doi: 10.1128/jvi.70.3.1384-1389.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croitoru-Lamoury J, Guillemin GJ, Boussin FD, Mognetti B, Gigout LI, Cheret A, Vaslin B, Le Grand R, Brew BJ, Dormont D. Expression of chemokines and their receptors in human and simian astrocytes: evidence for a central role of TNF alpha and IFN gamma in CXCR4 and CCR5 modulation. Glia. 2003;41(4):354–70. doi: 10.1002/glia.10181. [DOI] [PubMed] [Google Scholar]
- Darlington CL. Astrocytes as targets for neuroprotective drugs. Curr Opin Investig Drugs. 2005;6(7):700–3. [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Davis RL, Dertien J, Syapin PJ. Ethanol-induced modulation of inducible nitric-oxide synthase activity in human A172 astrocytoma cells. Alcohol Clin Exp Res. 2002;26(9):1404–11. doi: 10.1097/01.ALC.0000030841.92766.80. [DOI] [PubMed] [Google Scholar]
- Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, Ghorpade A. Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res. 2005;7(3):183–92. doi: 10.1007/BF03036448. [DOI] [PubMed] [Google Scholar]
- Dhillon NK, Peng F, Ransohoff RM, Buch S. PDGF synergistically enhances IFN-gamma-induced expression of CXCL10 in blood-derived macrophages: implications for HIV dementia. J Immunol. 2007a;179(5):2722–30. doi: 10.4049/jimmunol.179.5.2722. [DOI] [PubMed] [Google Scholar]
- Dhillon NK, Pinson D, Dhillon S, Tawfik O, Danley M, Davis M, Nemon O, Mayo M, Kumar A, Tsai YJ, et al. Bleomycin treatment causes enhancement of virus replication in the lungs of SHIV-infected macaques. Am J Physiol Lung Cell Mol Physiol. 2007b;292(5):L1233–40. doi: 10.1152/ajplung.00293.2006. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36(2):180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Giunta B, Obregon D, Hou H, Zeng J, Sun N, Nikolic V, Ehrhart J, Shytle D, Fernandez F, Tan J. EGCG mitigates neurotoxicity mediated by HIV-1 proteins gp120 and Tat in the presence of IFN-gamma: role of JAK/STAT1 signaling and implications for HIV-associated dementia. Brain Res. 2006;1123(1):216–25. doi: 10.1016/j.brainres.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38(5):755–62. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorny MK, Xu JY, Gianakakos V, Karwowska S, Williams C, Sheppard HW, Hanson CV, Zolla-Pazner S. Production of site-selected neutralizing human monoclonal antibodies against the third variable domain of the human immunodeficiency virus type 1 envelope glycoprotein. Proc Natl Acad Sci U S A. 1991;88(8):3238–42. doi: 10.1073/pnas.88.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorny MK, Xu JY, Karwowska S, Buchbinder A, Zolla-Pazner S. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J Immunol. 1993;150(2):635–43. [PubMed] [Google Scholar]
- Hardaker EL, Bacon AM, Carlson K, Roshak AK, Foley JJ, Schmidt DB, Buckley PT, Comegys M, Panettieri RA, Jr, Sarau HM, et al. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. Faseb J. 2004;18(1):191–3. doi: 10.1096/fj.03-0170fje. [DOI] [PubMed] [Google Scholar]
- Hiroi M, Ohmori Y. Constitutive nuclear factor kappaB activity is required to elicit interferon-gamma-induced expression of chemokine CXC ligand 9 (CXCL9) and CXCL10 in human tumour cell lines. Biochem J. 2003;376(Pt 2):393–402. doi: 10.1042/BJ20030842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4(3):307–18. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44(5):831–5. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J Neuroimmunol. 1999;93(1-2):172–81. doi: 10.1016/s0165-5728(98)00223-9. [DOI] [PubMed] [Google Scholar]
- Kolson DL. Neuropathogenesis of central nervous system HIV-1 infection. Clin Lab Med. 2002;22(3):703–17. doi: 10.1016/s0272-2712(02)00009-4. [DOI] [PubMed] [Google Scholar]
- Kolson DL, Pomerantz RJ. AIDS Dementia and HIV-1-Induced Neurotoxicity: Possible Pathogenic Associations and Mechanisms. J Biomed Sci. 1996;3(6):389–414. doi: 10.1007/BF02258044. [DOI] [PubMed] [Google Scholar]
- Kutsch O, Oh J, Nath A, Benveniste EN. Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 tat in astrocytes. J Virol. 2000;74(19):9214–21. doi: 10.1128/jvi.74.19.9214-9221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackmann M, Harpur AG, Oates AC, Mann RJ, Gabriel A, Meutermans W, Alewood PF, Kerr IM, Stark GR, Wilks AF. Biomolecular interaction analysis of IFN gamma-induced signaling events in whole-cell lysates: prevalence of latent STAT1 in high-molecular weight complexes. Growth Factors. 1998;16(1):39–51. doi: 10.3109/08977199809017490. [DOI] [PubMed] [Google Scholar]
- Lawrence DM, Seth P, Durham L, Diaz F, Boursiquot R, Ransohoff RM, Major EO. Astrocyte differentiation selectively upregulates CCL2/monocyte chemoattractant protein-1 in cultured human brain-derived progenitor cells. Glia. 2006;53(1):81–91. doi: 10.1002/glia.20261. [DOI] [PubMed] [Google Scholar]
- Lee C, Liu QH, Tomkowicz B, Yi Y, Freedman BD, Collman RG. Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways. J Leukoc Biol. 2003;74(5):676–82. doi: 10.1189/jlb.0503206. [DOI] [PubMed] [Google Scholar]
- Lee C, Tomkowicz B, Freedman BD, Collman RG. HIV-1 gp120-induced TNF-{alpha} production by primary human macrophages is mediated by phosphatidylinositol-3 (PI-3) kinase and mitogen-activated protein (MAP) kinase pathways. J Leukoc Biol. 2005;78(4):1016–23. doi: 10.1189/jlb.0105056. [DOI] [PubMed] [Google Scholar]
- Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10) J Exp Med. 1987;166(4):1084–97. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Zhou LZ, Chaturvedi P, Babcock G, Aras S, Ransohoff RM. p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J Immunol. 1998a;161(9):4736–44. [PubMed] [Google Scholar]
- Majumder S, Zhou LZ, Chaturvedi P, Babcock G, Aras S, Ransohoff RM. Regulation of human IP-10 gene expression in astrocytoma cells by inflammatory cytokines. J Neurosci Res. 1998b;54(2):169–80. doi: 10.1002/(SICI)1097-4547(19981015)54:2<169::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA, Jacobson LP, et al. Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993;43(11):2245–52. doi: 10.1212/wnl.43.11.2245. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Sacktor N, Selnes O. Human immunodeficiency virus-associated dementia. Semin Neurol. 1999;19(2):129–50. doi: 10.1055/s-2008-1040831. [DOI] [PubMed] [Google Scholar]
- Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol. 2000;67(6):885–93. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- Miller RJ, Meucci O. AIDS and the brain: is there a chemokine connection? Trends Neurosci. 1999;22(10):471–9. doi: 10.1016/s0166-2236(99)01408-3. [DOI] [PubMed] [Google Scholar]
- Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202(1-2):13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur AG, Barbieri G, Witthuhn BA, Schindler C, et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 1993;366(6451):129–35. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- Nath A. Pathobiology of human immunodeficiency virus dementia. Semin Neurol. 1999;19(2):113–27. doi: 10.1055/s-2008-1040830. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274(24):17098–102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–35. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- Nosheny RL, Ahmed F, Yakovlev A, Meyer EM, Ren K, Tessarollo L, Mocchetti I. Brain-derived neurotrophic factor prevents the nigrostriatal degeneration induced by human immunodeficiency virus-1 glycoprotein 120 in vivo. Eur J Neurosci. 2007;25(8):2275–84. doi: 10.1111/j.1460-9568.2007.05506.x. [DOI] [PubMed] [Google Scholar]
- Oh JW, Schwiebert LM, Benveniste EN. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol. 1999;5(1):82–94. doi: 10.3109/13550289909029749. [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Hamilton TA. Cooperative interaction between interferon (IFN) stimulus response element and kappa B sequence motifs controls IFN gamma- and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J Biol Chem. 1993;268(9):6677–88. [PubMed] [Google Scholar]
- Ohmori Y, Hamilton TA. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol. 1995;154(10):5235–44. [PubMed] [Google Scholar]
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood. 2001;97(2):352–8. doi: 10.1182/blood.v97.2.352. [DOI] [PubMed] [Google Scholar]
- Popik W, Hesselgesser JE, Pitha PM. Binding of human immunodeficiency virus type 1 to CD4 and CXCR4 receptors differentially regulates expression of inflammatory genes and activates the MEK/ERK signaling pathway. J Virol. 1998;72(8):6406–13. doi: 10.1128/jvi.72.8.6406-6413.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23(2):96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- Robichaud GA, Poulin L. HIV type 1 nef gene inhibits tumor necrosis factor alpha-induced apoptosis and promotes cell proliferation through the action of MAPK and JNK in human glial cells. AIDS Res Hum Retroviruses. 2000;16(18):1959–65. doi: 10.1089/088922200750054684. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. J Neurochem. 2003;86(5):1057–71. doi: 10.1046/j.1471-4159.2003.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders VJ, Pittman CA, White MG, Wang G, Wiley CA, Achim CL. Chemokines and receptors in HIV encephalitis. Aids. 1998;12(9):1021–6. [PubMed] [Google Scholar]
- Sasseville VG, Smith MM, Mackay CR, Pauley DR, Mansfield KG, Ringler DJ, Lackner AA. Chemokine expression in simian immunodeficiency virus-induced AIDS encephalitis. Am J Pathol. 1996;149(5):1459–67. [PMC free article] [PubMed] [Google Scholar]
- Schutze S, Wiegmann K, Machleidt T, Kronke M. TNF-induced activation of NF-kappa B. Immunobiology. 1995;193(2-4):193–203. doi: 10.1016/s0171-2985(11)80543-7. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Heidenreich KA, Meintzer MK, Dinarello CA. Role of p38 mitogen-activated protein kinase in HIV type 1 production in vitro. Proc Natl Acad Sci U S A. 1998;95(13):7422–6. doi: 10.1073/pnas.95.13.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapshak P, Duncan R, Minagar A, Rodriguez de la Vega P, Stewart RV, Goodkin K. Elevated expression of IFN-gamma in the HIV-1 infected brain. Front Biosci. 2004;9:1073–81. doi: 10.2741/1271. [DOI] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Lokensgard JR, Peterson PK. U50,488 inhibits HIV-1 Tat-induced monocyte chemoattractant protein-1 (CCL2) production by human astrocytes. Biochem Pharmacol. 2003;65(1):9–14. doi: 10.1016/s0006-2952(02)01480-6. [DOI] [PubMed] [Google Scholar]
- Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JE. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993;366(6455):580–3. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, et al. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol. 2004;164(5):1557–66. doi: 10.1016/S0002-9440(10)63714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, Nath A, Kolson D, Narayan O, Buch S. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23(4):957–64. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- Thompson KA, McArthur JC, Wesselingh SL. Correlation between neurological progression and astrocyte apoptosis in HIV-associated dementia. Ann Neurol. 2001;49(6):745–52. doi: 10.1002/ana.1011. [DOI] [PubMed] [Google Scholar]
- Tomura K, Narumi S. Differential induction of interferon (IFN)-inducible protein 10 following differentiation of a monocyte, macrophage cell lineage is related to the changes of nuclear proteins bound to IFN stimulus response element and kappaB sites. Int J Mol Med. 1999;3(5):477–84. doi: 10.3892/ijmm.3.5.477. [DOI] [PubMed] [Google Scholar]
- van Marle G, Henry S, Todoruk T, Sullivan A, Silva C, Rourke SB, Holden J, McArthur JC, Gill MJ, Power C. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329(2):302–18. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE. Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J Neuroimmunol. 1997;74(1-2):1–8. doi: 10.1016/s0165-5728(96)00160-9. [DOI] [PubMed] [Google Scholar]
- Westmoreland SV, Rottman JB, Williams KC, Lackner AA, Sasseville VG. Chemokine receptor expression on resident and inflammatory cells in the brain of macaques with simian immunodeficiency virus encephalitis. Am J Pathol. 1998;152(3):659–65. [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34(3):249–70. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]