Effects of a Saturated Fat and High Cholesterol Diet on Memory and Hippocampal Morphology in the Middle-Aged Rat (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 20.
Published in final edited form as: J Alzheimers Dis. 2008 Jun;14(2):133–145. doi: 10.3233/jad-2008-14202
Abstract
Diets rich in cholesterol and/or saturated fats have been shown to be detrimental to cognitive performance. Therefore, we fed a cholesterol (2%) and saturated fat (hydrogenated coconut oil, Sat Fat 10%) diet to 16-month old rats for 8 weeks to explore the effects on the working memory performance of middle-aged rats. Lipid profiles revealed elevated plasma triglycerides, total cholesterol, HDL, and LDL for the Sat-Fat group as compared to an iso-caloric control diet (12% soybean oil). Weight gain and food consumption were similar in both groups. Sat-Fat treated rats committed more working memory errors in the water radial arm maze, especially at higher memory loads. Cholesterol, amyloid-β peptide of 40 (A_β_40) or 42 (A_β_42) residues, and nerve growth factor in cortical regions was unaffected, but hippocampal Map-2 staining was reduced in rats fed a Sat-Fat diet, indicating a loss of dendritic integrity. Map-2 reduction correlated with memory errors. Microglial activation, indicating inflammation and/or gliosis, was also observed in the hippocampus of Sat-Fat fed rats. These data suggest that saturated fat, hydrogenated fat and cholesterol can profoundly impair memory and hippocampal morphology.
Keywords: Aging, cognitive impairment, dietary effects, memory, saturated fatty acids
INTRODUCTION
In Western Society, diets have gradually changed since World War II, with an increase in total caloric intake, saturated fat, and hydrogenated fat, leading to a decrease in the healthier, unsaturated fats. As a result, obesity and obesity-related diseases have increased exponentially, especially in the United States [37,50]. Although there is a significant genetic component to obesity, data suggest that the epidemic is primarily caused by an increase in dietary fat, caloric input, and a decrease in physical activity. The obesity epidemic is second only to tobacco use for the number of health care dollars spent and as a cause of premature mortality, highlighting the importance of this issue. During the last few years, there has been an increase in awareness among scientists and it is now recognized that this lifestyle can be severely detrimental, in particular to cardiovascular function, cancer rates, and cognitive function. As an example, Zhang and collaborators [66] found that high cholesterol intake is strongly correlated with poor performance in cognitive tests on school children (6−16 years of age). Other studies demonstrated that greater intake of cholesterol and saturated fat increased the risk for impaired cognitive function in middle-aged or aged populations [18,33,42,58]. Although we are aware of the relative risks of high cholesterol intake, we still do not know the biological mechanisms behind its actions in the central nervous system (CNS).
The CNS accounts for only 2% of body mass, but contains almost a quarter of the unesterified cholesterol in the body [16]. Moreover, the input of cholesterol into the CNS comes almost entirely from in situ synthesis, and there is currently little evidence for the net transfer of sterol from plasma into the adult brain. However, changes in cholesterol balance may cause alterations in sterol recycling and expression of apolipoprotein E (ApoE) in the brain, leading to detrimental effects on neurons and glial cells [16]. For example, it is well known that the sterol flux across the CNS is elevated in patients with Alzheimer's disease (AD), and correlates with degree of dementia [38]. However, the biological mechanisms for the consequences of a diet rich in cholesterol and saturated fatty acids upon brain markers are not well known. Even correlations of the high-fat diet with brain markers are not yet well described. Although transgenic mouse models (using the Tg2576 mouse model for AD) have shown that a high cholesterol diet gives rise to increased levels of brain cholesterol and amyloid plaque load (e.g., [51]), these mouse studies have not included a methodical examination of the consequence of this pathological change on learning and memory. However, other studies do suggest that a high fat diet in transgenic mice gives rise to decreased spatial memory function, although the mechanism for these alterations is unknown [36]. Furthermore, studies in transgenic mice that were fed a high-fat diet demonstrate that the mice were prone to atherosclerosis, which may provide one potential biological mechanism for cholesterol-induced cognitive impairment [36].
Cognitive studies have been performed on rats for a number of years in terms of the influence of specific diets on different aspects of memory (e.g., [5]). It has further been documented that malnutrition or altered lipid composition gives rise to enduring cognitive impairment when given during development [56]. In a series of elegant studies [26,27,59–62], it has been demonstrated that young mature rats, fed nutritionally adequate diets but with high levels of fat, were impaired on the radial arm maze, the Hebb-Williams complex maze series, and a variable-interval delayed alternation task. The diet in these studies contained different compositions of saturated or poly-unsaturated fats, but did not contain pure cholesterol, as utilized in our experiments here. Further, since those studies were performed in young adult rats, the consequence of this type of diet in older animals is as yet unknown. Based on these earlier findings, we have chosen to explore the effects of a high cholesterol/high saturated fatty acid diet (“Sat-Fat”) upon working and reference memory, as well as brain biochemistry and morphology in middle-aged male rats. The purpose for studying the effects of these diets on middle-aged instead of young rats was to explore whether the rate of age-related decline in memory function might be exacerbated by a diet rich in cholesterol and saturated fatty acids, since the decline in memory function is known to commence between 16 and 18 months of age in male rats. The aims of this study were to 1) explore if the Sat-fat diet is detrimental to memory functions compared to a diet consisting of the same amount (12%) of soybean oil (unsaturated fatty acids), and 2) to determine if the Sat-Fat diet alters hippocampal morphology.
MATERIALS AND METHODS
Dietary composition
The control diet contained 12% soybean oil (unsaturated fatty acids), and in exchange for the soybean oil, the experimental diet contained 2% cholesterol and 10% hydrogenated coconut oil, as used in previous studies (Table 1 [20,52]).
Table 1.
Diet composition. The soybean oil (12%) in control diet was replaced with 2% cholesterol + 10% hydrogenated coconut oil in the Sat-Fat group
| Ingredient | Control | Sat-Fat |
|---|---|---|
| Corn starch (CS) | 20.27% | 20.27% |
| Casein | 14.0% | 14.0% |
| Dextrinized corn starch | 15.5% | 15.5% |
| Sucrose | 10.0% | 10.0% |
| Soybean oil | 12.0% | 0 |
| Hydrogenated coconut oil | 0 | 10.0% |
| Mineral mix | 3.5% | 3.5% |
| Vitamin mix* | 1.0% | 1.0% |
| L Cysteine | 0.18% | 0.18% |
| t-Butyl hydroquinone | 0.0008% | 0.0008% |
| Alphacel | 23.55% | 23.55% |
| Cholesterol | 0.00% | 2.00% |
Animals
Male Fischer 344 rats were used (Harlan Laboratories, Illinois). They were 16 months at the beginning of the experiment, and 18 months at the end of the experiment. The animals were given one week to acclimate to the vivarium, after which they were weight-matched and randomly divided into two groups, receiving control or experimental diet. Ten animals were included in each of the different diet groups, of which 7 in each group were randomly selected for testing in the radial arm maze (see below), to ensure that testing itself did not interfere with potential diet effects on brain parameters. Weight and food intake was tested weekly throughout the experiment. All studies using experimental animals were carried out according to the regulations of the Office of Laboratory Animal Welfare (OLAW) and approved by the Medical University of South Carolina Institutional Care & Use Committee.
Behavioral testing
Working and reference memory were assessed simultaneously using the 8-arm water radial maze (RAM), which has been used routinely in our laboratory and other laboratories (see [6–10,30]). The animals were tested on week 7 and 8 after initiation of the high fat diet. The maze was constructed of galvanized steel, painted black, placed in a room with salient extra-maze cues, and filled with room temperature water. Escape platforms were hidden 1 cm below the water level in the ends of 4 of the 8 arms. The testing procedure has been described in detail previously [7,8]. Briefly, each subject had platform locations that remained fixed throughout the experiment. For each trial a subject was released from the start arm and had 3 min to locate a platform. If the subject had not located the platform in 3 min, the experimenter guided it to the closest platform. Once a platform was found, the trial was over, the subject remained on the platform for 15 s, and then was returned to its heated cage for a 30 s inter-trial interval (ITI). During the ITI, the just-chosen platform was removed from the maze. A daily session consisted of this sequence of events repeated until all 4 platforms were located, resulting in 4 trials per session. Each subject received 1 session per day for 12 consecutive days. An arm entry was counted when a subject's snout reached a mark delineated on the outside of the maze (11 cm into the arm).
Error quantification
As done previously with the water RAM, Working Memory Correct (WMC), Reference Memory (RM), and Working Memory Incorrect (WMI) errors were quantified using Jarrard's definitions of orthogonal working and reference memory errors [31]. WMC errors were first and repeat entries into an arm from which a platform had been removed during that session. RM errors were first entries into an arm that never contained a platform (i.e., a reference memory arm). WMI errors were repeat entries into an arm that never contained a platform (i.e., repeat entries into a reference memory arm). To determine when errors were made, the number of WMC or WMI errors committed during each trial within each session was determined. This allowed evaluation of differences in performance of the treatment groups as trials progressed and working memory load increased.
The data were blocked into initial (days 2−6) and latter (days 7−15) phases. The first day of testing was considered a training day and was not included in analyses. Each type of error (WMC, WMI, or RM) was analyzed separately. For WMC and WMI measures, the data for each phase were analyzed with a 1-Between (treatment) × 2-Within (days and trials) repeated measures ANOVA to evaluate two-group comparisons of interest. In the case of WMC, trial 1 was not included in the analysis, because it is not possible to make a WMC error on the first trial. For RM, each phase was analyzed with a 1-Between (treatment) × 1-Within (days) repeated measures ANOVA to evaluate two-group comparisons of interest. Two-tailed tests were used for all analyses. Pearson r correlation analyses were performed to determine if there were relationships between hippocampal staining density and memory errors.
Immunohistochemistry
The spatial memory tests utilized in the experiments herein are known to be hippocampal dependent in rat; therefore, the morphological studies were focused on this brain region. The right hemisphere was sectioned on a freezing microtome (Microm HM400) at 40 microns throughout the hippocampus and collected for immunohistochemistry in Tris-buffered saline (TBS) according to our previous protocols [2]. The other hemisphere was analyzed using ELISAs and Western blots (see below). Every 12th section through the hippocampus was selected for immunohistochemistry for each stain and incubated in sodium metaperiodate [2.139 g/100 ml Tris-buffered saline (TBS)] for 20 min to block endogenous peroxidase, washed in TBS 3 × 10 min, then placed in a blocking solution of 0.4% Triton X-100 (TX-100) and 10% normal goat serum (NGS) in TBS for one hr. Sections were then placed in the primary antibody solution: 0.4% TX-100, 3% NGS, and Map 2 (1:1000, Sigma), laminin (1:1000, Stratagene) or Iba1 (1:1000, Wako Pure Chemical Industries Ltd., Osaka, Japan) in TBS. Sections were incubated overnight at room temperature in this solution. Thereafter, sections were washed in TBS 3 × 10 min and then placed in the secondary antibody solution: biotinylated goat anti-rabbit IgG (Vector) and 3% NGS in TBS for one hr. Sections were washed 3 × 10 min in TBS, and then placed into the Elite ABC reagent (Vector) for one hr. Sections were washed 3 × 10 min in imidazole-acetate buffer, pH 7.4 (0.68 g imidazole/6.8 g sodium acetate trihydrate/1 l dH2O) and were thereafter incubated with 3,3’-diaminobenzidine (DAB) and nickel ammonium sulfate (NAS) (Fisher; 0.2 g/10 ml buffer). Brain sections were washed with the imidazole-acetate buffer (pH 7.4) for 3 × 10 min and mounted on subbed slides, air dried overnight, dehydrated, coverslipped with Permount, and examined with a light microscope (Nikon Optiphot). Staining controls consisted of omission of the primary antibody. When the words “immunoreactive” or “-positive” are used in the text, this always refers to “-like immunoreactivity”, since no direct evidence for location of molecules can be obtained with indirect immunohistochemical techniques. Sections from all groups were incubated in the same bath to avoid group inter-variability in staining.
Image analysis
A quantitative analysis of immunohistochemical optical density (OD) with the Map 2 antibody was achieved with the NIH Image software package, a Scion Frame Grabber card, and a Macintosh Quadra 840 AV computer. Every 12th section was examined for Map 2 immunohistochemistry throughout the longitudinal axis of the hippocampus. This system determines the integrated optical density of an image, with 0 representing a white image and 256 representing a black image. Images were acquired with a Cohu videocamera (4990 series) coupled to a Nikon Optiphot microscope. A 10X objective was used for the images presented in the figures, except when noted otherwise. The images were captured at the same illumination intensity, and were digitized as unaltered images by the computer. The background was measured in three places where no staining was present, and the average of these measurements was subtracted from each image. This image analysis system is used extensively in our laboratory [2,29]. Image analysis data were analyzed using ANOVA with Fisher's LSD a posteriori. The threshold for statistical significance was set at p < 0.05.
Serum cholesterol, LDL, HDL, and triglycerides
Cholesterol (CHOL), triglycerides, and HDL levels in serum from both groups were examined using the SYNCHRON® Systems kits [53]. CHOL reagent was used to measure cholesterol concentration by a timed-endpoint method. In the reaction, cholesterol esterase (CE) hydrolyzes cholesterol esters to free cholesterol and fatty acids. Free cholesterol was oxidized to cholestene-3-one and hydrogen peroxide by cholesterol oxidase (CO). Peroxidase catalyzed the reaction of hydrogen peroxide with 4-aminoantipyrine (4-AAP) and phenol to produce a colored quinoneimine product. The SYNCHRON® System(s) automatically proportioned the appropriate sample and reagent volumes into the cuvette. The ratio used was one part sample to 100 parts reagent. The system monitors the change in absorbance at 520 nanometers. This change in absorbance is directly proportional to the concentration of CHOL in the sample and is used by the System to calculate and express CHOL concentration.
Triglycerides GPO reagent (SYNCHRON) was used to measure the triglyceride serum concentration by a timed endpoint method [12]. Triglycerides in the sample were hydrolyzed to glycerol and free fatty acids by the action of lipase. A sequence of three coupled enzymatic steps using glycerol kinase (GK), glycerophosphate oxidase (GPO), and horseradish peroxidase (HPO) causes the oxidative coupling of 3,5-dichloro-2-hydroxybenzenesulfonic acid (DHBS) with 4-aminoantipyrine to form a red quinoneimine dye.
The SYNCHRON® System(s) automatically proportions the appropriate sample and reagent volumes into the cuvette. The ratio used is one part sample to 100 parts reagent. The system monitors the change in absorbance at 520 nanometers. This change in absorbance is directly proportional to the concentration of triglycerides in the sample and is used by the System to calculate and express the concentration of triglycerides. The lipid concentration is expressed as mg/dl calculated from the standard curve.
Amyloid peptide and nerve growth factor analysis
Upon sacrifice, the brain was divided into left hemisphere for biochemical measurements and right hemisphere for morphology. Brain regions were extracted using the alkaline extraction method for amyloid-β (A_β_) measurements using minor modifications of previous reports [47,49]. Briefly, cortex samples were dissected and collected without contamination from the striatum, weighed and homogenized in 10 volumes of phosphate-buffered saline (PBS: Invitrogen; Carlsbad, CA) and centrifuged at 100,000 xg/1h to collect membranes. The membrane pellets were then extracted in 10 volumes of an alkaline solution containing 0.2% diethylamine (DEA; Sigma-Aldrich, St Louis, MO), 50 mM NaCl, protease inhibitor cocktail (Roche, IN) and 5 mM [O-phenanthroline (Sigma-Aldrich, MO)]. The DEA extract (100 μ_l) was neutralized in 50 mM Tris-HCl, pH 6.8, centrifuged at 100,000 xg/1h and the supernatant was diluted in the ELISA buffer (EIA) for further analysis. The levels of A_β_40 and 42 were measured using a Sandwich ELISA using kits obtained from IBL-America (Minneapolis, MN). This Sandwich ELISA uses antibodies specific for the A_β_40 (1A10) and A_β_42 (affinity purified polyclonal) ends for capture and a monoclonal antibody (12B2) against A_β residues 11−28 coupled to horseradish peroxidase for detection. The target sequence is conserved in rodents (except residue 13) and the antibody detects rodent A_β_ peptide as efficiently as human A_β_ as reported by the manufacturer. Student's t-test determined the statistical significance of changes in A_β_ measurements per unit of brain.
Hippocampal tissue was also analyzed for nerve growth factor (NGF) levels using an ELISA, as described previously in our laboratory [1]. Hippocampal tissue pieces contained all layers of the CA1/CA2 excluding the alveus and the dentate gyrus. All tissue pieces were placed in pre-weighed Eppendorf tubes, weighed, and kept in −70°C until assayed. The tissue was analyzed for NGF levels using an ELISA kit (Promega, Madison, WI). The NGF ELISA kit has been tested for specificity [1] and has less than 3% cross-reactivity with other neurotrophins. Briefly, 96-well flat-bottom NUNC-Immuno maxisorp plates were incubated with carbonate-coating buffer containing polyclonal anti-NGF antibody (Promega) overnight at 4°C. Non-specific binding was blocked by incubating the plate with 1 X “block and sample” buffer for 1 hr at room temperature. A standard curve was generated from serial dilutions of known concentrations of NGF ranging from 0 to 500 pg/ml. Dissected brain tissue was homogenized in lysis buffer supplemented with protease inhibitors according to manufacturer's recommendations. Samples were placed in coated wells and the plate was incubated for 6 h at room temperature. A secondary anti-NGF antibody raised in rat was incubated overnight at 4°C, followed by an anti-rat IgG antibody conjugated to horseradish peroxidase for 2.5 hours at room temperature. A TMB/peroxidase solution was used as the chromogen to visualize the reaction product in the plate wells. The reaction was terminated with 1N HCl. Optical density was measured at 450 nm in an ELISA plate reader (Molecular Devices SpectraMax 340 PC) (Softmax Pro version 3.1.2).
Membrane cholesterol measurements
Since brain is rich in myelin, which contains high levels of cholesterol, we measured cholesterol (Free) and its ester (Total) in the hippocampus using the Amplex red kit (Invitrogen, Carlbad, CA) as described by the manufacturer.
RESULTS
Weight and food consumption
The animals were kept on the Sat-Fat or Control diets for 8 weeks. During the 8 weeks of treatment, there were no significant differences in weight between the two groups at any time point (Fig. 1A). A summary of the diet composition is shown in Table 1. The mean increase in weight from week 1 to week 7 was 22 ± 6 grams (S.E.M.; n = 10) in the control group, and 29 ± 5 grams in the Sat-Fat diet group (n = 10). This mean weight increase was not statistically different between the two groups. The food intake was also similar between the two groups (Fig. 1B, p > 0.1). The control group had a mean food intake of 220 ± 12 grams per day (calculated by cage with 2 rats/cage) and the Sat-Fat diet group had a weekly intake of 243 ± 3 grams per cage (n = 5 per group). Thus, the Sat-Fat diet does not appear to significantly alter eating patterns or increase weight after 8 weeks of treatment.
Fig. 1.

Physiological Changes with diet. A) Final weight of animals week 8 of the experiment. No difference was found between the groups. B) Food consumption (per cage with 2 animals per cage). No difference between the two groups was found for dL food consumption. C) Serum triglycerides (mg/dL) were significantly elevated in the Sat-Fat compared to the control group. D) HDL, LDL, and total cholesterol levels (mg/dL) were also significantly elevated in the Sat-Fat group (n = 10; *** = p < 0.0001).
Cholesterol, LDL, and HDL levels
Free (FC) and total cholesterol (TC) measured in hippocampal membranes did not change significantly in the Sat-Fat diet (p = 0.89 and 0.45, respectively; Fig. 3A), consistent with other studies indicating that cholesterol does not cross the blood brain barrier and is independently regulated in the brain [16]. At the end of the experiment, the Sat-Fat group exhibited elevated total serum cholesterol levels (Sat-Fat: 122 ± 6 mg/dL, controls: 47 ± 2 mg/dL, n = 9 per group; F = 160.236; p < 0.0001), as well as HDL (59 ± 4 vs. 36 ± 1 mg/dL; F = 29.2; p < 0.0001) and LDL (63 ± 6 vs. 11 ± 2 mg/dl; p < 0.0001) levels. In addition, triglyceride levels were significantly elevated (374 ± 32 vs. 181 ± 12.7 mg/dl; F = 31.06; p < 0.0001). The Sat-Fat diet also altered the calculated LDL:HDL ratio, from 0.31 in controls to 1.07 in the Sat-Fat group. Thus, 8 weeks on this diet more than tripled the LDL:HDL ratio (p < 0.001). Interestingly, there was a significant correlation (p < 0.0006) between total serum cholesterol levels and memory errors in the control group, but not the Sat-Fat group (Fig. 3B), suggesting that serum cholesterol is related to memory performance in animals on a normal diet as well. The lack of correlation in the Sat-Fat group was most likely due to the saturation of effects of hypercholesterolemia in all Sat-Fat treated subjects resulting in a lack of variability.
Fig. 3.
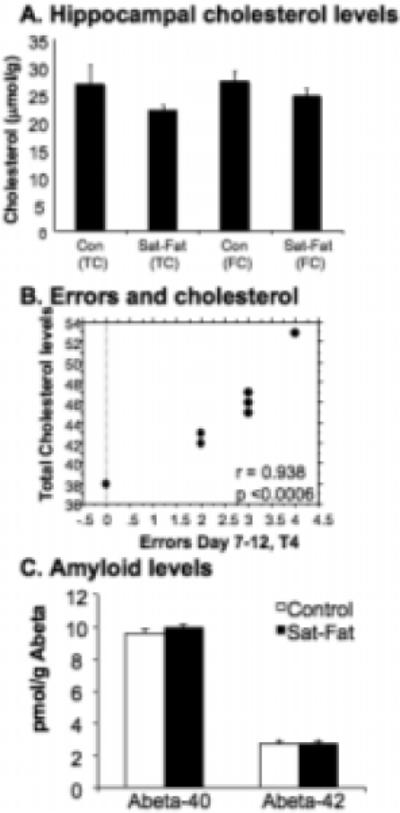
Brain Cholesterol, Behavior and A_β_ levels. A) Brain cholesterol was measured in extracts of the hippocampus as described in materials and methods and presented as micromol/g (mM). Consistent with previous studies, brain cholesterol levels are not altered despite the large increase in plasma cholesterol. Note: Total and free cholesterol measurements indicate that the levels of esterified cholesterol in the hippocampus are very small and remain unaltered with the diet. B) There was a significant correlation between WMI errors (Day 7−12, trial 4) and plasma cholesterol levels in the control group (r = 0.938; p < 0.0006). Thus, more WMI errors were found in rats with the highest cholesterol levels. The lines in the graph represent 95% confidence bands (for slope). C) No difference was found between the Sat-Fat group and animals fed a control diet in terms of A_β_40 or A_β_42 levels, as measured in cerebral cortex samples.
Behavior
During treatment week 7 and 8, animals were tested in the water version of the radial arm maze (RAM) as previously described [8]. Errors were quantified for each daily session using Jarrad's [31] orthogonal measures of working and reference memory errors, and as done previously in studies using the water RAM [8, 29]. Seven animals in each treatment group were randomly selected to undergo behavioral testing. By assessing different types of errors in the maze, we analyzed reference memory (RM), working memory incorrect (WMI), and working memory correct (WMC) errors (see Methods above). We expressed the errors as mean errors either total per day (days 2−12) or analyzed by trial within each day and collapsed across the different days (trial 2−4, where 4 represents the highest memory load). We divided the days into two blocks (day 2−6 and day 7−12) based on the learning phase and the asymptotic phase of the task, as previously reported [27]. There were no significant differences between the groups in terms of the RM portion of the water RAM testing, either when data were analyzed as errors across all the testing days [1,14; F = 1.635; p = 0.218], or when data were analyzed for days 2−6 [1,14; F = 0.112; p = 0.7429] or days 7−12 [1,14; F = 2.049; p = 0.1743] separately. However, Sat-Fat animals made more WMC (Fig. 2A) and WMI (Fig. 2C) errors than animals fed the control diet. The Sat-Fat group performed more WMC errors (all trials within each day) during Day 7−12 of testing [1,14; F = 5.671; p < 0.032] and also specifically in trial 4 (the last trial) during days 7−12, when the task had the highest working memory demand [Fig. 2B; 1,14; F = 11.07; p < 0.0050]. For WMI, errors across all days 2−12, there was a marginal difference between the groups, with the Sat-Fat group making more errors [1.14; F = 3.579; p < 0.0794]. There were no group differences for WMI for Day 2−6 [F = 0.648; p < 0.4344]. However, the Sat-Fat group made significantly more errors (collapsed across all trials within a day) during days 7−12 [1,14; F = 5.437; _p_ < 0.0353; see Fig. 2C]. Further analysis of the WMI data (Fig. 2D) demonstrated that the Sat-Fat group made more errors on the trial with the highest working memory demand (trial 4, Fig. 2D) compared to the control fed group [1,14; F = 4.564; p < 0.050], indicating that the saturated fat diet impaired the ability to handle an increased working memory load also in terms of WMI.
Fig. 2.

A and B) Working Memory Correct errors (repeat entry into an arm that previously had a platform) were higher in animals given the Sat-Fat diet on days 7−12, when all trials were collapsed (A; * = p < 0.05) and especially on trial 4, across days 7−12 (B; ** = p < 0.005). B depicts mean errors analyzed by trial for day 7−12, demonstrating that animals with a Sat-Fat diet made disproportionately more errors as memory load increased. C and D) Working Memory Incorrect errors were also significantly elevated on days 7−12 in the Sat-Fat compared to the control group (* = p <0.03) and when errors were analyzed by trials, Days 7−12 (T4 = * = p <0.05, D). Thus, middle-aged control rats were better able to handle increasing memory load than Sat-Fat rats.
Aβ and NGF levels
Since A_β_40 and A_β_42 levels increase upon cholesterol treatment of cells in culture, and these changes are believed to play an important role in memory loss in AD, their levels were analyzed by specific sandwich ELISA assays using the cortex of the left hemisphere, as described previously [47,49]. As shown in Fig. 3C, there were no differences in either cortical A_β_40 or A_β_42 levels between the Sat-Fat and the control group. ELISA analysis of hippocampal levels of NGF, a key neurotrophic factor altered upon aging and in AD, did not reveal differences between the two groups. NGF levels in the control group were 3.83 ± 0.19 ng/mg tissue in the control group (n = 10), and 3.89 ± 0.42 (n = 10) in the Sat-Fat group. Thus, it does not appear that hippocampal NGF levels were altered as a function of the Sat-Fat diet, at least not using the parameters examined here.
Immunohistochemistry
Immunohistochemistry for the dendritic marker Map2, the vascular marker laminin, and the microglial marker Iba1, was performed on every 12th hippocampal section from the right hemisphere. Map 2 is an integral protein which is used to evaluate dendritic integrity [24], and labels microtubule associated protein 2. After 8 weeks on the Sat-Fat diet, the middle-aged rats had an observable reduction in Map 2 immunoreactivity in the CA1 region of the hippocampus, as evidenced in Fig. 4A and B. Densitometry measurements of Map 2 staining in this brain region revealed a marginal decrease in Map 2 density between the two groups (p < 0.08; n = 8 per group, see Fig. 5A). Further, there was a significant correlation between CA1 Map 2 immunodensity and WMC errors in the Sat-Fat group, such that lower staining density was correlated with more errors (Fig. 5B, r = − 0.904; p < 0.0008). The image analysis represented measurements from every 12th section of the CA1 as described in the Methods section. Map 2 staining density in the dentate was also significantly correlated to WMC d8−12 errors (r = − 0.72, p < 0.04) in the Sat-Fat group but not the control group (data not shown). Immunohistochemical evaluation revealed altered morphology of hippocampal microglial cells in the Sat-Fat group compared to controls, evaluated with antibodies directed against the microglial marker Iba1 (Fig. 4C and D). The Iba1 immunostaining demonstrated increased cell body size, associated with stubbier and shorter, thicker processes in microglial cells in Sat-Fat hippocampal sections (Fig. 4C and D). This morphology has been classified as activation of microglia in prior studies. Thus, microtubule-associated proteins are reduced and microglial markers are elevated in the hippocampal formation of animals given a diet rich in saturated and hydrogenated fats for 8 weeks during middle age.
Fig. 4.
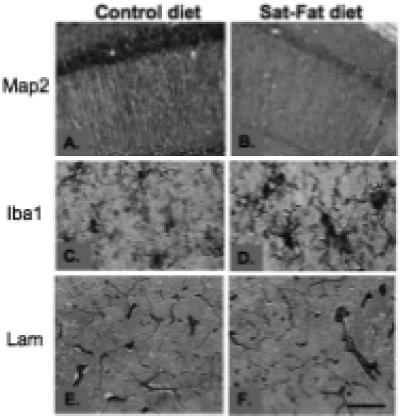
Immunohistochemical staining in the hippocampal formation. A and B) Map 2 staining demonstrated a significant difference in dendrite distribution in the hippocampal CA1, with fewer dendrites observed and weaker staining per dendrite in the Sat-Fat group (B) compared to controls (A). C and D) A microglial stain (Iba1) revealed increased activation of microglial cells in the Sat-Fat group (D) compared to controls (C). Laminin immunohistochemistry (E and F) did not reveal observable differences in terms of density or structure of microvessels in the hippocampal formation. Shown is the molecular layer of the CA1 in both micrographs. Scale bar in F represents 150 microns in A and B, 50 microns in C and D, and 200 microns E and F.
Fig. 5.
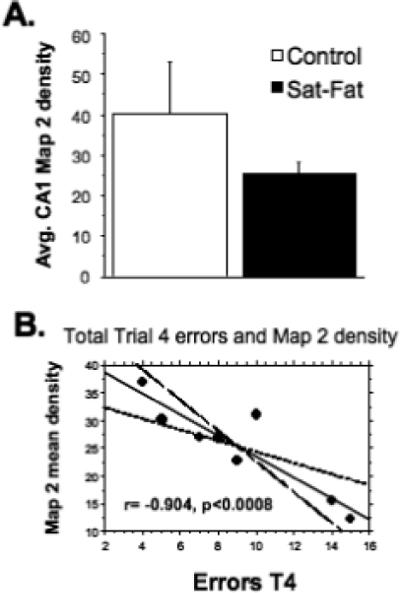
High-Fat diets reduce MAP-2 immunoreactivity. Mean Map 2 densitometry revealed a marginal effect of the Sat-Fat diet on the dendritic marker mean density (A, p < 0.08) but a highly significant correlation between WMC errors (Trial 4, collapsed over days 7−12) and Map 2 density (r = −0.904, p < 0.0008) in the Sat-Fat but not the control group. Figure 5B depicts a bivariate scattergram with regression (95% confidence bands) of WMC errors in trial 4 (days 7−12) and Map2 CA1 density respectively.
One possible mechanism by which these diets may exert their effects on memory performance is via altered perfusion or vascular function in the CNS. Therefore, we stained sections from each group with the vascular marker laminin. We did not detect observable alterations in laminin distribution between the two groups; Fig. 4E and F depict laminin immunohistochemistry in the hippocampal CA1 region from a control brain (4E) and a Sat-Fat brain (4F). Thus, more detailed vascular studies are needed to determine if other factors, such as perfusion rate or blood-brain barrier penetration, are altered by the Sat-Fat diet and therefore could lay grounds for altered memory performance in these animals.
DISCUSSION
The powerful influence of diet on brain function has been known for quite some time (for review, see [40, 48]). However, the specific mechanism by which fatty acids influence brain function and morphology are less well defined. This is the first study, to our knowledge, correlating memory loss with brain morphology in animals on a Sat-Fat/cholesterol diet. The present study demonstrated that saturated and hydrogenated fatty acids supplemented in the form of hydrogenated coconut oil plus cholesterol in the diet (12%) gave rise to alterations in behavior and brain morphology of middle-aged male rats compared to rats fed a low-cholesterol diet with equal levels of unsaturated fat (soybean oil). Our findings demonstrated a significant increase in the number of working memory errors, reduced Map 2 staining and increased microglial markers in the hippocampal formation. Further, the Sat-Fat diet gave rise to increased total cholesterol, triglyceride and LDL serum levels. The LDL:HDL ratio was tripled in the group receiving the Sat-Fat diet, compared to the control group, which received soybean oil. The fact that the saturated fat diet also increased HDL somewhat may not be surprising, since there is a significant strain-dependent difference in rats in terms of HDL and LDL/VLDL response to high fat diets (see [17]). It will be interesting in future studies to explore the lipoprotein and apolipoprotein parameters when the rats are consuming this diet, to examine, for example, if there will be alterations with aging in this response. Even though this was not the focus of the present study, it is possible that HDL in the strain utilized here (Fischer 344) has a lower cholesterol-carrying capacity.
The control group in the present study, receiving 12% of soybean oil in the diet, had significantly lower levels of serum cholesterol than the Sat-Fat group. Further, we found that serum cholesterol levels correlated with working memory errors in the control group. This further justifies the middle-aged rat as a suitable model for dietary studies and cognition, since serum cholesterol levels have also been found to correlate with memory measures in middle-aged and aged humans [21]. Further, other studies in humans have found that statins (cholesterol-lowering agents) can, at least moderately, reduce age-associated cognitive decline [4], which further supports a relationship between cholesterol and memory loss in normal aging. The fact that we did not find alterations in terms of reference memory in the present study is interesting and warrants further investigation. These findings are in parallel with previous studies from our laboratory and others, both in terms of memory loss with normal aging, and induced by pathology in transgenic mice [8,29]. The water RAM is more taxing when it comes to the working memory components rather than the reference memory, and it is possible that with prolonged exposure to the Sat-Fat diet, we would also see significant alterations in this module, similar to what we have previously observed in a mouse model for Down's syndrome [29]. However, the specific mechanisms for this effect of serum cholesterol levels on brain function are not yet fully understood. Nonetheless, the middle-aged rat appears to be an excellent model to examine these relationships, since the rat is sensitive to cholesterol-rich diets and appears to exhibit elevations in triglyceride and LDL:HDL levels on the Sat-Fat diet tested here.
One proposed mechanism for cholesterol-induced neuronal dysfunction is the increase in production and deposition of A_β_ (both A_β_40 and A_β_42 [51,54,63]). It was interesting to note in the present study that A_β_40 and A_β_42 levels were not altered by the Sat-Fat diet, and, further, did not correlate with any of the memory measures. These results differ from those obtained in the rabbit (e.g. [35] and a transgenic mouse model of AD [51]), where high fat or high cholesterol diets give rise to elevated amyloid production and plaque formation. Of course, the rats used are not transgenic and therefore express A_β_ with a different sequence, and do not deposit amyloid plaques. Moreover, the findings of increases in A_β_ upon feeding on a high-cholesterol diet remain controversial, and several authors using various strains of transgenic mice have reported that changes may or may not be seen depending on the sex and strain of the animal model [28,46]. However, we would like to note that while we do not observe large changes in A_β_ with diet, the treatment interval is rather short and small incipient regionally localized changes in A_β_ may be masked by the presence of large pools of the amyloid-β protein precursor (A_β_PP) and A_β_ in the brain. This will be the focus of future studies, since most of the hippocampus was utilized for morphology in the present study. In addition, the fact that we did not see elevated amyloid levels does not preclude abnormal processing of APP or formation of A_β_ oligomers as a cause of the decline in the middle-aged rat, as these parameters were not examined in detail. The strain of rat and other experimental parameters such as diet or physiological conditions may play an unclear role, as in contrast to these findings, other studies [34] have recently described AD-like amyloid deposits in rats fed a high-cholesterol diet. These investigators found that rats fed this diet exhibited increased hippocampal cholesterol biosynthesis and impaired long-term potentiation (LTP), and proposed that biological cholesterol homeostasis regulation may play a key role in the deficits observed in the animals fed a high cholesterol diet. They also found that cholesterol imbalance may cause hippocampal alteration of neural processes and the appearance of tau protein pathology in rats, substantiating our findings in this study where we found altered Map 2 distribution in the Sat-Fat group. Map 2 is a microtubule associated protein which has been shown to be decreased in animal models of AD (see [24]) and is displaced in the hippocampal formation of patients with AD [22]. The distribution of this protein appears to have a bearing on integrity of hippocampal neurons, in particular the CA1-CA3 region. Interestingly, the Map 2 staining density correlated with memory errors in the present study, suggesting indeed that this hippocampal pathology is related to memory function. It is possible that the failure to observe changes in A_β_ is due to the strain and/or sex of the animals used in this study, and possibly the time and extent of treatment. Nevertheless, failure to observe these changes indicates that A_β_ accumulation may be a secondary change that occurs after inflammatory changes that influence the neuronal activity leading to behavior deficits. These findings are also consistent with the failure to see significant changes in NGF levels, which has been previously correlated with the extent of cognitive deficits in AD patients and aging rats (see [29,35]).
The Sat-Fat diet gave rise to altered microglial morphology in the hippocampal formation, as evidenced by Iba1 immunohistochemistry, in the present study. Other investigators have previously demonstrated that cholesterol and its break down products can activate microglial cells in the brain and in tissue culture experiments (e.g. [15,55]). In one study, investigators found that individuals with dementia combined with heart disease had elevated microglial activation in their brains, compared to those with only a dementia diagnosis, suggesting that the cholesterol levels recorded could directly be connected to the microglial activation [55]. Further, studies have shown that LDL and other scavenger receptor (SR) ligands can affect microglia's ability for amyloid peptide uptake, perhaps interfering with removal of amyloid in individuals with AD [45]. Even though a connection between high-fat diets and microglial activation has thus been shown, the continued pathological process towards memory loss and hippocampal neuropathology has not been established. Future studies will include more in-depth examination of microglial activation and other inflammatory cascades in the brain of Sat-Fat treated rats.
Another possible mechanism for the hippocampal pathology observed is an interaction between growth factor systems and the Sat-Fat diet. Other investigators have demonstrated that high-fat diets give rise to lower levels of brain-derived neurotrophic factor (BDNF) in the hippocampal formation [41] and in cerebral cortex [64]. BDNF is essential for learning and memory processes (e.g. [65]), and has also been shown to have direct anti-inflammatory effects in the CNS (e.g. [32]). Conversely, Tanaka and collaborators [57] found that lipopolysaccharide injection into the hippocampus reduced BDNF levels and led to memory impairment without actual neuronal loss in the hippocampus, further suggesting a direct connection between BDNF and inflammatory processes in this brain region. Interestingly, we did not find alterations in hippocampal NGF levels in the Sat-Fat treated rats. However, future studies will focus on BDNF levels since this growth factor is more important for normal hippocampal function and has been shown to be involved in hippocampal-dependent learning as well as regulation of appetite and food intake [25].
The question still remains how circulating cholesterol is able to affect brain function, since cholesterol does not pass the blood-brain barrier (e.g. [11]). Consistently, the behavior changes observed are not accompanied by detectable changes in the levels of cholesterol in the hippocampus. However, we cannot rule out cholesterol-specific effects in the brain induced by dietary cholesterol. Some cholesterol is known to be transcytosed to the brain, which may lower endogenous synthesis or increase turnover to maintain homeostasis [14,16]. This may result in reduced synthesis of isoprenoids, which are implicated in inflammation as well as amyloid production [67]. It is also possible that the distribution of cholesterol in the brain will change in response to high cholesterol diets although overall homeostasis is maintained, as exemplified by a report showing that cholesterol in neurons is increased in high fat diets [23]. Moreover, side-chain oxidized cholesterol metabolites such as 24S-hydroxycholesterol and 27-hydroxycholesterol are able to pass the blood-brain barrier [11] and a significant net uptake of 27-hydroxycholesterol by the brain from the circulation has been described. These investigators also found that patients with AD have increased brain levels of 27-hydroxycholesterol, which may affect the production of A_β_ in the brain. The levels of 27-hydroxycholesterol in the circulation were correlated with the levels of cholesterol and others [11,39] have proposed that the flux of 27-hydroxycholesterol into the brain could be the missing link between hypercholesterolemia and AD. As mentioned in the introduction, studies in transgenic mice demonstrated that mice on a high-fat diet were prone to atherosclerosis, which may provide one potential biological mechanism for cholesterol-induced cognitive impairment [36]. Based on these studies, we examined the distribution of blood vessels in the hippocampus of both groups, utilizing the basal membrane marker laminin. As can be seen in Fig. 4, there were no obvious differences in the vascular tree between the two groups, although it cannot be excluded that microcirculation may be affected. Perfusion studies and more detailed vascular examination utilizing specific markers for blood-brain barrier integrity etc. would further elucidate the role of vascularization in the observed effects.
Recent data from human and animal studies demonstrate that it is not simply a high-fat diet that is damaging for the brain, but that the type of fatty acids that are ingested matters greatly (e.g. [27]). For example, Baran and colleagues [3] found that a high-fat versus low-fat diet in itself did not render dendritic alterations in the hippocampal formation of rats, but when the high-fat diet was combined with stress they found significant atrophy of CA3 dendrites. In the present study, we tested isocaloric diets, with the same fatty acid content (12%), which consisted of either soybean oil (unsaturated fatty acids) or hydrogenated coconut oil (saturated and hydrogenated fatty acids). In continued studies, we will explore whether saturated or hydrogenated fatty acids are more damaging to memory and hippocampal integrity, since the hydrogenated coconut oil here contains both types of fatty acids. The degree of saturation of fatty acids and the position of the first double bond in essential fatty acids are definitely the critical factors determining the effect of dietary fats on the risk of AD, with unsaturated fats and n-3 double bonds conferring protection and an overabundance of saturated fats or n-6 double bonds increasing the risk (see [13]). The hydrogenated fatty acids have received significant attention in recent years, due to the fact that these artificial oils significantly increase the LDL:HDL ratio, a feature associated with a strongly increased risk of both brain and cardiovascular system disease [43, 44]. However, there have been no systematic animal studies performed on these fatty acids to scientifically predict their long-term effects on the human brain. Meanwhile, the American Food and Drug Administration (FDA) has demanded full disclosure of fatty acid content in all foods as of January 2006 [19], and the debate over hydrogenated oils (“trans-fats”) will surely continue until more definite data are obtained.
In conclusion, the present study is the first to demonstrate significant effects of hydrogenated coconut oil on both memory function and hippocampal morphology in the middle-aged rat. Importantly, the serum levels of cholesterol correlated significantly to working memory errors, suggesting that the older rat may be a suitable model for examining diet-related effects of different fatty acids on memory. Further studies will be necessary to determine the biological mechanisms for these significant dietary effects on brain function, and perhaps help in clarifying the role of multiple pathways associated with changes in plasma cholesterol levels on cognition.
ACKNOWLEDGMENTS
This work was made possible by a grant from the South Carolina Nutrition Council and by NIH RO1 AG022103. Thanks are due to Ms. Claudia Umphlet, Latasha Amisial and Shannon Taylor for expert technical assistance.
References
- 1.Albeck DS, Backman C, Veng L, Friden P, Rose GM, Granholm A. Acute application of NGF increases the firing rate of aged rat basal forebrain neurons. Eur J Neurosci. 1999;11:2291–2304. doi: 10.1046/j.1460-9568.1999.00644.x. [DOI] [PubMed] [Google Scholar]
- 2.Backman C, Rose GM, Hoffer BJ, Henry MA, Bartus RT, Friden P, Granholm AC. Systemic administration of a nerve growth factor conjugate reverses age-related cognitive dysfunction and prevents cholinergic neuron atrophy. J Neurosci. 1996;16:5437–5442. doi: 10.1523/JNEUROSCI.16-17-05437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baran SE, Campbell AM, Kleen JK, Foltz CH, Wright RL, Diamond DM, Conrad CD. Combination of high fat diet and chronic stress retracts hippocampal dendrites. Neuroreport. 2005;16:39–43. doi: 10.1097/00001756-200501190-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernick C, Katz R, Smith NL, Rapp S, Bhadelia R, Carlson M, Kuller L. Statins and cognitive function in the elderly: the Cardiovascular Health Study. Neurology. 2005;65:1388–1394. doi: 10.1212/01.wnl.0000182897.18229.ec. [DOI] [PubMed] [Google Scholar]
- 5.Bickford PC, Gould T, Briederick L, Chadman K, Pollock A, Young D, Shukitt-Hale B, Joseph J. Antioxidantrich diets improve cerebellar physiology and motor learning in aged rats. Brain Res. 2000;866:211–217. doi: 10.1016/s0006-8993(00)02280-0. [DOI] [PubMed] [Google Scholar]
- 6.Bimonte HA, Denenberg VH. Estradiol facilitates performance as working memory load increases. Psychoneuroendocrinology. 1999;24:161–173. doi: 10.1016/s0306-4530(98)00068-7. [DOI] [PubMed] [Google Scholar]
- 7.Bimonte HA, Hyde LA, Hoplight BJ, Denenberg VH. In two species, females exhibit superior working memory and inferior reference memory on the water radial-arm maze. Physiol Behav. 2000;70:311–317. doi: 10.1016/s0031-9384(00)00259-6. [DOI] [PubMed] [Google Scholar]
- 8.Bimonte HA, Nelson ME, Granholm AC. Age-related deficits as working memory load increases: relationships with growth factors. Neurobiol Aging. 2003;24:37–48. doi: 10.1016/s0197-4580(02)00015-5. [DOI] [PubMed] [Google Scholar]
- 9.Bimonte-Nelson HA, Nelson ME, Granholm AC. Progesterone counteracts estrogen-induced increases in neurotrophins in the aged female rat brain. Neuroreport. 2004;15:2659–2663. doi: 10.1097/00001756-200412030-00021. [DOI] [PubMed] [Google Scholar]
- 10.Bimonte-Nelson HA, Francis KR, Umphlet CD, Granholm AC. Progesterone reverses the spatial memory enhancements initiated by tonic and cyclic oestrogen therapy in middle-aged ovariectomized female rats. Eur J Neurosci. 2006;24:229–242. doi: 10.1111/j.1460-9568.2006.04867.x. [DOI] [PubMed] [Google Scholar]
- 11.Bjorkhem I, Heverin M, Leoni V, Meaney S, Diczfalusy U. Oxysterols and Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:43–49. doi: 10.1111/j.1600-0404.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- 12.Bucolo G, David H. Quantitative determination of serum triglycerides by the use of enzymes. Clin Chem. 1973;19:476–482. [PubMed] [Google Scholar]
- 13.Cooper JL. Dietary lipids in the aetiology of Alzheimer's disease: implications for therapy. Drugs Aging. 2003;20:399–418. doi: 10.2165/00002512-200320060-00001. [DOI] [PubMed] [Google Scholar]
- 14.Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol. 1997;138:877–889. doi: 10.1083/jcb.138.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med. 2003;198:1729–1740. doi: 10.1084/jem.20030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Doucet C, Flament C, Sautier C, Lemonnier D. Effect of an hypercholesterolemic diet on the level of several serum lipids and apolipoproteins in nine rat strains. Reprod Nutr Dev. 1987;27:897–906. doi: 10.1051/rnd:19870702. [DOI] [PubMed] [Google Scholar]
- 18.Elias PK, Elias MF, D'Agostino RB, Sullivan LM, Wolf PA. Serum cholesterol and cognitive performance in the Framingham Heart Study. Psychosom Med. 2005;67:24–30. doi: 10.1097/01.psy.0000151745.67285.c2. [DOI] [PubMed] [Google Scholar]
- 19.Eller FJ, List GR, Teel JA, Steidley KR, Adlof RO. Preparation of spread oils meeting U.S. Food and Drug Administration Labeling requirements for trans fatty acids via pressure-controlled hydrogenation. J Agric Food Chem. 2005;53:5982–5984. doi: 10.1021/jf047849+. [DOI] [PubMed] [Google Scholar]
- 20.Feoli AM, Roehrig C, Rotta LN, Kruger AH, Souza KB, Kessler AM, Renz SV, Brusque AM, Souza DO, Perry ML. Serum and liver lipids in rats and chicks fed with diets containing different oils. Nutrition. 2003;19:789–793. doi: 10.1016/s0899-9007(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 21.Foster TC. Biological markers of age-related memory deficits: treatment of senescent physiology. CNS Drugs. 2006;20:153–166. doi: 10.2165/00023210-200620020-00006. [DOI] [PubMed] [Google Scholar]
- 22.Geddes JW, Tekirian TL, Soultanian NS, Ashford JW, Davis DG, Markesbery WR. Comparison of neuropathologic criteria for the diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S99–S105. doi: 10.1016/s0197-4580(97)00063-8. [DOI] [PubMed] [Google Scholar]
- 23.Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99:438–449. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- 24.Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O. Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus. 2003;13:905–914. doi: 10.1002/hipo.10130. [DOI] [PubMed] [Google Scholar]
- 25.Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O'Rahilly S, Farooqi IS. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenwood CE, Winocur G. Learning and memory impairment in rats fed a high saturated fat diet. Behav Neural Biol. 1990;53:74–87. doi: 10.1016/0163-1047(90)90831-p. [DOI] [PubMed] [Google Scholar]
- 27.Greenwood CE, Winocur G. Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav Neurosci. 1996;110:451–459. doi: 10.1037//0735-7044.110.3.451. [DOI] [PubMed] [Google Scholar]
- 28.Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, Maeda N, Siman R, Greenberg BD, Scott RW, Flood DG. Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem. 1998;273:16576–16582. doi: 10.1074/jbc.273.26.16576. [DOI] [PubMed] [Google Scholar]
- 29.Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann Neurol. 2004;56:675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- 30.Hyde LA, Hoplight BJ, Denenberg VH. Water version of the radial-arm maze: learning in three inbred strains of mice. Brain Res. 1998;785:236–244. doi: 10.1016/s0006-8993(97)01417-0. [DOI] [PubMed] [Google Scholar]
- 31.Jarrard LE, Okaichi H, Steward O, Goldschmidt RB. On the role of hippocampal connections in the performance of place and cue tasks: comparisons with damage to hippocampus. Behav Neurosci. 1984;98:946–954. doi: 10.1037//0735-7044.98.6.946. [DOI] [PubMed] [Google Scholar]
- 32.Joosten EA, Houweling DA. Local acute application of BDNF in the lesioned spinal cord anti-inflammatory and anti-oxidant effects. Neuroreport. 2004;15:1163–1166. doi: 10.1097/00001756-200405190-00016. [DOI] [PubMed] [Google Scholar]
- 33.Kalmijn S, van Boxtel MP, Ocke M, Verschuren WM, Kromhout D, Launer LJ. Dietary intake of fatty acids and fish in relation to cognitive performance at middle age. Neurology. 2004;62:275–280. doi: 10.1212/01.wnl.0000103860.75218.a5. [DOI] [PubMed] [Google Scholar]
- 34.Koudinov AR, Koudinova NV. Cholesterol, synaptic function and Alzheimer's disease. Pharmacopsychiatry. 2003;36(Suppl 2):S107–S112. doi: 10.1055/s-2003-43055. [DOI] [PubMed] [Google Scholar]
- 35.Sparks DL. Cholesterol, copper, and accumulation of thioflavine S-reactive Alzheimer's-like amyloid beta in rabbit brain. J Mol Neurosci. 2004;24:97–104. doi: 10.1385/jmn:24:1:097. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. Am J Pathol. 2003;163:2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Bowerman S, Heber D. Health ramifications of the obesity epidemic. Surg Clin North Am. 2005;85:681–0701. doi: 10.1016/j.suc.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K, Heun R. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41:195–198. [PubMed] [Google Scholar]
- 39.Lutjohann D. Cholesterol metabolism in the brain: importance of 24S-hydroxylation. Acta Neurol Scand Suppl. 2006;185:33–42. doi: 10.1111/j.1600-0404.2006.00683.x. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP. Neuroprotective signaling and the aging brain: take away my food and let me run. Brain Res. 2000;886:47–53. doi: 10.1016/s0006-8993(00)02790-6. [DOI] [PubMed] [Google Scholar]
- 41.Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gomez-Pinilla F. Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience. 2004;123:429–440. doi: 10.1016/j.neuroscience.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 42.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology. 2004;62:1573–1579. doi: 10.1212/01.wnl.0000123250.82849.b6. [DOI] [PubMed] [Google Scholar]
- 43.Mozaffarian D, Katan MB, Ascherio A, Stampfer MJ, Willett WC. Trans fatty acids and cardiovascular disease. N Engl J Med. 2006;354:1601–1613. doi: 10.1056/NEJMra054035. [DOI] [PubMed] [Google Scholar]
- 44.National Academies Press [April 7, 2008];Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Proteins and Amino Acids (Macronutrients) doi: 10.1016/s0002-8223(02)90346-9. http://www.nap. edu/openbook.php?/isbn=0309085373&page=423, [DOI] [PubMed]
- 45.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 46.Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I. Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–643. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 47.Petanceska SS, Nagy V, Frail D, Gandy S. Ovariectomy and 17beta-estradiol modulate the levels of Alzheimer's amyloid beta peptides in brain. Exp Gerontol. 2000;35:1317–1325. doi: 10.1016/s0531-5565(00)00157-1. [DOI] [PubMed] [Google Scholar]
- 48.Peters R. Ageing and the brain. Postgrad Med J. 2006;82:84–88. doi: 10.1136/pgmj.2005.036665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinnix I, Musunuru U, Tun H, Sridharan A, Golde T, Eckman C, Ziani-Cherif C, Onstead L, Sambamurti K. A novel gamma -secretase assay based on detection of the putative C-terminal fragment-gamma of amyloid beta protein precursor. J Biol Chem. 2001;276:481–487. doi: 10.1074/jbc.M005968200. [DOI] [PubMed] [Google Scholar]
- 50.Polk SL. Definitions and demographics of obesity: diagnosis and risk factors. Anesthesiol Clin North Am. 2005;23:397–403. doi: 10.1016/j.atc.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 51.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 52.Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2001;8:890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 53.Roeschlau P, Bernt E, Gruber W. Enzymatic determination of total cholesterol in serum. Z Klin Chem Klin Biochem. 1974;12:226. [PubMed] [Google Scholar]
- 54.Sparks DL, Martin TA, Gross DR, Hunsaker JC., 3rd. Link between heart disease, cholesterol, and Alzheimer's disease: a review. Microsc Res Tech. 2000;50:287–290. doi: 10.1002/1097-0029(20000815)50:4<287::AID-JEMT7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 55.Streit WJ, Sparks DL. Activation of microglia in the brains of humans with heart disease and hypercholesterolemic rabbits. J Mol Med. 1997;75:130–138. doi: 10.1007/s001090050097. [DOI] [PubMed] [Google Scholar]
- 56.Strupp BJ, Levitsky DA. Enduring cognitive effects of early malnutrition: a theoretical reappraisal. J Nutr. 1995;125:2221S–2232S. doi: 10.1093/jn/125.suppl_8.2221S. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, Yoshida T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006;83:557–566. doi: 10.1002/jnr.20752. [DOI] [PubMed] [Google Scholar]
- 58.Teunissen CE, De Vente J, von Bergmann K, Bosma H, van Boxtel MP, De Bruijn C, Jolles J, Steinbusch HW, Lutjohann D. Serum cholesterol, precursors and metabolites and cognitive performance in an aging population. Neurobiol Aging. 2003;24:147–155. doi: 10.1016/s0197-4580(02)00061-1. [DOI] [PubMed] [Google Scholar]
- 59.Winocur G, Greenwood CE. High-fat diets impair conditional discrimination learning in rats. Psychobiol. 1993;21:286–292. [Google Scholar]
- 60.Winocur G, Greenwood CE. The effects of high fat diets and environmental influences on cognitive performance in rats. Behav Brain Res. 1999;101:153–161. doi: 10.1016/s0166-4328(98)00147-8. [DOI] [PubMed] [Google Scholar]
- 61.Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26(Suppl 1):46–49. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 62.Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory impairment in obese Zucker rats: an investigation of cognitive function in an animal model of insulin resistance and obesity. Behav Neurosci. 2005;119:1389–1395. doi: 10.1037/0735-7044.119.5.1389. [DOI] [PubMed] [Google Scholar]
- 63.Wolozin B. Cholesterol and Alzheimer's disease. Biochem Soc Trans. 2002;30:525–529. doi: 10.1042/bst0300525. [DOI] [PubMed] [Google Scholar]
- 64.Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119:365–375. doi: 10.1016/s0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
- 65.Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Hebert JR, Muldoon MF. Dietary fat intake is associated with psychosocial and cognitive functioning of school-aged children in the United States. J Nutr. 2005;135:1967–1973. doi: 10.1093/jn/135.8.1967. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Y, Suram A, Venugopal C, Prakasam A, Lin S, Su Y, Li B, Paul SM, Sambamurti K. Geranylgeranyl pyrophosphate stimulates gamma-secretase to increase the generation of Abeta and APP-CTFgamma. FASEB J. 2008;22:47–54. doi: 10.1096/fj.07-8175com. [DOI] [PMC free article] [PubMed] [Google Scholar]