Picomolar Amyloid-β Positively Modulates Synaptic Plasticity and Memory in Hippocampus (original) (raw)
Abstract
Amyloid-β (Aβ) peptides are produced in high amounts during Alzheimer's disease, causing synaptic and memory dysfunction. However, they are also released in lower amounts in normal brains throughout life during synaptic activity. Here we show that low picomolar concentrations of a preparation containing both Aβ42 monomers and oligomers cause a marked increase of hippocampal long-term potentiation, whereas high nanomolar concentrations lead to the well established reduction of potentiation. Picomolar levels of Aβ42 also produce a pronounced enhancement of both reference and contextual fear memory. The mechanism of action of picomolar Aβ42 on both synaptic plasticity and memory involves α7-containing nicotinic acetylcholine receptors. These findings strongly support a model for Aβ effects in which low concentrations play a novel positive, modulatory role on neurotransmission and memory, whereas high concentrations play the well known detrimental effect culminating in dementia.
Keywords: amyloid-β, synaptic plasticity, memory, hippocampus, α-7 nicotinic receptors, Alzheimer's disease
Introduction
Amyloid-β (Aβ) peptides constitute the major component of amyloid plaques in Alzheimer's disease (AD). Aβ peptides derive from cleavage of the amyloid-β precursor protein (APP) (Kametani, 2008). APP is initially cleaved by α- or β-secretases, generating large, soluble, secreted fragments (sAPPα and sAPPβ) and membrane-associated Carboxy-terminal fragments (CTFs). Aβ peptides of various lengths (e.g., Aβ42 and Aβ40) are produced after β-secretase cleavage, followed by γ-secretase cleavage. Because of the relevance of the amyloid hypothesis in AD, toxic effects of high Aβ levels have been widely investigated during the last 20 years. Abnormally high amounts of Aβ have been shown to cause synaptic and memory dysfunction (Haass and Selkoe, 2007). However, Aβ is normally produced in the brain, where the in vivo concentration in the rodent brain has been estimated to be in the picomolar range (Cirrito et al., 2003). Nonetheless, most researchers have held the view that it is just a “garbage” product of APP metabolism generated during the production of other, biologically important APP fragments, with the exception of two studies suggesting that picomolar levels of Aβ40 play a neurotrophic role in cell cultures (Yankner et al., 1990; Plant et al., 2003), and another work in which Aβ42 increased the number of newborn neurons in cultured neural stem cells (López-Toledano and Shelanski, 2004).
A positive role of Aβ in synaptic plasticity and memory in normal brain is supported by the observation that APP knock-out (KO) mice show long-term potentiation (LTP) and memory impairment (Dawson et al., 1999; Phinney et al., 1999; Seabrook et al., 1999). The knock-out approach, however, has precluded a clear assessment of the physiological role of Aβ because of the possibility that other APP fragments and APP itself might also be biologically important. For instance, studies on APP fragment function have demonstrated that the sAPP fragments may have neurotrophic properties and enhance synaptic plasticity and memory (Araki et al., 1991; Mattson, 1994; Mucke et al., 1994; Smith-Swintosky et al., 1994; Furukawa et al., 1996; Ishida et al., 1997; Meziane et al., 1998), and the intracellular CTF may regulate gene transcription, calcium signaling, synaptic plasticity, and memory (Cao and Südhof, 2001; Gao and Pimplikar, 2001; Kimberly et al., 2001; Leissring et al., 2002; Ma et al., 2007). Another important link between Aβ, synaptic plasticity, and memory has been suggested by studies in which the loss of presenilin function, the enzymatic subunit of the multicomponent γ-secretase protein complex, has been found to impair LTP and memory (Saura et al., 2004; Dewachter et al., 2006). Likewise, suppression of β-secretase function in BACE1 knock-out mice also impaired synaptic plasticity and memory (Laird et al., 2005). However, because of the diverse substrates and pathways activated by the secretases in addition to APP, it remains to be determined through what mechanism(s), Aβ or otherwise, loss of secretase function causes these effects. Because of these findings, we set out to investigate whether low amounts of Aβ42, in the picomolar range, as in the normal brain, enhance synaptic plasticity and memory.
Materials and Methods
Animals.
Mice were maintained on a 12 h light/dark cycle in temperature- and humidity-controlled rooms of the animal facility of Columbia University. Animals were killed by cervical dislocation followed by decapitation. Three- to 4-month-old male wild-type (WT) mice (C57BL/6) were obtained from a breeding colony kept in the animal facility of Columbia University. α7-KO mice and their WT littermates were obtained by crossing heterozygous animals purchased from Jackson Laboratories (#003232, B6.129S7-Chrna7/J). Mice from the 7-null mutation line were genotyped as follows: 2 mm tails from the heterozygous breedings were digested and the DNA was extracted using lysis buffer containing 1 m Tris-HCl, 0.5 m EDTA, 10% SDS, 5 m NaCl, proteinase K in dH2O. Jackson Laboratories supplied the sequence of primers used to identify either the neo-cassette of the null mutation or the wild-type allele, for use with the PCR: forward, 5′CCTGGTCCTGCTGTGTTAAACTGCTTC-3′; reverse WT(α7+), 5′-CTGCTGGGAAATCCTAGGCACACTTGAG-3′; reverse Neo(α7−), 5′-GACAAGACCGGCTTCCATCC-3′. Thermocycling conditions were as follows: 95°C for 4 min; 35 cycles of 5°C for 30 s, 56°C for 60 s, 72C for 90 s; 72°C for 10 min; store at 4°C. PCR products were run on a 2% agarose gel, using ethidium bromide UV detection of bands at 440 bp (α7+) or 750 bp (α7−).
Aβ preparation.
Aβ42 was prepared as previously described (Puzzo et al., 2005). Briefly, the lyophilized peptide (American Peptide) was suspended in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (Sigma-Aldrich) to 1 mm. HFIP was allowed to evaporate, and the resulting clear peptide film was stored at 20°C. Twenty-four hours before use, the film was added to DMSO (Sigma-Aldrich) and sonicated for 10 min. Aβ42-DMSO was diluted into the bath solution, vortexed for 30 s, and incubated at 4°C for 24 h. The biochemistry of this aged synthetic Aβ was routinely characterized using Western blot analysis in which Aβ samples were resolved by Tris-tricine PAGE under nondenaturing/nonreducing conditions, and then transferred onto a nitrocellulose membrane. After membrane incubation with the anti-human Aβ monoclonal antibody 6E10 (Covance Research Products), horseradish peroxidase chemiluminescence revealed the presence of both monomers and oligomers (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Scramble Aβ42 was purchased from AnaSpec Inc. Concentrations of Aβ42 were calculated based on the molecular weight of its monomeric peptide.
Electrophysiological measurements.
Brain slices (400 μm) from C57BL/6 mice were cut and maintained in an interface chamber at 29°C for 90 min before recording, as previously reported (Puzzo et al., 2005). The bath solution consisted of the following (in mm): 124.0 NaCl, 4.4 KCl, 1.0 Na2HPO4, 25.0 NaHCO3, 2.0 CaCl2, 2.0 MgSO4, and 10.0 glucose. The flow rate of the perfusion was 1 ml/min. The stimulating electrode, a bipolar tungsten electrode, was placed at the level of the Schaeffer collateral fibers, whereas the recording electrode, a glass electrode filled with bath solution, was placed at the level of CA1 stratum radiatum. Basal synaptic transmission (BST) was assayed by plotting the stimulus voltages against slopes of field EPSP. For LTP experiments, a 15 min baseline was recorded every minute at an intensity that evokes a response ∼35% of the maximum evoked response. LTP was induced using θ-burst stimulation (4 pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus including three 10-burst trains separated by 15 s). For posttetanic potentiation (PTP) measurements, three 10-burst trains similar to those used to produce LTP were applied in the presence of 50 μm d-APV. Mecamylamine (MCL) and α-bungarotoxin (α-BgTx) were purchased from Sigma-Aldrich.
For patch-clamp experiments, 350 μm hippocampal slices were cut with a vibratome and maintained in a submerged chamber at 29°C, perfused with artificial cerebrospinal fluid containing (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2HPO4, 25 NaHCO3, 2 CaCl2, 1.4 MgCl2, 25 glucose, pH 7.4 (95% O2, 5% CO2). Slices were permitted to recover from cutting for at least 90 min before recordings. For recordings, neurons were voltage-clamped throughout the experiment. Patch pipettes (4–6 MΩ) were pulled from thick-walled borosilicate glass tubing and filled with a solution containing (in mm): 117.5 Cs-methylsulfonate, 17.5 CsCl2, 4 NaCl, 0.1 EGTA, 10 HEPES, 5 QX-314·Cl, 4 MgATP, 0.3 Na2GTP, 10 phosphocreatine-Tris, pH adjusted to 7.3 with CsOH, osmolarity adjusted to 290 mOsm with sucrose. Currents were recorded with a Warner amplifier (PC-501A) and filtered at 1 kHz (holding potential, −70 mV). The input resistance was determined from the current at the end of a 5 mV, 10 ms hyperpolarization voltage step. To eliminate artifacts resulting from variation of the seal properties, the access resistance was monitored for constancy throughout all experiments. Synaptic input was evoked by Schaeffer collateral pathway stimulation of 150 μs pulses at a frequency of 0.1 Hz using concentric bipolar electrodes. Ten minutes of stable access resistance was required before beginning measurements of the EPSC amplitude. The amplitude was measured automatically by using the Clampfit program (version 10.1) from Molecular Devices. For the I–V experiments, 100 μm picrotoxin was added to the bath. To isolate the synaptic currents, membrane currents recorded at each membrane potential in the absence of a Schaeffer collateral stimulation were subtracted from the evoked synaptic responses. The AMPAR/NMDAR receptor ratio was calculated by dividing the amplitude of the AMPAR current measured at the peak response at −70 mV by the NMDAR current measured 30 ms after the peak at +50 mV. Miniature EPSCs were recorded at −60 mV in the presence of 1 μm tetrodotoxin. PTP was induced using the same stimulation protocol as in the extracellular recordings. All of the electrophysiological experiments were performed using polypropylene tubing, which has been shown not to alter the ratio of different Aβ fragments (Lewczuk et al., 2006).
Aβ measurements.
Brain regions were dissected, flash-frozen, homogenized, and extracted, as previously described (Citron et al., 1997), before sandwich ELISA using the mouse β-amyloid 1-42 kit from Invitrogen to quantitate Aβ42.
Infusion technique.
After anesthesia with 20 mg/kg Avertin, mice were implanted with a 26-gauge guide cannula into the dorsal part of the hippocampi (coordinates: posterior = 2.46 mm, lateral = 1.50 mm to a depth of 1.30 mm) (Paxinos, 1998). The cannulas were fixed to the skull with acrylic dental cement (Paladur). After 6–8 d, we bilaterally injected 200 pm Aβ42, or 200 pm scramble Aβ42, or vehicle in a final volume of 1 μl over 1 min (0.903 pg) through infusion cannulas that were connected to a microsyringe by a polyethylene tube. For the Morris water maze, mice were injected 20 min before performing each session and the probe trial, whereas for fear conditioning, mice received a single injection 20 min before the training. Mice were handled once a day for 3 d before behavioral experiments. During infusion, animals were handled gently to minimize stress. After infusion, the needle was left in place for another minute to allow diffusion. After behavioral testing, a solution of 4% methylene blue was infused into the cannulas. Animals were killed and their brains were removed, frozen, and then cut at −20° with a cryostat for histological localization of infusion cannulas.
Behavioral studies.
For the Morris water maze, mice were trained in two daily sessions (4 h apart), each consisting of three trials (1 min each), for 3 d. Time required to reach the hidden platform (latency) was recorded. During this time, the location of the platform was kept constant. The training was followed by four probe trials with the platform moved to test the retention of the spatial memory. The maze was divided into four quadrants. The percentage of time spent in the quadrant that previously contained the platform was recorded and analyzed with a video tracking system (HVS 2020; HVS Image). After the probe trials, visual, motor, and motivation skills were also tested using a visible platform to measure the time and the speed needed to reach a visible platform placed within the pool by means of the video tracking system. No difference in the time and the speed needed to reach the platform was observed among the different groups of mice.
For fear conditioning, mice were placed in a conditioning chamber for 2 min before the onset of a tone [conditioned stimulus (CS)] (a 30 s, 85 dB sound at 2800 Hz). In the last 2 s of the CS, mice were given a 2 s, 0.45 mA foot shock (unconditioned stimulus) through the bars of the floor. Then, the mice were left in the conditioning chamber for another 30 s. Freezing behavior, defined as the absence of movement except for that needed for breathing, was scored using Freezeview software. Contextual fear learning was evaluated 24 h after training by measuring freezing for 5 min in the chamber in which the mice were trained. Cued fear learning was evaluated 24 h after contextual testing by placing mice in a novel context for 2 min (pre-CS test), after which they were exposed to the CS for 3 min (CS test), and freezing was measured. Sensory perception of the shock was determined through threshold assessment. Briefly, the electric current (0.1 mA for 1 s) was increased at 30 s intervals by 0.1–0.7 mA. Threshold to flinching (first visible response to shock), jumping (first extreme motor response), and screaming (first vocalized distress) was quantified for each animal by averaging of the shock intensity at which each animal manifested a behavioral response of that type to the foot shock. No difference in the sensory threshold assessment was observed among different groups of mice in experiments in which fear conditioning was tested. Moreover, different groups of mice had similar exploratory behavior, as demonstrated by a similar percentage of time spent in the center compartment and the number of entries into the center compartment in the open-field test.
Statistics.
For all experiments, mice were coded by “blind” investigators with respect to treatment and genotype. Data are expressed as mean ± SEM. Statistical analysis was performed with two-way ANOVA (with multiple comparisons or with repeated measures for LTP), Student's t test (pairwise comparisons). The level of significance was set for p < 0.05.
Results
Picomolar concentrations of Aβ42 enhance hippocampal long-term potentiation
Given that extracellular Aβ levels are likely to be regulated by synaptic activity (Kamenetz et al., 2003; Cirrito et al., 2005), we speculated that Aβ is normally involved in hippocampal LTP, the dominant model of activity-dependent synaptic plasticity. However, lack of information regarding range of Aβ assemblies present in human brain places a challenge in examining the role of Aβ in normal brain. As a first step toward the understanding of the putative physiological role of Aβ, we reasoned that a straightforward strategy was to perform a dose–response curve for the effect of Aβ on LTP. As previously demonstrated (Haass and Selkoe, 2007), when hippocampal slices were perfused with an aged Aβ42 preparation at 200 nm for 20 min, we observed an impairment of LTP at the synapses between Schaeffer collateral fibers and CA1 neurons (n = 7 slices from 7 mice, two-way ANOVA F(1,17) = 27.56, p < 0.0001 compared with 12 vehicle-treated slices from 12 mice) (Fig. 1A). Surprisingly, when we perfused slices with lower concentrations of Aβ42, the first portion of the concentration–response curve showed an enhancement of LTP with a maximal effect around 200 pm (n = 6–10 slices from 6–10 mice per Aβ42 concentration) (Fig. 1A). Application of 200 pm human Aβ42 for 20 min before tetanization increased CA1-LTP (n = 10 slices from 10 mice, F(1,20) = 7.20, p = 0.014 compared with 12 vehicle-treated slices from 12 mice) (Fig. 1B). Aβ42 alone did not affect basal transmission (n = 6 slices from 6 mice, F(1,12) = 0.05, p = 0.814 compared with 8 vehicle-treated slices from 8 mice) (Fig. 1B), nor did application of Aβ42 for 20 min immediately after the tetanus (n = 5 slices from 5 mice, F(1,8) = 0.34, p = 0.573 compared with 5 vehicle-treated slices from 5 mice) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). As a control for nonspecific effects of Aβ42, a peptide consisting of scramble Aβ42 sequence failed to enhance LTP (n = 6 slices from 6 mice, F(1,14) = 0.44, p = 0.514) or baseline transmission (n = 4 slices from 4 mice, F(1,10) = 0.02, p = 0.918) (Fig. 1B). Interestingly, previous studies have reported endogenous brain concentrations of Aβ42 in the picomolar range (Rozmahel et al., 2002; Phinney et al., 2003; Pawlik et al., 2004; Yao et al., 2004; Mastrangelo et al., 2005; Schmidt et al., 2005). When we measured the levels of Aβ42 in normal mouse brain using sandwich ELISA, cerebellum, cortex, and hippocampus also showed values in the picomolar range (n = 4 mice per group) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, the concentration of 200 pm suggested by our dose–response curve is likely to approximate the endogenous levels of Aβ42.
Figure 1.
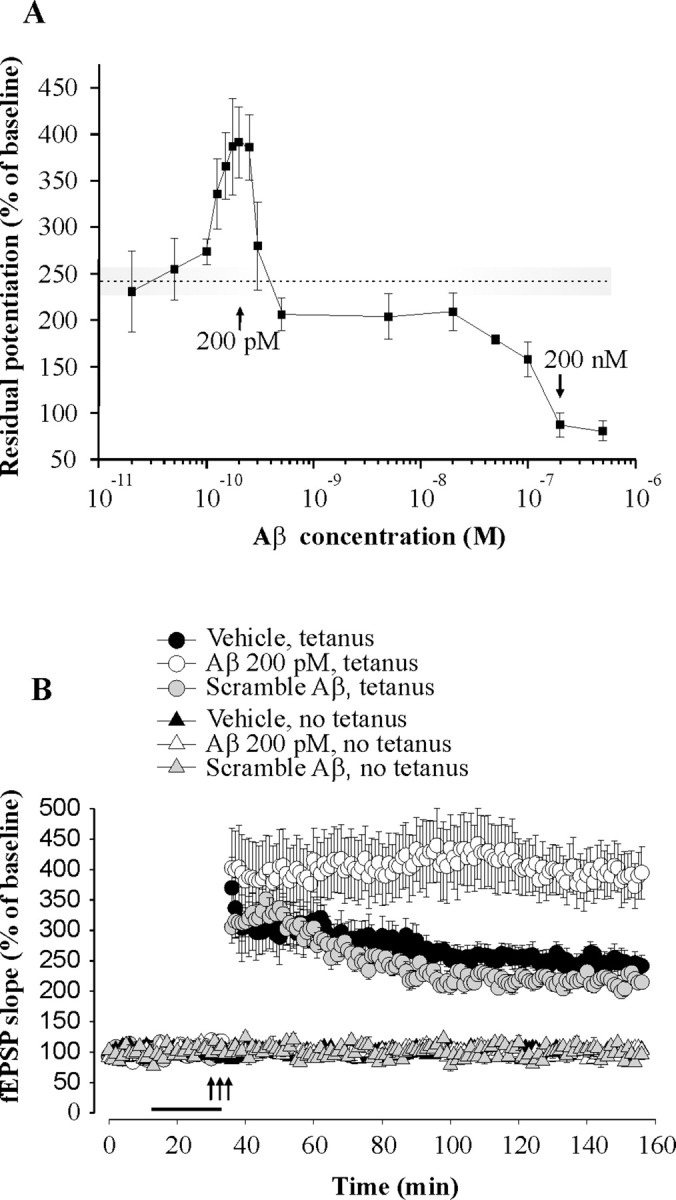
Aβ42 has two opposite effects on LTP in hippocampus. A, Concentration–response curve for the effect of Aβ42 on CA1-LTP indicating that the peptide has an enhancing effect with a peak around 200 pm, whereas it has an opposite detrimental effect above 20 nm. The dotted line and the shaded area around it correspond to the amount of potentiation and the SE range in vehicle-treated slices. The residual potentiation was calculated by averaging the last 5 min of LTP. B, Perfusion of hippocampal slices with a preparation containing human Aβ42 (200 pm), but not scramble Aβ42 (200 pm), for 20 min before a theta-burst stimulation increases LTP without affecting baseline transmission. The horizontal bar indicates the period during which Aβ was added to the bath solution. Each bar denotes the mean ± SEM in this and the following figures.
Picomolar concentrations of Aβ42 enhance hippocampal-dependent memory
Given that LTP is thought to be associated with learning and memory, we next assessed the effects of low doses of Aβ42 on memory. Cannulas were implanted bilaterally into the mouse dorsal hippocampi (Fig. 2A). After 6–8 d, animals were infused with 200 pm Aβ42, 200 pm scramble Aβ42, or vehicle, and after 20 min tested for reference and contextual fear memory.
Figure 2.

Picomolar concentrations of Aβ42 enhance hippocampal-dependent memory. A, Schematic representation showing cannulas implanted bilaterally into the dorsal hippocampi. B, Bilateral injections of human Aβ42 (200 pm), but not scramble Aβ42 (200 pm), into dorsal hippocampi, 20 min before the session improve the performance with the Morris water maze task both as the mice search for the hidden platform (C) and for the probe test. D, Bilateral injections of human Aβ42 (200 pm), but not scramble Aβ42 (200 pm), into dorsal hippocampi, 20 min before training, enhance contextual conditioning performance as the mice are exposed to the context after 24 h. The asterisks indicate statistical significance in this and the following figures.
Reference memory can be studied with the Morris water maze, a widely used spatial learning test known to require hippocampal function (Schenk and Morris, 1985). Mice were trained to find a hidden platform beneath the surface of the water. Mice that had received Aβ42 needed less time to find the platform after six sessions (Fig. 2B) compared with vehicle-infused mice. Statistical analysis revealed a significant difference in the overall performance of Aβ-treated mice compared with that of vehicle-treated animals (n = 15/14 mice; F(1,27) = 6.66, p = 0.016) (Fig. 2B). Planned comparisons of latency on each individual session revealed a significant difference between Aβ-treated mice and vehicle-treated mice in the sixth session (t(27) = 2.3, p = 0.029). Thus, we also assessed reference memory with the probe trial, another test of spatial long-term memory (Schenk and Morris, 1985). This task is performed after the sixth hidden-platform session. The platform is removed from the water and the animals are allowed to search for 60 s. The amount of time spent in each quadrant of the maze can be used to evaluate the spatial bias of an animal's search pattern. We found that Aβ-treated mice spent more time than vehicle-treated mice in the target quadrant (TQ), where the platform had been located during training (t(27) = 2.22, p = 0.035) (Fig. 2C). The time spent in the TQ by the vehicle-treated mice was also verified by the single proportion test to ensure that WT mice had learning ability. These mice had a significant probability to spend >25% of the given time in the TQ (64%, p = 0.002). Moreover, they spent significantly more time in the TQ compared with other quadrants (p = 0.022). In interleaved experiments, infusion with scramble Aβ42 did not affect mouse performance during both the six sessions with the hidden platform and the probe trials (n = 12 mice; hidden platform: t(24) = 0.42, p = 0.676; probe trials: t(24) = 0.33, p = 0.743; probability to spend >25% of the given time in the TQ: 83%, p = 0.0001; comparison TQ vs other quadrants: p = 0.026) (Fig. 2B,C). A visible platform trial performed after the probe trials did not reveal any significant difference in the time to reach the platform among the three groups during the four sessions of the task (time at the fourth session: 11.21 ± 1.79 s for Aβ42-infused mice; 12.45 ± 1.75 s for vehicle-treated mice; 12.74 ± 2.14 s for scramble-treated mice; F(2,37) = 0.19, p = 0.825) (data not shown).
Next, we studied contextual fear memory (Phillips and LeDoux, 1992), a form of associative learning for which hippocampal function is indispensable. Mice were trained to associate neutral stimuli with an aversive one. We found no difference in the freezing behavior among the three groups of mice during the training phase of the fear conditioning (F(2,48) = 0.04, p = 0.961) (Fig. 2D). However, when fear learning was assessed 24 h later by measuring freezing behavior, the absence of all movement except for that necessitated by breathing, in response to representation of the context, Aβ42-treated mice showed an enhancement of freezing (n = 19 mice, t(34) = 2.36, p = 0.024 compared with 17 vehicle-treated mice) (Fig. 2D). In contrast, scramble Aβ42 did not affect freezing (n = 15 mice; t(30) = 0.33, p = 0.743 compared with vehicle-injected mice) (Fig. 2D). Finally, when cued fear learning, a type of learning that depends on amygdala function (Phillips and LeDoux, 1992), was assessed on the same animals 24 h after contextual learning by measuring freezing in response to representation of the auditory cue within a completely different context, we found no difference between Aβ42-infused mice and vehicle-infused mice (t(34) = 0.20, p = 0.838) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), indicating that behavioral changes produced by Aβ42 were the result of a selective hippocampus-dependent effect on fear learning. Together, these experiments indicate that low doses of Aβ42 cause a long-lasting increase in synaptic strength and enhance memory.
Picomolar concentrations of Aβ42 enhance posttetanic potentiation
Our next goal was to identify the mechanism of the Aβ-induced enhancement in LTP and memory. Given that both NMDA and AMPA receptors are known to play a key role in CA1-LTP (Lisman and Raghavachari, 2006), we assessed the current-voltage (I–V) relationships for NMDA and AMPA receptor currents before and after perfusion of the slices with 200 pm Aβ42 for 20 min. Aβ did not affect either I–V relationships for the two currents or AMPA/NMDA receptor current ratios (1.06 ± 0.14 in Aβ-treated slices vs 1.02 ± 0.09 in vehicle-treated slices, n = 3 slices from 3 mice per group) (Fig. 3A–C). Moreover, Aβ did not alter amplitude (n = 6 slices from 6 mice, F(1,7) = 0.41, p = 0.542 compared with 3 vehicle-treated slices from 3 mice) (Fig. 3D) or amplitude distribution (Fig. 3E) of AMPA receptor-mediated EPSCs. These findings suggest that the enhanced LTP was not caused by postsynaptic changes in NMDA and AMPA receptor currents.
Figure 3.

Picomolar concentrations of Aβ42 enhance PTP without affecting NMDA and AMPA receptor currents. A, Example of EPSCs evoked at holding potentials ranging from −70 mV to +50 mV with a step of 20 mV. Dotted lines represent time points at which AMPA receptor (AMPAR) and NMDA receptor (NMDAR) currents were measured. B, C, Perfusion of hippocampal slices with human Aβ42 (200 pm) does not affect current–voltage relationship for NMDAR current (I-NMDAR) (B) and AMPAR current (I-AMPAR) (C). D, Perfusion of hippocampal slices with human Aβ42 (200 pm) for 20 min does not affect EPSC amplitude. Each point of the trace is the average of six consecutive recordings. The horizontal bar indicates the period of perfusion with Aβ. E, Perfusion of hippocampal slices with human Aβ42 (200 pm) does not affect EPSC amplitude distributions. Data from one cell before and after perfusion with Aβ42 are shown. F, G, Perfusion of hippocampal slices with human Aβ42 (200 pm) for 20 min does not affect mEPSC frequency (F) and amplitude (G). Each point represents the average of a 2 min period. Frequency and amplitude were normalized to the average value during the 6 min before Aβ application. The horizontal bar indicates the period of perfusion with Aβ. H, Perfusion of hippocampal slices with human Aβ42 (200 pm) in d-APV (50 μm) enhances PTP. The horizontal bars indicate the period during which Aβ and/or APV were added to the bath solution.
To further investigate the mechanism of the Aβ-induced enhancement in LTP and memory, we recorded spontaneous miniature EPSCs (mEPSCs) in the presence of tetrodotoxin (1 μm) to block sodium channels. Neither the mean frequency nor the mean amplitude of mEPSCs was affected by the treatment with 200 pm human Aβ42 for 20 min (n = 3 slices from 3 mice both for the Aβ42-treated group and the vehicle-treated group; frequency: F(1,4) = 0.40, p = 0.557; amplitude: F(1,4) = 0.44, p = 0.541) (Fig. 3F,G), suggesting that Aβ does not affect spontaneous release of neurotransmitter.
An alternative mechanism for the long-lasting enhancement in synaptic strength might be an increase of transmitter release during the tetanus. To test this possibility, we examined PTP in the presence of the NMDA antagonist d-APV (50 μm) to block LTP inductive mechanisms. PTP corresponds to a transient increase of glutamate release from the presynaptic terminal that results from brief periods of high-frequency stimulation with Ca2+ buildup within the terminal that triggers mechanisms of short-term synaptic plasticity (Zucker and Regehr, 2002). Perfusion of the slices with 200 pm human Aβ42 for 20 min increased PTP (n = 11 slices from 11 mice; t(20) = 2.18, p = 0.041) (Fig. 3H). We obtained similar results when we used a patch-clamp technique to examine PTP with and without Aβ and found no additional synaptic currents (n = 3 slices from 3 mice) (data not shown), suggesting that Aβ enhances transmitter release during the tetanus.
The enhancement of synaptic plasticity and memory induced by picomolar concentrations of Aβ42 involves α7-nicotinic ACh receptors.
Aβ is known to act through multiple targets (Small et al., 2001). Given that Aβ42 has been shown to activate α7-nicotinic ACh receptors (nAChRs) at presynaptic nerve endings of synaptosomes when administered in the low picomolar range (Dougherty et al., 2003), we tested whether α7-nAChRs are involved in the increase in PTP by picomolar levels of Aβ42. When hippocampal slices were perfused with the nonselective nAChR blocker MCL (3 μm for 20 min), the Aβ-induced PTP increase was blocked in slices that had previously shown an enhancement of PTP when perfused with Aβ alone (n = 11 slices from 11 mice; t(20) = 2.24, p = 0.036 comparing Aβ to Aβ + MCL) (Fig. 4A). In interleaved experiments, MCL alone did not affect PTP (n = 9 slices from 9 mice; t(16) = 0.55, p = 0.589) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Moreover, the selective α7-nAChR blocker, α-BgTx (0.1 μm for 20 min) also blocked the Aβ-induced PTP increase (n = 10 slices from 10 mice; t(18) = 5.59, p < 0.0001 comparing Aβ to Aβ + α-BgTx) (Fig. 4B). The slices were still capable of displaying the Aβ-induced PTP enhancement if perfused again with Aβ alone after washout of both α-BgTx and Aβ (t(18) = 3.99, p = 0.001) (Fig. 4B). Furthermore, both the PTP increase by Aβ alone and its block by α-BgTx were still present during perfusion with the GABA receptor antagonist picrotoxin (50 μm) (Aβ, 407.56 ± 26.99%; Aβ + α-BgTx, 233.57 ± 15.72%; n = 7 slices from 7 mice, t(12) = 5.29, p < 0.0001) (data not shown), suggesting that α7-nAChRs located in inhibitory interneurons (Freedman et al., 1993) are not involved in the Aβ-induced enhancement of transmitter release occurring during tetanic stimulation. Finally, in interleaved experiments, α-BgTx alone did not affect PTP (n = 7 slices from 7 mice; t(12) = 2.26, p = 0.410) (supplemental Fig. 5_b_, available at www.jneurosci.org as supplemental material).
Figure 4.
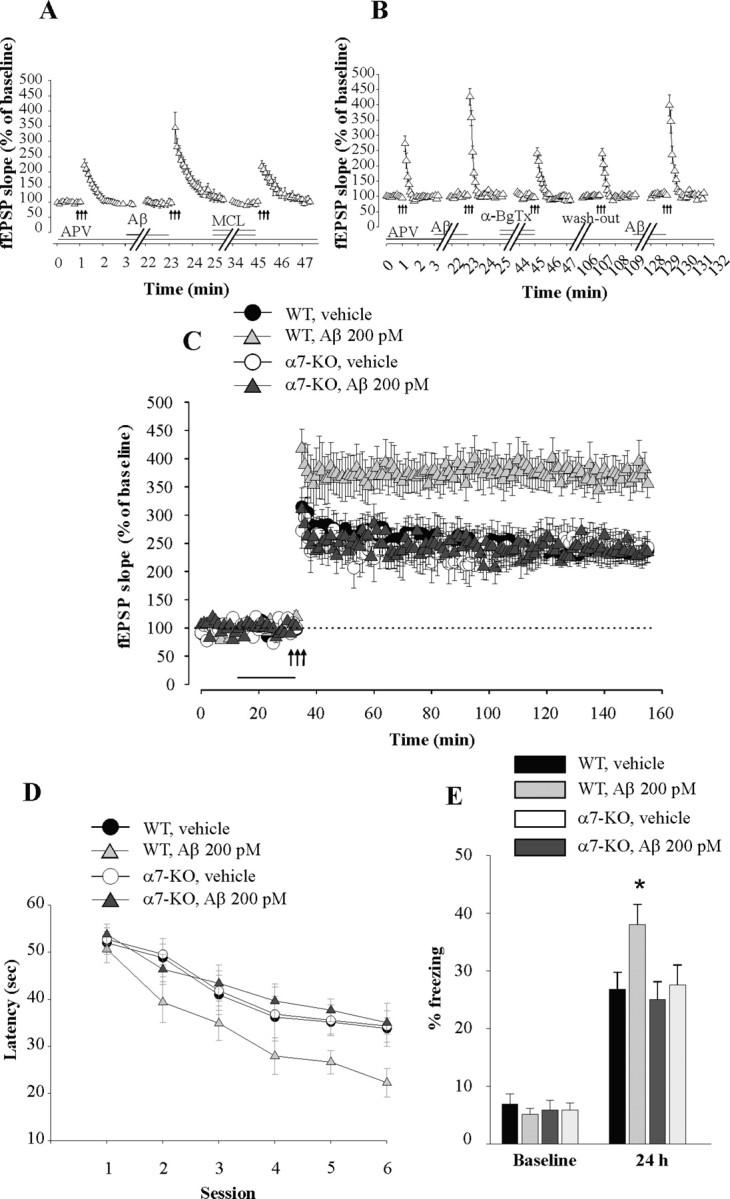
The enhancement of synaptic plasticity and memory by picomolar concentrations of Aβ42 involves α7-nAChRs. A, Hippocampal slices perfused with MCL (3 μm) concurrent with human Aβ42 (200 pm) and d-APV (50 μm) no longer display the Aβ-induced PTP enhancement. The horizontal bars indicate the period of perfusion with MCL, Aβ, and APV. B, Hippocampal slices perfused with α-BgTx (0.1 μm) concurrent with human Aβ42 (200 pm) and D-APV (50 μm) no longer display the Aβ-induced PTP enhancement. The enhancement is still present after washout of α-BgTx if slices are perfused again with Aβ42 (200 pm). The horizontal bars indicate the period of perfusion with α-BgTx, Aβ, and APV. C, Perfusion of hippocampal slices with human Aβ42 (200 pm) for 20 min before tetanus does not increase LTP in slices from α7-KO mice, whereas it still enhances potentiation in slices from WT littermates. BST was normal in the α7-KO mice (data not shown). The horizontal bar indicates the period of perfusion with Aβ. D, E, Bilateral injections of human Aβ42 (200 pm) into dorsal hippocampi, 15 min before training, do not enhance reference or contextual memory in α7-KO mice, whereas they still enhance memory in WT littermates.
To provide genetic evidence for the involvement of α7-nAChRs in Aβ42-induced synaptic plasticity increase, we performed additional experiments using α7-nAChR KO mice. When hippocampal slices from these animals were perfused with 200 pm Aβ42 for 20 min before inducing LTP, the peptide failed to enhance LTP (n = 8 slices from 8 mice, F(1,15) = 0.001, p = 0.975 compared with 9 vehicle-treated slices from 9 KO mice) (Fig. 4C). In contrast, Aβ42 was still capable of enhancing LTP in slices from wild-type littermates (n = 7 slices from 7 mice, F(1,12) = 11.42, p = 0.005 compared with 7 vehicle-treated slices from 7 wild-type mice) (Fig. 4C). Aβ42 alone without tetanus did not affect baseline transmission either in α7-KO or WT slices (106.85 ± 4.05% and 98.53 ± 4.49%, n = 4 slices from 4 mice for both groups; F(1,6) = 0.10, p = 0.757 and F(1,6) = 0.70, p = 0.433) (data not shown).
To determine whether the effect of Aβ42 on memory shares the same molecular mechanism as that on synaptic plasticity, we also sought to demonstrate that the enhancement of memory by picomolar levels of Aβ42 involves α7-nAChRs. Hippocampal infusion of 200 pm Aβ42 failed to enhance reference memory in α7-KO mice (n = 10 mice vs 9 vehicle-treated KO mice; t(17) = 0.15, p = 0.881 at the six hidden sessions) (Fig. 4D), whereas it still increased memory in WT littermates (n = 10 mice vs 9 vehicle-treated WT mice, t(17) = 2.40, p = 0.028) (Fig. 4D). Probe trials also did not show an increase in memory in α7-KO mice, whereas WT-littermates still showed an increase in the time spent in the TQ (data not shown). Similarly, contextual fear memory in α7-KO mice was not enhanced (n = 7 mice; t(12) = 0.54, p = 0.599 compared with 7 vehicle-treated KO mice) (Fig. 4E), whereas WT-littermates still showed the enhancement (n = 9 mice; t(16) = 2.42, p = 0.028 compared with 9 vehicle-treated WT mice) (Fig. 4E). Cued fear learning showed no difference between the four groups of mice (WT-vehicle, 52.45 ± 5.04%; WT-Aβ, 51.83 ± 5.44%; α7-KO-vehicle, 49.54 ± 5.86%; α7-KO-Aβ, 50.24 ± 8.18%; t(12) = 0.07, p = 0.945 comparing α7-KO-vehicle to α7-KO-Aβ) (data not shown). Together, these findings indicate that both synaptic plasticity and memory rely on α7-nAChR involvement for the enhancing effect of Aβ.
Discussion
Growing evidence suggests that APP and its derivatives (i.e., the products of the α-, β-, γ-, and ε-secretase actions) display regulatory effects on synaptic transmission before the onset of neurodegeneration (Arancio and Chao, 2007). Our data provide robust evidence that an aged preparation of human Aβ42, when used at low concentrations, presumably neighboring those found in the normal brain (Rozmahel et al., 2002; Phinney et al., 2003; Pawlik et al., 2004; Yao et al., 2004; Mastrangelo et al., 2005; Schmidt et al., 2005), enhances LTP and memory. These effects are opposite to those (well established) caused by high levels of Aβ42, namely an impairment of LTP and memory.
We found that picomolar concentrations of an aged Aβ42 preparation containing both monomers and oligomers enhance synaptic plasticity and memory. However, it is impossible to know the molarities of different oligomers and monomers to which slices and hippocampi were actually exposed because Aβ can easily change its conformation by the time it reaches the brain tissue after its initial preparation. Moreover, scramble Aβ does not share the same capability of forming oligomers as normal Aβ. Furthermore, injected Aβ used in our behavioral experiments would add to the baseline levels of endogenous murine Aβ, which might increase concentrations beyond those identified as LTP-enhancing in the acute slice studies. Finally, there is uncertainty on the molarities of monomers and different oligomeric forms of Aβ existing at synapses. Thus, to address the issue of the concentration of Aβ responsible for its effects, we expressed the molarity of Aβ based on the molecular weight of its monomeric peptide. In addition, careful monitoring of our preparation among different experiments using PAGE analysis did not show a large variability from batch to batch in the relative percentage of different Aβ species. We kept constant the temperature of the bath solution in our electrophysiological experiments because temperature is known to affect Aβ conformation. We also used polypropylene tubing, which has been shown not to alter the ratio of different Aβ fragments (Lewczuk et al., 2006). Finally, as previously demonstrated (for instance, Puzzo et al., 2005), our preparation was capable of impairing LTP at a concentration of 200 nm. Nonetheless, we still cannot rigorously define the concentration and form(s) of Aβ42 responsible for the enhancing effects on synaptic plasticity and memory and whether scramble Aβ is present in the same species and concentration as normal Aβ. Nevertheless, the main observation of this manuscript, showing that Aβ42 enhances synaptic plasticity and memory, is clear and has important implications for mechanisms regulating the strength of synaptic connections and memory in hippocampus. It should also be noted that our results are consistent with loss of function studies in mice lacking APP, presenilin, or BACE1 (Dawson et al., 1999; Phinney et al., 1999; Seabrook et al., 1999; Saura et al., 2004; Laird et al., 2005; Dewachter et al., 2006). The enhancement of synaptic plasticity and memory with our gain of function approach specific to Aβ is likely, therefore, to be relevant to normal physiology.
Previous studies on a putative physiological function of Aβ have suggested that it selectively depresses excitatory synaptic transmission (Kamenetz et al., 2003). However, they relied on preparations exposed to high amounts of Aβ derived from overexpression of APP. Given that high levels of Aβ are known to impair synaptic function and memory (Haass and Selkoe, 2007), the discrepancy with our findings may be reconciled by the high amount of Aβ used in these previous studies, which might have produced a toxic effect on synaptic function. In contrast, we have carefully monitored the total concentration of Aβ by exogenously applying the peptide, and demonstrating that we could produce both depression and enhancement of synaptic plasticity according to the concentration used in the experiment.
Analysis of baseline transmission and AMPA receptor currents revealed no changes in these parameters during perfusion with Aβ. Thus, we also investigated NMDA receptor currents, because they are known to play a key role in synaptic plasticity. However, similar to AMPA receptor currents, exposure to Aβ did not affect NMDA receptor currents, and therefore, we had to exclude this possibility. As a next step, we investigated the miniature release of neurotransmitter before and after perfusion with Aβ42 and found no change in mEPSC frequency and amplitude, suggesting that Aβ alone does not affect these parameters. Finally, we investigated PTP, a phenomenon dependent on Ca2+ buildup within the presynaptic terminal, and found that picomolar concentrations of Aβ42 enhance neurotransmitter release during high-frequency stimulation. Thus, we believe that the most likely explanation for the enhancing effect of Aβ is that the increase in Ca2+ levels produced by Aβ paired with a single stimulus is not sufficient to overcome a threshold above which neurotransmitter release is enhanced. Aβ, in turn, would need to be associated with high-frequency stimulation for Ca2+ buildup within the presynaptic terminal above a given threshold and activation of mechanisms of synaptic plasticity. Because activation of α7-nAChRs is necessary for the Aβ-induced increase of synaptic plasticity and memory, we propose a model whereby Aβ released by neuronal activity during vesicle exocytosis (Cirrito et al., 2005) under normal conditions may modify glutamate release with a mechanism dependent upon activation of α7-nAChRs. This results in an increase of synaptic plasticity and memory (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). This model is consistent with the observation that Aβ42 might bind selectively and with picomolar affinity to α7-nAChRs (Wang et al., 2000), or it might be responsible for regulation of nAChR function through strong binding with membrane lipids (Small et al., 2007). Moreover, it is also consistent with the observation that Aβ42 activates α7-nAChRs at presynaptic nerve endings of synaptosomes when administered in the low picomolar range (Dougherty et al., 2003) [whereas it would act on non-α7-nAChRs at nanomolar concentrations (Dougherty et al., 2003)]. In addition, this model is consistent with the finding that activation of α7-nAChRs is involved in diverse brain functions including synaptic plasticity and memory (Levin and Simon, 1998; Jones et al., 1999) and enhances transmitter release in several brain regions including the hippocampus (Gray et al., 1996; Radcliffe and Dani, 1998), the spinal cord dorsal horn (Genzen and McGehee, 2003), the olfactory bulb, and the amygdala (Girod et al., 2000). Finally, this model is consistent with the observation that nicotinic activity at pyramidal neurons boosts LTP induction (Ji et al., 2001). Clearly, although we find that α7-nAChRs are involved in the Aβ-induced enhancement of synaptic plasticity and memory, it remains to be determined whether these effects are mediated by a direct physical interaction of the peptide with the α7-nAChR. Indeed, the effects of Aβ42 might be more complex than those accounted for by bona fide α7-nAChR agonists, such as acetylcholine and nicotine. Notwithstanding these findings, the involvement of α7-nAChRs we report here is likely to provide an important contribution to the enhancing effect of Aβ42 at picomolar concentrations.
Major drug discovery efforts are ongoing to develop strategies to decrease Aβ load (Haass and Selkoe, 2007). Our work does not challenge the amyloid hypothesis. However, our lack of understanding of the physiological role of Aβ may present important issues when designing effective and safe approaches to AD therapy. Our findings strongly support the possibility that Aβ42 itself may be an important modulator of synaptic plasticity and memory in the normal brain. Indeed, and paradoxically, the use of drugs that mimic Aβ structure or are targeted to the receptor(s) on which Aβ acts under normal physiological conditions, or even of Aβ itself or Aβ derivatives, might serve to enhance memory at appropriate concentrations and conditions.
Footnotes
This work was supported by National Institutes of Health Grant NS049442 (to O.A.) and Alzheimer's Association Grant NIRG-07-59597 (to D.P.). We thank Cristina Alberini, Rusiko Bourtchouladze, Moses V. Chao, Gilbert Di Paolo, Ana Garcia-Osta, Paul M. Mathews, Ipe Ninan, Filippo Palermo, Marina Picciotto, Lorna Role, and David Talmage for helpful comments and discussion, Benjamin Sherman for the genotyping of α7-nAChR KO mice, Gakuji Hashimoto and Fahad Aziz for the Aβ measurements, and the rest of the Arancio laboratory for numerous discussions.
References
- ArakiW, Kitaguchi N, Tokushima Y, Ishii K, Aratake H, Shimohama S, Nakamura S, Kimura J (1991) Trophic effect of beta-amyloid precursor protein on cerebral cortical neurons in culture. Biochem Biophys Res Commun 181:265–271. [DOI] [PubMed] [Google Scholar]
- ArancioO, Chao MV (2007) Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol 17:325–330. [DOI] [PubMed] [Google Scholar]
- CaoX, Südhof TC (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293:115–120. [DOI] [PubMed] [Google Scholar]
- CirritoJR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM (2003) In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci 23:8844–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CirritoJR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM (2005) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48:913–922. [DOI] [PubMed] [Google Scholar]
- CitronM, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, et al (1997) Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 3:67–72. [DOI] [PubMed] [Google Scholar]
- DawsonGR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ (1999) Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90:1–13. [DOI] [PubMed] [Google Scholar]
- DewachterI, Ris L, Croes S, Borghgraef P, Voets T, Nilius B, Godaux E, Van Leuven F (2006) Differential affects of presenilin-1 deficiency and mutant presenilin-1 on synaptic activity. In: Alzheimer's Association 10th International Conference on Alzheimer's Disease and Related Disorders p S58, O53-03-08. Madrid. Chicago: Alzheimer's Association.
- DoughertyJJ, Wu J, Nichols RA (2003) β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 23:6740–6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FreedmanR, Wetmore C, Strömberg I, Leonard S, Olson L (1993) α-Bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. J Neurosci 13:1965–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FurukawaK, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP (1996) Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem 67:1882–1896. [DOI] [PubMed] [Google Scholar]
- GaoY, Pimplikar SW (2001) The gamma-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci U S A 98:14979–14984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GenzenJR, McGehee DS (2003) Short- and long-term enhancement of excitatory transmission in the spinal cord dorsal horn by nicotinic acetylcholine receptors. Proc Natl Acad Sci U S A 100:6807–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GirodR, Barazangi N, McGehee D, Role LW (2000) Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology 39:2715–2725. [DOI] [PubMed] [Google Scholar]
- GrayR, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383:713–716. [DOI] [PubMed] [Google Scholar]
- HaassC, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112. [DOI] [PubMed] [Google Scholar]
- IshidaA, Furukawa K, Keller JN, Mattson MP (1997) Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport 8:2133–2137. [DOI] [PubMed] [Google Scholar]
- JiD, Lape R, Dani JA (2001) Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 31:131–141. [DOI] [PubMed] [Google Scholar]
- JonesS, Sudweeks S, Yakel JL (1999) Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci 22:555–561. [DOI] [PubMed] [Google Scholar]
- KamenetzF, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925–937. [DOI] [PubMed] [Google Scholar]
- KametaniF (2008) Epsilon-secretase: reduction of amyloid precursor protein epsilon-site cleavage in Alzheimer's disease. Curr Alzheimer Res 5:165–171. [DOI] [PubMed] [Google Scholar]
- KimberlyWT, Zheng JB, Guénette SY, Selkoe DJ (2001) The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 276:40288–40292. [DOI] [PubMed] [Google Scholar]
- LairdFM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25:11693–11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeissringMA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Müller U, Saftig P, De Strooper B, Wolfe MS, Golde TE, LaFerla FM (2002) A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci U S A 99:4697–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LevinED, Simon BB (1998) Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) 138:217–230. [DOI] [PubMed] [Google Scholar]
- LewczukP, Beck G, Esselmann H, Bruckmoser R, Zimmermann R, Fiszer M, Bibl M, Maler JM, Kornhuber J, Wiltfang J (2006) Effect of sample collection tubes on cerebrospinal fluid concentrations of tau proteins and amyloid beta peptides. Clin Chem 52:332–334. [DOI] [PubMed] [Google Scholar]
- LismanJ, Raghavachari S (2006) A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE 2006:re11. [DOI] [PubMed] [Google Scholar]
- López-ToledanoMA, Shelanski ML (2004) Neurogenic effect of β-amyloid peptide in the development of neural stem cells. J Neurosci 24:5439–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MaH, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, Younkin S, Forster C, Sergeant N, Delacourte A, Vassar R, Citron M, Kofuji P, Boland LM, Ashe KH (2007) Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A 104:8167–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MastrangeloP, Mathews PM, Chishti MA, Schmidt SD, Gu Y, Yang J, Mazzella MJ, Coomaraswamy J, Horne P, Strome B, Pelly H, Levesque G, Ebeling C, Jiang Y, Nixon RA, Rozmahel R, Fraser PE, St George-Hyslop P, Carlson GA, Westaway D (2005) Dissociated phenotypes in presenilin transgenic mice define functionally distinct gamma-secretases. Proc Natl Acad Sci U S A 102:8972–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MattsonMP (1994) Secreted forms of beta-amyloid precursor protein modulate dendrite outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons. J Neurobiol 25:439–450. [DOI] [PubMed] [Google Scholar]
- MezianeH, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, Ungerer A (1998) Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci U S A 95:12683–12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MuckeL, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR (1994) Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res 666:151–167. [DOI] [PubMed] [Google Scholar]
- PawlikM, Sastre M, Calero M, Mathews PM, Schmidt SD, Nixon RA, Levy E (2004) Overexpression of human cystatin C in transgenic mice does not affect levels of endogenous brain amyloid beta peptide. J Mol Neurosci 22:13–18. [DOI] [PubMed] [Google Scholar]
- PaxinosG (1998) Mouse brain in stereotaxic coordinates, Ed 2. New York: Academic.
- PhillipsRG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106:274–285. [DOI] [PubMed] [Google Scholar]
- PhinneyAL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M (1999) No hippocampal neuron or synaptic bouton loss in learning-impaired aged beta-amyloid precursor protein-null mice. Neuroscience 90:1207–1216. [DOI] [PubMed] [Google Scholar]
- PhinneyAL, Drisaldi B, Schmidt SD, Lugowski S, Coronado V, Liang Y, Horne P, Yang J, Sekoulidis J, Coomaraswamy J, Chishti MA, Cox DW, Mathews PM, Nixon RA, Carlson GA, St George-Hyslop P, Westaway D (2003) In vivo reduction of amyloid-beta by a mutant copper transporter. Proc Natl Acad Sci U S A 100:14193–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PlantLD, Boyle JP, Smith IF, Peers C, Pearson HA (2003) The production of amyloid β peptide is a critical requirement for the viability of central neurons. J Neurosci 23:5531–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PuzzoD, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O (2005) Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci 25:6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RadcliffeKA, Dani JA (1998) Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci 18:7075–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RozmahelR, Huang J, Chen F, Liang Y, Nguyen V, Ikeda M, Levesque G, Yu G, Nishimura M, Mathews P, Schmidt SD, Mercken M, Bergeron C, Westaway D, St George-Hyslop P (2002) Normal brain development in PS1 hypomorphic mice with markedly reduced gamma-secretase cleavage of betaAPP. Neurobiol Aging 23:187–194. [DOI] [PubMed] [Google Scholar]
- SauraCA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ 3rd, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42:23–36. [DOI] [PubMed] [Google Scholar]
- SchenkF, Morris RG (1985) Dissociation between components of spatial memory in rats after recovery from the effects of retrohippocampal lesions. Exp Brain Res 58:11–28. [DOI] [PubMed] [Google Scholar]
- SchmidtSD, Nixon RA, Mathews PM (2005) ELISA method for measurement of amyloid-beta levels. Methods Mol Biol 299:279–297. [DOI] [PubMed] [Google Scholar]
- SeabrookGR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG (1999) Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38:349–359. [DOI] [PubMed] [Google Scholar]
- SmallDH, Mok SS, Bornstein JC (2001) Alzheimer's disease and Abeta toxicity: from top to bottom. Nature reviews 2:595–598. [DOI] [PubMed] [Google Scholar]
- SmallDH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, Mehrani H, Unabia S, Azari MF, Loiacono R, Aguilar MI, Chebib M (2007) The beta-amyloid protein of Alzheimer's disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J Neurochem 101:1527–1538. [DOI] [PubMed] [Google Scholar]
- Smith-SwintoskyVL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP (1994) Secreted forms of beta-amyloid precursor protein protect against ischemic brain injury. J Neurochem 63:781–784. [DOI] [PubMed] [Google Scholar]
- WangHY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB (2000) beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275:5626–5632. [DOI] [PubMed] [Google Scholar]
- YanknerBA, Duffy LK, Kirschner DA (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250:279–282. [DOI] [PubMed] [Google Scholar]
- YaoJ, Petanceska SS, Montine TJ, Holtzman DM, Schmidt SD, Parker CA, Callahan MJ, Lipinski WJ, Bisgaier CL, Turner BA, Nixon RA, Martins RN, Ouimet C, Smith JD, Davies P, Laska E, Ehrlich ME, Walker LC, Mathews PM, Gandy S (2004) Aging, gender and APOE isotype modulate metabolism of Alzheimer's Abeta peptides and F-isoprostanes in the absence of detectable amyloid deposits. J Neurochem 90:1011–1018. [DOI] [PubMed] [Google Scholar]
- ZuckerRS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. [DOI] [PubMed] [Google Scholar]