Identification of ALK as the Major Familial Neuroblastoma Predisposition Gene (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 22.
Published in final edited form as: Nature. 2008 Aug 24;455(7215):930–935. doi: 10.1038/nature07261
SUMMARY
Survival rates for the childhood cancer neuroblastoma have not substantively improved despite dramatic escalation in chemotherapy intensity. Like most human cancers, this embryonal malignancy can be inherited, but the genetic etiology of familial and sporadically occurring neuroblastoma was largely unknown. Here we show that germline mutations in the anaplastic lymphoma kinase gene (ALK) explain the majority of hereditary neuroblastomas, and that activating mutations can also be somatically acquired. We first identified a significant linkage signal at the short arm of chromosome 2 (maximum nonparametric LOD=4.23 at rs1344063) using a whole-genome scan in neuroblastoma pedigrees. Resequencing of regional candidate genes identified three separate missense mutations in the tyrosine kinase domain of ALK (G1128A, R1192P and R1275Q) that segregated with the disease in eight separate families. Examination of 491 sporadically occurring human neuroblastoma samples showed that the ALK locus was gained in 22.8%, and highly amplified in an additional 3.3%, and that these aberrations were highly associated with death from disease (_P_=0.0003). Resequencing of 194 high-risk neuroblastoma samples showed somatically acquired mutations within the tyrosine kinase domain in 12.4%. Nine of the ten mutations map to critical regions of the kinase domain and were predicted to be oncogenic drivers with high probability. Mutations resulted in constitutive phosphorylation consistent with activation, and targeted knockdown of ALK mRNA resulted in profound growth inhibition of 4 of 4 cell lines harboring mutant or amplified ALK, as well as 2 of 6 wild type for ALK. Our results demonstrate that heritable mutations of ALK are the major cause of familial neuroblastoma, and that germline or acquired activation of this cell surface kinase is a tractable therapeutic target for this lethal pediatric malignancy.
Neuroblastoma is a cancer of early childhood that arises from the developing autonomic nervous system. It is the most common malignancy diagnosed in the first year of life and shows a wide range of clinical phenotypes with some patients having tumors that regress spontaneously, whereas the majority of patients have aggressive metastatic disease1. These latter neuroblastoma cases have survival probabilities of less then 40% despite intensive chemoradiotherapy, and the disease continues to account for 15% of childhood cancer mortality1,2. Tumors from patients with an aggressive phenotype often show amplification of the MYCN oncogene3, and/or deletions of chromosome arms 1p and 11q4. However, because MYCN is so aberrantly dysregulated, and no putative tumor suppressor gene at 1p and 11q has been shown to harbor inactivating mutations in more than a small percentage of cases, no tractable molecular target approaches currently exist for this disease.
Like most human cancers, a small subset of neuroblastoma cases are inherited in an autosomal dominant manner5-7. A family history of the disease is found in about 1-2% of newly diagnosed cases, with a standardized incidence ratio of 9.7 for siblings of index cases8. Neuroblastoma pedigrees show striking heterogeneity in the type of tumors that arise, with both benign and malignant forms occurring in the same family9. Familial neuroblastoma patients differ from those with sporadic disease in that they are diagnosed at an earlier age and/or with multiple primary tumors, clinical characteristics that are hallmarks of cancer predisposition syndromes. Because of the lethality of the condition prior to reproductive age, previous genetic linkage scans have been underpowered and results difficult to replicate10-12. Remarkably, neuroblastoma can occur with a spectrum of disorders related to abnormal development of neural crest derived tissues including central congenital hypoventilation syndrome and Hirschsprung disease. Missense or nonsense mutations in PHOX2B, a homeobox gene that is a master regulator of normal autonomic nervous system development, were recently shown to predispose to this rare field defect of the sympathicoadrenal lineage tissues13-15. However, PHOX2B mutations explain only a small subset of hereditary neuroblastoma, are almost exclusive to cases with associated disorders of neural crest-derived tissues, and are not somatically acquired in tumors16,17, leaving the genetic etiology for the majority of familial neuroblastoma cases unknown.
Identification of germline ALK mutations
To identify the location of a hereditary neuroblastoma predisposition gene, we performed a genome-wide scan for linkage at ∼6000 single nucleotide polymorphisms (SNPs) in 20 neuroblastoma families. Because of the rarity of the condition, the genome-wide scan included pedigrees with varying degrees of confidence of actual heritability. Eight families had three or more affected individuals of close relation (high confidence), whereas 6 families consisted of only two individuals of first-degree relation (moderate confidence), and 6 families also only consisted of two affected individuals, but of more distant relationship (low confidence). We discovered a significant linkage signal at chromosome 2p with a maximum nonparametric LOD score of 4.23 at rs1344063 in 18 of the families (two excluded due to insufficient DNA). This refined a region previously reported for one of the pedigrees studied here10. By mapping informative recombination events, we defined a predisposition locus at chromosome bands 2p23-p24 delimited by SNPs rs1862110 and rs2008535 with 104 genes including the known neuroblastoma oncogene, MYCN3,18, and the ALK oncogene located 13.2 Mb centromeric. Despite previous work showing that forced overexpression of MYCN to the murine neural crest causes neurobastoma18, resequencing of the MYCN coding region and 18 Kb of surrounding genomic DNA in probands from each linked family showed no disease-causal sequence variations.
We next focused on the anaplastic lymphoma kinase gene (ALK) because our group and others had previously identified ALK as a potential oncogene in neuroblastoma through somatically acquired amplification of the genomic locus19,20. In addition, oncogenic fusion proteins leading to constitutive activation of the ALK kinase domain occur in many human cancers including anaplastic large cell lymphoma21, inflammatory myofibroblastic tumors22, squamous cell carcinomas23, and non-small cell lung cancers24, 25. Resequencing of the 29 ALK coding exons identified three separate single base substitutions within the ALK tyrosine kinase domain in eight of the probands screened (Figure 1, Table 1). These DNA sequence alterations were not present in single nucleotide polymorphism (dbSNP; www.ncbi.nlm.nih.gov/projects/SNP/) or somatic mutation (COSMIC; www.sanger.ac.uk/genetics/CGP/cosmic/) databases, and were not detected in direct sequencing of the ALK tyrosine kinase domain in 218 normal control alleles. Each substitution was subsequently shown to segregate with the disease within each family (Figure 1). The sequence variation in FNB12 (R1275Q) appears to have been acquired de novo in the affected father, and non-paternity was excluded by analysis of inheritance of genotypes within this pedigree. There are several asymptomatic obligate carriers identified (FNB2, FNB13, FNB32, FNB52, FNB56), suggesting that the incomplete penetrance of this disease may be due to lack of the acquisition of a second hit, or alternatively spontaneous regression following malignant transformation in at least a subset of cases. Notable is the very large multiplex family (FNB52) with discordance in twins and multiple unaffected carriers that segregates a unique germline mutation (G1128A).
Figure 1. Eight neuroblastoma pedigrees with ALK mutations.

All family members with DNA available for genotyping indicated with either wild type (wt) for ALK, or with mutation in the ALK tyrosine kinase domain (R1192P, R1275Q, G1128A). Individuals affected by neuroblastoma indicated by filled symbol.
Table 1.
ALK mutations in neuroblastoma
| Mutation | cDNA Variation | Type/Frequency | Region1 | Probability Activating Mutation2 |
|---|---|---|---|---|
| G1128A | c.3383G>C | Germline (1/8) | P-Loop | 0.95 |
| R1192P | c.3575G>C | Germline (2/8) | β4 Strand | 0.96 |
| R1275Q | c.3824G>A | Germline (5/8) | Activation Loop | 0.91 |
| Somatic (8/24) | ||||
| D1091N | c.3271G>A | Somatic (1/24) | N-Terminal | 0.29 |
| M1166R | c.3497T>G | Somatic (1/24) | C-Helix | 0.79 |
| I1171N | c.3512T>A | Somatic (2/24) | C-Helix | 0.85 |
| F1174I | c.3520T>A | Somatic (1/24) | End of C-Helix | 0.92 |
| F1174L | c.3522C>A | Somatic (8/24) | End of C-Helix | 0.96 |
| F1245C | c.3734T>G | Somatic (1/24) | Catalytic Loop | 0.94 |
| F1245V | c.3733T>G | Somatic (1/24) | Catalytic Loop | 0.91 |
| I1250T | c.3749T>C | Somatic (1/24) | Catalytic Loop | 0.87 |
ALK sequence variations occurred only in the families with high or moderate degrees of confidence for harboring a predisposing allele. Six of the eight families with three or more affected individuals had ALK missense alterations. The two families that did not have ALK sequence alterations identified were each shown by us to harbor mutations in the sympathicoadrenal lineage specific PHOX2B neurodevelopmental gene14,16. Two of the six families consisting of only two affected individuals, but of first-degree relation, had ALK sequence variations. Each of these families carried the R1275Q alteration, and in FNB12 we showed that the mutation arose de novo in the affected father, whereas in FNB56 the alteration was inherited from an unaffected father (Figure 1). None of the six families with two distant relations affected with neuroblastoma showed ALK alterations, suggesting that the occurrence of an additional case of this relatively rare disease in an extended family member was likely a chance occurrence. Since there are several families who share identical mutations, we looked to see if these families shared a common haplotype around the ALK gene and showed that the affected individuals with the same mutations did not share haplotypes, arguing against a founder effect.
Because ALK functions as an oncogene in other human cancers, we predicted that the sequence variations discovered in the neuroblastoma pedigrees would result in constitutive activation. We therefore utilized a support vector machine-based statistical classifier to map the putative mutations and determine the probability that they would act as drivers of an oncogenic process26,27. Each of the germline alterations occurred at regions of the ALK kinase domain that have been shown to be major targets for cancer driver mutations in other oncogenic kinases (Table 1, Figure 2). The R1275Q mutation was present in the germline DNA of affected individuals from five pedigrees (Figure 1), and falls within the kinase activation loop in a region strongly associated with activating mutations in many different protein kinases, such as BRAF28. This amino acid substitution results in an electropositive residue being replaced by a more electronegative one, possibly mimicking activating phosphorylation events. The R1192P mutation occurred at the beginning of the β4 strand of the kinase domain, and although it is predicted to be a driver mutation with high confidence (Table 1) the mechanism for activation is not yet clear27. The G1128A was seen only in the large pedigree with affected individuals in a single generation. The variation falls at the third glycine of the glycine loop, and identical mutations of this glycine to alanine in BRAF have been shown to increase kinase activity29.
Figure 2. Germline and somatic ALK mutations.
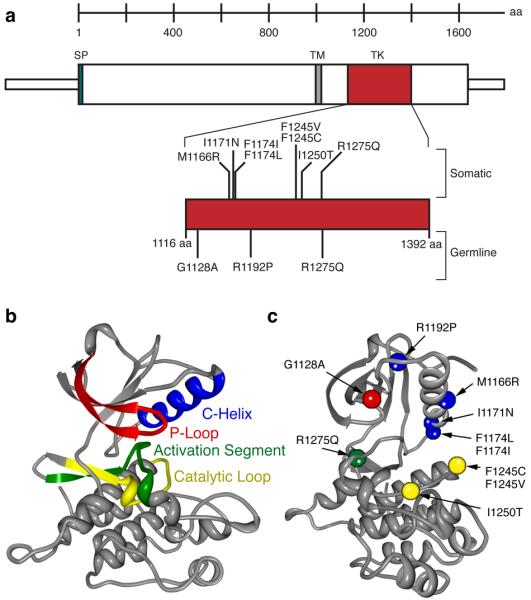
a. Schematic indicating protein structure of ALK, with mutations discovered in constitutional DNAs of familial cases (germline) and primary tumors from sporadic cases (somatic) indicated. All but one sequence alteration mapped to the tyrosine kinase domain (D1091N was just N-terminal and is not indicated here). Of the three germline mutations discovered, only the R1275Q was found in the tumor DNA samples. Conversely, the I1250T mutation discovered in the tumor set was also present in the matched germline DNA of that patient, while all of the other mutations studied here were somatically acquired. b. Homology model of wild-type ALK with each major subdomain indicated26, 27. c. ALK mutations mapped onto homology model (orientation different to show all mutations) with colors indicating subdomain in which the mutation resides (e.g. the R1275Q mutation falls within the activation segment, indicated in green).
Identification of somatic ALK mutations
Having shown that heritable mutations in the ALK tyrosine kinase domain are associated with a highly penetrant predisposition to develop neuroblastoma, we next sought to determine if ALK activation might also be somatically acquired. We examined a representative set of 491 sporadically occurring primary neuroblastoma samples acquired from children at the time of diagnosis on a 550K SNP-based microarray to assess for genome-wide copy number alterations. A total of 112 cases (22.8%) showed unbalanced gain of a large genomic region at 2p including the ALK locus (partial trisomy), and an additional 16 cases (3.3%) showed high-level focal amplification of ALK (Figure 3). Each of the high-level amplifications co-occurred with MYCN amplification and/or other regions at 2p, except one case with an ALK amplicon only. The presence of aberrant ALK copy number status (gain or amplification) was highly associated with an aggressive clinical phenotype such as metastasis at diagnosis (P<0.0001) and death from disease (_P_=0.0003).
Figure 3. Representative ALK copy number alterations in three neuroblastoma primary tumors.
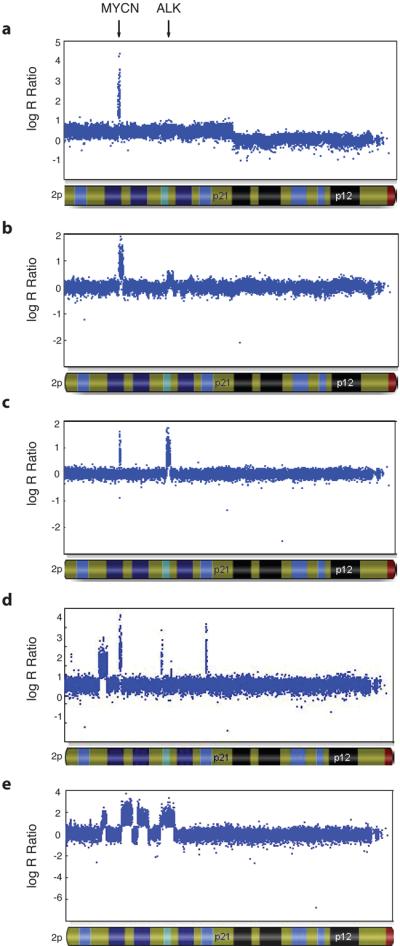
Hybridization intensity reflecting copy number for all SNPs along chromosome 2p in three primary tumors from patients with sporadically occurring disease is shown, represented on a logarithmic scale. MYCN amplification is present in all tumors. a. Regional gain (trisomy) of chromosome 2p, including the ALK locus. b. Focal gain of the ALK locus. c. Focal amplification of the ALK locus. d and e. Complex rearrangement of the 2p locus, showing various focal amplicons, including MYCN and ALK.
Because of the association of ALK gain and amplification with high-risk disease features, we next examined a subset of 167 tumor samples from high-risk patients, and 27 human neuroblastoma-derived cell lines (all from high-risk patients), for sequence alterations in the ALK tyrosine kinase domain. Fourteen of the 167 tumor (8.4%) and 10 out of 27 cell line (35.7%) samples showed single base substitutions consistent with activating mutations (Figure 2). Eight separate single base substitutions were identified, with the R1275Q mutation being the only mutation also seen in the germline DNA of the families studied. Again, none of the sequence variations discovered here were in SNP databases or were identified in our resequencing of the ALK tyrosine kinase domain in 109 control subjects (218 alleles). Mutations were equally distributed between cases with and without MYCN amplification. Only one case had a co-occurrence of an ALK mutation (F1174L) and genomic amplification of the ALK locus, and in this case the mutated allele was amplified (data not shown). Germline DNA was available for 9/14 patients with ALK mutations, and in one of these cases the sequence alteration (I1250T) was also present in the germline suggesting a hereditary predisposition that may or may not be de novo in this case.
Using the same statistical classifier employed for the germline mutations, we next showed that all but one of the sequence variations discovered in the tumor tissues were predicted to be activating mutations (Table 1), and the one that shows a low probability (D1091N) was outside of the core kinase domain. The vast majority of the somatically acquired mutations fell into either the catalytic loop or C-helix kinase domains, both frequent sites for oncogenic activating mutations (Figure 2). Catalytic loop mutants, especially I1250T, may promote oncogenesis by altering substrate binding or, more likely, alter packing of the HRD and DFG motifs towards an activated conformation30. The mutations observed in the ALK C-helix domain occurred at positions within the kinase domain previously observed to be mutated in other tumors. I1171N falls at an equivalent weakly oncogenic position in MET (M1149T)31, and the M1166R, F1174I and F1174L mutants fall at equivalent positions mutated in ErbB2 (D769, V777) and EGFR (D761, V769)32-34.
Functional consequences of ALK mutations
We had previously shown that ALK is differentially expressed in human primary neuroblastomas with higher expression generally seen in the most aggressive subset of tumors35. We confirmed that ALK is highly expressed in all but one of 20 human neuroblastoma-derived cell lines using quantitative RT-PCR. ALK expression was higher in neuroblastoma cells compared to developing fetal brain, and cell lines harboring ALK mutations (N=6) expressed the mRNA at significantly higher copy number than ALK wild-type cell lines (N=14, Figure 4a). Analysis of protein lysates from a panel of neuroblastoma cell lines showed constitutive phosphorylation of the tyrosine residue at codon 1604 in each of the cell lines harboring mutations, with weak phosphostaining in two wild-type cell lines (Figure 4b).
Figure 4. ALK is highly expressed and the kinase is phosphorylated in neuroblastoma cell lines harboring activating mutations.

a. Relative ALK expression of neuroblastoma cell lines and fetal brain is shown and was determined using the 2-ΔΔCt method49. Statistical significance was determined by unpaired T-test. b. Immunoblots showing differential ALK expression in neuroblastoma cell lines with phosphorylation of the tyrosine 1604 codon restricted to cell lines with mutations (the wild-type lines NBEC1 and NB1771 show faint phosphostaining).
To determine if ALK activation via mutation and/or amplification is functionally relevant in models of high-risk neuroblastoma, and thus might offer a tractable therapeutic target, we examined consequences of disrupting ALK signaling via knockdown of messenger RNA. We transiently transfected siRNAs directed against ALK (Dharmacon, Lafayette, CO) into 10 neuroblastoma cell lines and screened for inhibition of substrate adherent growth. We demonstrated knockdown of the mRNA and protein in all lines studied, but showed differential effect on cellular proliferation (Figure 5a-l). Each of the cells harboring ALK mutation or amplification showed profound inhibition of proliferation to ALK knockdown. In addition, 2/6 of the ALK wild-type cell lines showed significant inhibition of growth with ALK knockdown and each of these had shown weak evidence for phosphorylation at tyrosine 160 (Figure 4b), suggesting an alternative mechanism may have resulted in ALK kinase activation in these two cell lines.
Figure 5. Inhibition of growth in ALK mutated or amplified neuroblastoma cell lines.

Substrate adherent cellular growth was monitored and recorded every 30 minutes for at least 120 hours in ten neuroblastoma cell lines that were untreated, transfected with siRNAs against ALK, GAPDH (negative control), a non-targeting control (NTC, negative control) or the polo-like kinase gene (PLK1; positive control). The x-axis is time in hours after transfection, and differs based on the growth kinetics of each cell line. The y-axis is percent growth normalized to the siRNA against GAPDH. The normalized average percent growth is shown for siRNAs against GAPDH (red) and ALK (blue). In each experiment the siRNA NTC did not differ significantly from the siRNA against GAPDH, and in each case the siRNA against PLK1 showed profound growth inhibition, but only the siRNA against GAPDH and ALK are shown here for ease of visualization. Each experiment was plated in triplicate, and each experiment was repeated at least once with concordant results (data not shown). The replicates for each siRNA curve showed little variation with standard deviations <5% in all lines (except the siRNA against GAPDH in SKNDZ and NB1643 which were <10%). The cell lines, their mutation status and percent ALK mRNA knockdown at 48 hours are as follows: a. KELLY (F1174L) 78% mRNA knockdown (kd); b. SKNSH (F1174L), 21% mRNA kd; c. NB1643 (R1275Q), 48% mRNA kd; d. IMR5 (AMPLIFIED), 60% mRNA kd; e. NBEBC1 (WT), 74% mRNA kd; f. NB1771 (WT), 68% mRNA kd; g. SKNAS (WT), ALK not expressed at detectable levels; h. SKNDZ (WT), 41% mRNA kd; i. SKNBE2C (WT), 62% mRNA kd; j. NGP (WT), 86% mRNA kd. k. Summary of percentage growth inhibition with ALK siRNA knockdown by ALK mutational and allelic status; l. Immunoblot showing ALK protein knockdown at 24, 48 and 72 hours following siRNA transfection in the representative neuroblastoma cell lines KELLY (F1174L mutation and showing growth inhibition) and SKNDZ (wild-type ALK sequence and no growth inhibition).
DISCUSSION
Knudson and Strong predicted that neuroblastoma, like the analogous embryonal cancer retinoblastoma, would follow a two-hit model explaining hereditary and sporadic cases5. This model has proven to be correct for the majority of childhood and adult hereditary cancers and the susceptibility genes are typically tumor suppressors where the two hits are sequential inactivation of both alleles. Discovery of heritable mutations in oncogenes as the etiology of multiple endocrine neoplasia 1 cancers (RET), papillary renal carcinoma (MET) and gastrointestinal stromal tumors (KIT) challenged this paradigm, but it is now clear that somatically acquired duplication or amplification of the mutant allele provides the second hit36. We have now shown that heritable mutations in ALK are the cause of the majority of hereditary neuroblastoma cases, providing the first example of a pediatric cancer arising due to mutations in an oncogene. Taken together with our recent report that common variations at chromosome band 6p22 predispose to the development of sporadic neuroblastoma,37 the genetic etiology of this disease is now being defined. The discovery of highly penetrant heritable ALK mutations as the cause of hereditary neuroblastoma are of immediate relevance to patients with a family history as screening with noninvasive techniques such as ultrasonography and measurement of urinary catecholamine metabolites should likely be implemented for unaffected children carrying an ALK mutation. In addition, ongoing efforts to characterize the full spectrum of mutations in the entire ALK coding sequence, as well as determining the frequency of mutations across all neuroblastoma disease subsets, will be required to formulate genetic screening recommendations for newly diagnosed neuroblastoma patients with or without a family history.
ALK is an orphan tyrosine kinase transmembrane receptor with homology to neurotrophin receptors and the MET oncogene. Expression is restricted to the developing nervous system with a postulated role in participating in the regulation of neuronal differentiation38. It is now clear that many human cancers activate ALK signaling by creating unique oncogenic fusions of ALK with a variety of partners through chromosomal translocation events39. Previous work had shown that a substantial percentage of human-derived neuroblastoma cell lines express ALK transcripts and ALK protein 40, but no definitive role for this oncogene had been proven 19, 41-43. McDermott and colleagues recently identified ALK as a molecular target in neuroblastoma through a screen of human cancer cell lines with pharmacologic antagonists of the ALK kinase domain.44 Our current report provides the first evidence for oncogenic activation of ALK via mutation of the kinase domain, and these data provide the genetic basis for the observation of sensitization to ALK kinase inhibition. In addition, our discoveries in neuroblastoma may lead to future resequencing efforts in other malignancies, especially those where oncogenic fusion proteins have recently been discovered. The data presented here clearly establish ALK as critical neuroblastoma oncogene and should increase efforts to identify the ligand for this receptor and understand if _ALK_-mediated signaling can be activated by mechanisms other than direct mutation and/or amplification of ALK alleles. Finally, receptor tyrosine kinases provide tractable targets for pharmacologic inhibition, and this work should provide the impetus for developing therapeutic strategies aimed at inhibiting _ALK_-mediated signaling.
METHODS SUMMARY
Twenty probands with neuroblastoma and a family history of the disease were identified for study. These pedigrees were considered in three categories based on the likelihood that a highly penetrant cancer susceptibility allele was present in each family: eight pedigrees had 3 or more affected individuals (mean 4.5; range 3-7); six pedigrees contained only two affected individuals, but of first degree relation (one of these families consisted of a sibling pair affected with neuroblastoma, and a distant relation also with the disease); and six pedigrees consisted of only two affected individuals, but of second (N=1), third (N=2), or ≥ fourth (N=3) degree relationship. Because of the rarity of the condition, available individuals from all pedigrees were genotyped genome-wide using the Illumina Linkage IVb SNP panel. We used simulation to estimate the power of linkage analysis on this platform in our collection of pedigrees. Given the observed pedigree structures and disease phenotypes, we simulated marker data under a model of genetic homogeneity and autosomal dominant inheritance of the disease, and analyzed the data using an affected-only approach that does not make assumptions on the unknown disease penetrance and is thus comparable to the model-free approach we used in the actual linkage analysis. We estimated that the expected maximum lod-score attainable at a locus perfectly linked to the disease locus ranged from 1.7 to 5.6 depending on the map information content, assumed to be in the range of 0.5 to 0.9 as from our empirical data. The observed maximum lod-score of 4.23 at the chromosome 2p locus is within this range and consistent with the high information content of the markers in that genomic region. A total of 176 individuals (49 affected with neuroblastoma) were genotyped, and two families were excluded due to insufficient DNA for genome-wide genotyping. Genotype data were checked for Mendelian inconsistencies using PedStats45, and analyzed for linkage using Merlin46 and Lamp47.
Regional candidates were re-sequenced using Sanger methodology focused on all coding exons including at least 50 basepairs of surrounding intronic DNA, and 1000 basepairs of DNA immediately adjacent to the terminal coding exons. Predictions on the probability that DNA sequence alterations encode a mutant protein were performed using a support vector machine-based statistical classifier26, 27. A total of 491 primary neuroblastoma tumor samples from patients with sporadic disease, and 27 human neuroblastoma-derived cell lines were used for whole genome SNP-array analyses (550K) to determine copy number alterations. mRNA knockdown of ALK and control targets (GAPDH-negative control and _PLK1_-positive control) was achieved with a pool of four separate siRNAs against each target, and %knockdown was quantified for each experiment. siRNA knockdown effects on substrate adherent growth was quantified with the RT-CES™ microelectronic cell sensor system (ACEA, San Diego, CA)48. Whole cell lysates were collected from the neuroblastoma cell lines KELLY, NB1643, SKNSH, NBEBC1, NB1771, SKNAS, SKNDZ, SKNBE2C and NGP (IMR5 lysates not available) and from 10 nM siALK and siNTC (non targeting control siRNA) treated KELLY and SKNDZ cells at 24, 48 and 72 hours after transfection. Proteins were separated by SDS PAGE gels and immunoblotted according to standard Western blotting procedures using 1:1000 primary antibodies for ALK (Cell Signalling, #3333) and Phospho-ALK (Cell Signalling, #3341) and 1:5000 for Actin (Santa Cruz, sc-2352).
METHODS
Research subjects and samples
Families with a history of neuroblastoma in at least one other relative were eligible to participate. Only germline DNA from the neuroblastoma pedigrees was studied, as no tumor tissue from affected members was available. Sporadic neuroblastoma tumor samples with matched constitutional DNA from peripheral blood mononuclear cells were acquired from the Children's Oncology Group Neuroblastoma Tumor Bank. The Children's Hospital of Philadelphia Institutional Review Board approved this research.
Linkage Analysis
A genome-wide linkage scan was done using the Illumina Linkage IVb SNP panel. Genotype data were checked for Mendelian inconsistencies using PedStats45, and analyzed for linkage using Merlin46 and Lamp47. The genome-wide screen for linkage was performed with both maximum likelihood allele frequency estimates and model-free analyses. Since the pattern of inheritance is complex, we used a model-free approach so as not to assume any specific mode of inheritance. We used a standard threshold to declare significance in linkage analysis, that is a lod-score of >3, corresponding to a p-value of <0.0001. The p-value associated with our most significant NPL of 4.23 was 0.00001. All other regions of the genome had NPLs ≤ 2.05, or p-values > 0.001.Model-based analyses were performed for all SNPs included in the critical region under a dominant mode of inheritance, assuming four different gene frequencies (0.0001, 0.001, 0.01, and 0.1) and dominant transmission with varying penetrance of the disease across a broad range, from 0.0001 to 0.68. Model-based linkage analysis was also performed using the method implemented in LAMP, assuming a prevalence of the disease of 0.000143 (1/7000), and maximizing the lod-scores over all possible disease models (MOD score analysis). The LAMP program does not require specification of an “a priori” genetic model but only an estimate of the trait prevalence. In every analysis the critical interval was defined as the region with associated lod-scores greater than the maximum lod-score minus 3. Since linkage disequilibrium (LD) among markers is known to inflate the lod-scores from linkage analysis in the presence of missing founders, its impact on the lod-scores was assessed at the chromosome 2p critical interval. To model LD, we organized markers into clusters by means of Merlin, which uses population haplotype frequencies derived from the HapMap project (http://www.hapmap.org/). Alternatively, Merlin can search for markers for which r2 is > 0.1 and define clusters including each identified pair and the intervening markers.
DNA sequencing
Shotgun resequencing from templates generated by long PCR for an 18 kilobase region surrounding the MYCN locus was performed using a 454 Life Sciences instrument (Branford, CT) after bi-directional sequencing of the three coding exons showed no disease causal sequence variations in the neuroblastoma pedigrees. Bi-directional sequencing of ALK coding sequence (primer sequences available upon request) was performed in the following distinct sample sets: 1) constitutional DNA from the proband and an unaffected first degree relative from the twenty neuroblastoma pedigrees, with repeat sequencing of amplicons containing any DNA sequence variations and sequencing of amplicons containing confirmed variations in remaining family members; 2) 27 human neuroblastoma-derived cell line DNAs maintained at the Children's Hospital of Philadelphia (available upon request); 3) tumor DNA from 167 sporadic neuroblastomas from the Children's Oncology Group tumor bank; and 4) 109 normal constitutional DNAs from the SNP500Cancer Resource panel purchased from the Coriell Institute for Medical Research (Camden, NJ). In order to verify neuroblastoma cell line integrity, all lines were routinely genotyped (AmpFLSTR Identifiler kit; Applied Biosystems, Foster City, CA), and mycoplasma tested.
Mutation Prediction
Cancer mutant predictions and analysis were performed as described by Torkamani and Schork27. Briefly, a support vector machine was trained upon common SNPs (presumed neutral) and congenital disease causing SNPs characterized by a variety of sequence, structural, and phylogenetic parameters. Training and predictions were performed using somatic mutations occurring within and outside of the kinase catalytic core separately. The support vector machine-based method was then applied to the ALK mutants, and the probability that each mutant is a driver was computed via the support vector machine. The threshold taken for calling a SNP a driver was taken to be 0.49 for catalytic domain mutations, and 0.53 for all other mutations26. For comparison to previously observed cancer mutations, ALK mutants were mapped to positions of a catalytic core alignment generated with characteristic site motifs, and previously observed cancer mutants mapping to the same positions were noted27.
Tumor copy number analysis
Tumor samples were assayed on the Illumina Infinium™ II HumanHap550 BeadChip technology (Illumina, San Diego, CA), at the Center for Applied Genomics at the Children's Hospital of Philadelphia. A total of 750 nanograms of genomic DNA was used as input in each case, and the assay was performed and data analyzed following the manufacturers recommendations and as previously described.37
Quantitative mRNA Expression
Total RNA was isolated using RNeasy (Qiagen, Valencia, CA). First-strand cDNA was synthesized from 2 μg using the SuperScript First Strand cDNA synthesis kit (Invitrogen, Carlsbad, CA). The resultant cDNA was diluted 1:4 and 2 μL was used for each real-time PCR reaction. Each sample was run in triplicate using pre-validated TaqMan gene expression assays from Applied Biosystems (Foster City, CA) in 20 μL reactions under standard conditions and data collected using an Applied Biosystems 7900-HT sequence detection system. Baselines, thresholds and replicates were evaluated using SDS 2.3 analysis software (Applied Biosystems) and outliers manually excluded from further analysis. Relative ALK expression was determined using the 2-ΔΔCt method,49 using GAPDH as the endogenous control and using the second dCT as the lowest expressed cell line.
ALK siRNA knockdown in neuroblastoma cell lines
A total of 1-5 × 104 neuroblastoma cells were plated in triplicate overnight in antibiotic-free complete media in the 96 well RT-CES™ microelectronic cell sensor system (ACEA, San Diego, CA)48. The cells were then transiently transfected with 200 μL containing 50 nM of pooled siRNAs (four separate siRNAs per transcript targeted) against ALK (catalog #M-003103-02), GAPDH (catalog #D-001140-01-20) negative control, non targeting negative control, or PLK1 (catalog #M-003290-01) positive control (siGENOME SMARTpool siRNA, Dharmacon, Lafayette, CO) using 0.1% v/v Dharmafect I, according to the manufacturer's protocol (Dharmacon, Lafayette, CO). In brief, 35 μL of 1 μM siRNA and 35 μL of serum-free media were combined with 0.7 μL Dharmafect I in 70 μL of serum-free media and incubated for 20 minutes at room temperature, and then 560 μL of antibiotic-free complete media was added. The culture media was gently removed from the plated cells and replaced by 200 μL of fresh media containing the siRNA, mock or complete media. Cell growth was monitored continuously and recorded as a cell index (CI) every 30 minutes for a minimum of 96 hours. The “Cell Index” (CI) is derived from the change in electrical impedance as the living cells interact with the biocompatible microelectrode surface in the microplate well effectively measuring cell number, shape and adherence. Forty-eight hours after siRNA transfection, total RNA was extracted from the cells that had been plated in a parallel 96-well plate using the Qiagen (Valencia, CA) mini extraction kit, with DNAase treatment. Two hundred ng of total RNA was oligodT primed and reverse transcribed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA). ALK, HPRT, GAPDH and PPIB expression levels were measured by quantitative RT-PCR using Taqman® gene expression assays (ABI, Foster City, CA), quantified on corresponding standard curves and normalized to the geometric mean of the three housekeeping genes. Two independent experiments were performed in triplicate. Growth inhibition of the neuroblastoma cell lines was determined by comparing the siRNA against ALK growth curve to that against GAPDH at the time of maximum cell index (CImax):
%growth inhibition=(1−ClsiALK∕ClsiGAPDH)×100.
ALK and GAPDH knockdown was determined by comparing the relative ALK expression:
%knockdown=(1−ALKsiALK∕ALKsiCONTROL)×100.
The average % knockdown of ALK across all cell lines was 60% (range 21%-86%). The average % knockdown of GAPDH was 75% (range 61%-95%).
ALK protein and phosphoprotein detection
The neuroblastoma cell lines KELLY, NB1643, SKNSH, NBEBC1, NB1771, SKNAS, SKNDZ, SKNBE2C and NGP were grown in T75 flasks under standard cell culturing conditions (IMR5 lysates not available). For KELLY and SKNDZ lysates collected from the siRNA knockdown experiments, cells were plated in T25s, transfected with 10 nM siRNA (as above) and collected at 24, 48 and 72 hours after transfection. At 60-80% confluency (or the appropriate time point), the cells were collected, pelleted and washed twice with ice cold PBS. Whole cell lysates were extracted with 100uL Cell Extraction buffer (Invitrogen FNN011) containing protease inhibitors (Sigma, P-2714) and PMSF, briefly sonicated and rotated for 1 hour at 4C. After a 30 minute centrifugation at 4C, the supernatant was removed and protein quantification was performed using the Bradford method. Lysates (50 ug for siRNA experiment and 100 ug for native cell lines) were separated on 4-12% Bis-Tris gradient gels and transferred to PVDF membranes. Membranes were then incubated and washed according to the Cell Signaling Western protocol with 1:1000 ALK (Cell Signalling, #3333) and Phospho-ALK (Cell Signalling, #3341) and 1:5000 actin (Santa Cruz, sc-2352).
ACKNOWLEDGEMENTS
The authors acknowledge the families and children that participated in this research study, and the Children's Oncology Group for providing specimens. The authors thank Dr. Wendy London for providing statistical analyses related to the Children's Oncology Group tumor set, Halfdan Rydbeck for his assistance with the linkage analysis, Mariusz Wasik for technical assistance, and Drs Judy Felgenhauer, Nadine Van Roy and Carmel McConville for providing neuroblastoma pedigrees. This work was supported in part by NIH Grants K08-111733 (YPM), R01-CA78454 (JMM), R01-CA87847 (JMM), an American Society of Clinical Oncology Career Development Award (YPM), the Foerderer-Murray Fund (YPM), the Carly Hillman Fund (YPM), the Alex's Lemonade Stand Foundation (JMM), the Giulio D'Angio Endowed Chair (JMM), the Italian Neuroblastoma Foundation (LL), the Center for Applied Genomics at the Joseph Stokes Research Institute (HH), Scripps Genomic Medicine (AT, NJS), the Scripps Dickinson Scholarship (AT), and the Abramson Family Cancer Research Institute (JMM).
Footnotes
All sequence variations have been deposited to GenBank under accession numbers EU660517 to EU660527.
The authors declare no competing financial interests.
REFERENCES
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, et al. Children's Cancer Group Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. New England Journal of Medicine. 1999;341:1165–73. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 3.Schwab M, et al. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature. 1984;308:288–91. doi: 10.1038/308288a0. [DOI] [PubMed] [Google Scholar]
- 4.Attiyeh EF, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243–53. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- 5.Knudson AGJ, Strong LC. Mutation and cancer: Neuroblastoma and pheochromocytoma. American Journal of Human Genetics. 1972;24:514–522. [PMC free article] [PubMed] [Google Scholar]
- 6.Kushner BH, Gilbert F, Helson L. Familial neuroblastoma. Case reports, literature review, and etiologic considerations. Cancer. 1986;57:1887–1893. doi: 10.1002/1097-0142(19860501)57:9<1887::aid-cncr2820570931>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Maris JM, et al. Molecular genetic analysis of familial neuroblastoma. Eur. J. Cancer. 1997;33:1923–1928. doi: 10.1016/s0959-8049(97)00265-7. [DOI] [PubMed] [Google Scholar]
- 8.Friedman DL, et al. Increased risk of cancer among siblings of long-term childhood cancer survivors: a report from the childhood cancer survivor study. Cancer Epidemiol Biomarkers Prev. 2005;14:1922–7. doi: 10.1158/1055-9965.EPI-05-0066. [DOI] [PubMed] [Google Scholar]
- 9.Maris JM, Brodeur GM. In: Neuroblastoma. Cheung N-KV, Cohn SL, editors. Springer; Berlin, Heidelberg, New York: 2005. pp. 21–26. [Google Scholar]
- 10.Longo L, et al. Genetic predisposition to familial neuroblastoma: identification of two novel genomic regions at 2p and 12p. Hum Hered. 2007;63:205–11. doi: 10.1159/000099997. [DOI] [PubMed] [Google Scholar]
- 11.Maris JM, et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. 2002;62:6651–6658. [PubMed] [Google Scholar]
- 12.Perri P, et al. Weak linkage at 4p16 to predisposition for human neuroblastoma. Oncogene. 2002;21:8356–60. doi: 10.1038/sj.onc.1206009. [DOI] [PubMed] [Google Scholar]
- 13.Amiel J, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–61. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 14.Mosse YP, et al. Germline PHOX2B Mutation in Hereditary Neuroblastoma. Am J Hum Genet. 2004;75:727–30. doi: 10.1086/424530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trochet D, et al. Germline Mutations of the Paired-Like Homeobox 2B (PHOX2B) Gene in Neuroblastoma. Am J Hum Genet. 2004;74:761–4. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raabe EH, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27:469–76. doi: 10.1038/sj.onc.1210659. [DOI] [PubMed] [Google Scholar]
- 17.van Limpt V, et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene. 2004;23:9280–8. doi: 10.1038/sj.onc.1208157. [DOI] [PubMed] [Google Scholar]
- 18.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO Journal. 1997;16:2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osajima-Hakomori Y, et al. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol. 2005;167:213–22. doi: 10.1016/S0002-9440(10)62966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George RE, et al. Genome-Wide Analysis of Neuroblastomas using High-Density Single Nucleotide Polymorphism Arrays. PLoS ONE. 2007;2:e255. doi: 10.1371/journal.pone.0000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris SW, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 22.Griffin CA, et al. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59:2776–80. [PubMed] [Google Scholar]
- 23.Jazii FR, et al. Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J Gastroenterol. 2006;12:7104–12. doi: 10.3748/wjg.v12.i44.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 25.Rikova K, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Torkamani A, Schork NJ. Accurate prediction of deleterious protein kinase polymorphisms. Bioinformatics. 2007;23:2918–25. doi: 10.1093/bioinformatics/btm437. [DOI] [PubMed] [Google Scholar]
- 27.Torkamani A, Schork NJ. Prediction of cancer driver mutations in protein kinases. Cancer Res. 2008;68:1675–82. doi: 10.1158/0008-5472.CAN-07-5283. [DOI] [PubMed] [Google Scholar]
- 28.Ikenoue T, et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63:8132–7. [PubMed] [Google Scholar]
- 29.Ikenoue T, et al. Different effects of point mutations within the B-Raf glycine-rich loop in colorectal tumors on mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase and nuclear factor kappaB pathway and cellular transformation. Cancer Res. 2004;64:3428–35. doi: 10.1158/0008-5472.CAN-03-3591. [DOI] [PubMed] [Google Scholar]
- 30.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–72. doi: 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 31.Jeffers M, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A. 1997;94:11445–50. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balak MN, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 33.Lee JW, et al. ERBB2 kinase domain mutation in the lung squamous cell carcinoma. Cancer Lett. 2006;237:89–94. doi: 10.1016/j.canlet.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 34.Lee JW, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006;12:57–61. doi: 10.1158/1078-0432.CCR-05-0976. [DOI] [PubMed] [Google Scholar]
- 35.Wang Q, et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006;66:6050–62. doi: 10.1158/0008-5472.CAN-05-4618. [DOI] [PubMed] [Google Scholar]
- 36.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 37.Maris JM, et al. Chromosome 6p22 Locus Associated with Clinically Aggressive Neuroblastoma. N Engl J Med. 2008 doi: 10.1056/NEJMoa0708698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwahara T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–49. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 39.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 40.Lamant L, et al. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000;156:1711–21. doi: 10.1016/S0002-9440(10)65042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci. 2004;117:3319–29. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- 42.Miyake I, et al. Activation of anaplastic lymphoma kinase is responsible for hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene. 2002;21:5823–34. doi: 10.1038/sj.onc.1205735. [DOI] [PubMed] [Google Scholar]
- 43.Dirks WG, et al. Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer. 2002;100:49–56. doi: 10.1002/ijc.10435. [DOI] [PubMed] [Google Scholar]
- 44.McDermott U, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 45.Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–7. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- 46.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 47.Li M, Boehnke M, Abecasis GR. Joint modeling of linkage and association: identifying SNPs responsible for a linkage signal. Am J Hum Genet. 2005;76:934–49. doi: 10.1086/430277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu N, et al. Real-time monitoring of morphological changes in living cells by electronic cell sensor arrays: an approach to study G protein-coupled receptors. Anal Chem. 2006;78:35–43. doi: 10.1021/ac051695v. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]