Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers (original) (raw)
Abstract
Accumulating evidence suggests that obstructive sleep apnea is associated with alterations in glucose metabolism. Although the pathophysiology of metabolic dysfunction in obstructive sleep apnea is not well understood, studies of murine models indicate that intermittent hypoxemia has an important contribution. However, corroborating data on the metabolic effects of intermittent hypoxia on glucose metabolism in humans are not available. Thus the primary aim of this study was to characterize the acute effects of intermittent hypoxia on glucose metabolism. Thirteen healthy volunteers were subjected to 5 h of intermittent hypoxia or normoxia during wakefulness in a randomized order on two separate days. The intravenous glucose tolerance test (IVGTT) was used to assess insulin-dependent and insulin-independent measures of glucose disposal. The IVGTT data were analyzed using the minimal model to determine insulin sensitivity (SI) and glucose effectiveness (SG). Drops in oxyhemoglobin saturation were induced during wakefulness at an average rate of 24.3 events/h. Compared with the normoxia condition, intermittent hypoxia was associated with a decrease in SI [4.1 vs. 3.4 (mU/l)−1·min−1; P = 0.0179] and SG (1.9 vs. 1.3 min−1×10−2, P = 0.0065). Despite worsening insulin sensitivity with intermittent hypoxia, pancreatic insulin secretion was comparable between the two conditions. Heart rate variability analysis showed the intermittent hypoxia was associated with a shift in sympathovagal balance toward an increase in sympathetic nervous system activity. The average R-R interval on the electrocardiogram was 919.0 ms during the normoxia condition and 874.4 ms during the intermittent hypoxia condition (P < 0.04). Serum cortisol levels after intermittent hypoxia and normoxia were similar. Hypoxic stress in obstructive sleep apnea may increase the predisposition for metabolic dysfunction by impairing insulin sensitivity, glucose effectiveness, and insulin secretion.
Keywords: sleep apnea, diabetes, insulin resistance, glucose intolerance
the global prevalence of Type 2 diabetes mellitus is increasing in epidemic proportions. It is estimated that Type 2 diabetes affects ∼150 million people worldwide and that its prevalence is likely to almost double by the year 2025 (19). Over the last decade, considerable progress has been made in our understanding of the factors that increase the propensity for Type 2 diabetes. It is now abundantly clear that insulin resistance and pancreatic β-cell dysfunction are common by the time hyperglycemia develops (34). Risk factors for Type 2 diabetes include age, race, family history, obesity, and sedentary lifestyle (1). In recent years, there has been increasing recognition that obstructive sleep apnea may also be an independent risk factor for altered glucose metabolism. Indeed, a number of epidemiological studies have shown that, independent of confounders such as obesity, obstructive sleep apnea is associated with insulin resistance, glucose intolerance, and Type 2 diabetes (29, 32, 35). Although the evidence for a causal link is still fairly limited, the underlying mechanisms are likely to include intermittent hypoxemia and fragmentation of sleep (28).
Upper airway obstruction during sleep is typically associated with a decrease in oxyhemoglobin saturation and a concurrent arousal from sleep. Therefore it is not surprising that data obtained from human studies, while highly suggestive of an independent association between obstructive sleep apnea and metabolic dysfunction, are unable to segregate the effects of intermittent hypoxemia from sleep fragmentation. Only by separately inducing each component of the disease process can the complex tapestry of metabolic abnormalities in obstructive sleep apnea be unraveled. Experiments in animal models, which provide a unique opportunity to study the effects of specific exposures, have shown that intermittent hypoxemia can decrease insulin sensitivity and induce glucose intolerance (16, 26, 37). While such studies have helped advance our understanding of the metabolic impairment in obstructive sleep apnea, the effects of intermittent hypoxemia on glucose metabolism in humans are not known. Specifically, uncertainty remains as to whether intermittent hypoxemia in humans alters glucose disposal by decreasing insulin sensitivity, insulin secretion, or both. Thus the overall objective of this study was to investigate whether short-term exposure to intermittent hypoxia would alter glucose metabolism. To accomplish this objective, the frequently sampled intravenous glucose tolerance test (IVGTT) (5, 6) was used to assess alterations induced by intermittent hypoxia in insulin-dependent and insulin-independent mechanisms of glucose disposal in healthy volunteers. Using minimal model analysis (7, 9) of the IVGTT data, we sought to examine whether intermittent hypoxia would decrease insulin sensitivity and diminish glucose effectiveness, the ability of glucose to facilitate its disposal independent of an insulin response. Because the IVGTT also allows a simultaneous assessment of insulin secretion, a secondary objective was to determine whether intermittent hypoxia would alter pancreatic β-cell insulin secretion. It was hypothesized that, in healthy volunteers, exposure to short-term intermittent hypoxia would decrease insulin sensitivity, glucose effectiveness, and insulin secretion.
MATERIALS AND METHODS
Study sample.
Healthy adult volunteers were recruited from the local community by advertisements. All potential volunteers were initially screened for the presence of medical problems by a telephone interview. Exclusion criteria included a history of physician-diagnosed asthma or other respiratory illness, hypertension, hepatic or renal dysfunction, cardiovascular or neurological disease, or a hematological disorder. In addition, habitual sleep duration of <7 h, usual bed time after midnight, circadian sleep disorder as determined by a clinical interview and actigraphy, shift work, and current smoking excluded participation. Eligible volunteers without any medical problems were then screened with a full montage polysomnogram to identify and exclude volunteers with undiagnosed obstructive sleep apnea. Habitual sleep patterns were objectively assessed with wrist actigraphy (Actigraph AW-64, MiniMitter, Bend, OR) for at least 5 days to exclude volunteers with habitually short sleep duration (<7 h/night) or irregular sleep-wake patterns. Serum chemistries, including a fasting glucose level, were required to be within normal limits, and the hematocrit had to exceed 30% for further eligibility. Finally, measurements of forced expiratory volume in the first second (FEV1), forced vital capacity (FVC), and the single-breath diffusing capacity of the lung (DlCO) were measured, and volunteers below their predicted values were excluded. After qualifying for enrollment, volunteers were required to repeat actigraphy monitoring of habitual sleep patterns to confirm that they maintained a regular sleep schedule before each of the two experimental days. Body fat mass was determined using a Hologic QDR-4500A DEXA scanner several days before the experiment. Informed consent was obtained from all volunteers, and the study protocol was approved by the local institutional review committee on human research.
Study protocol and procedures.
The study was performed in a single-blind fashion and consisted of 2 days with an interval of 1 wk. On one of the days, the subject was exposed to intermittent hypoxia for 8 h, and on the other day the subject was exposed to ambient air for 8 h as a control. The procedures were carried out in random order. Before each condition, volunteers were counseled to target at least 7 h of sleep per night, consume 250 g or more of carbohydrates per day, and come to the laboratory at ∼7:30 AM after a 12-h overnight fast. On arrival at the laboratory, measurements of weight, height, and waist circumference were obtained. Intravenous lines were inserted in both antecubital veins for blood sampling and were kept patent with a continuous infusion of 0.9% saline. During the course of the experiment, each subject remained in a semirecumbent position and was occupied by watching television, listening to music, or reading. Sleep was not allowed, and the electroencephalogram (C3-M2 and O2-M1) was continuously monitored with a computerized polysomnographic system (Embla N7000 and Somnologica Studio 3.0 Software, Embla, Broomfield, CO). The recording montage also included the following physiological signals: the right and left electrooculograms, chin electromyogram, chest and abdominal excursion, electrocardiogram, and oxyhemoglobin saturation by pulse oximetry (Ohmeda 3700; Englewood, CO).
A well-fitted and comfortable full face mask was applied, and the inspiratory line was attached to a three-way Hans Rudolph valve so that inspiration could be from one of two pressurized cylinders with the following gases: ambient air (21% O2) or hypoxic air (95% N2 and 5% O2). Figure 1 shows a schematic of the experimental set-up. Airflow from each cylinder was initially adjusted using a pneumotachometer (model 3700 A, Hans Rudolph, Kansas City, MO) to target a flow rate that was judged to be the most comfortable by the subject. Alveolar hypoxia was established by inspiration of the hypoxic N2-O2 gas mixture until the oxyhemoglobin saturation dropped to 85%. The three-valve was then switched to the cylinder containing pressurized ambient air until the oxyhemoglobin saturation returned to the preexposure baseline value or was at least 95%. Intermittent hypoxia was simulated by manually alternating the three-way valve between the hypoxia and normoxia gas cylinders for the entire 8-h duration to target ∼25 hypoxic events/h as to simulate the degree of intermittent hypoxemia seen in patients with moderate obstructive sleep apnea. For the control condition, the cylinder with hypoxic gas was substituted with that containing pressurized ambient air. As with the intermittent hypoxia condition, airflow from both normoxia cylinders was matched and adjusted for optimal subject comfort. Although the oxyhemoglobin saturation under the normoxia condition remained unchanged throughout, the three-way valve was alternated nonetheless between the two gas cylinders at a frequency comparable to the intermittent hypoxia condition (∼25/h). After 5 h of exposure to intermittent hypoxia or normoxia, the frequently sampled IVGTT, which lasted 3 h, was conducted as outlined below. Exposure to either condition was, however, maintained throughout for a cumulative duration of 8 h.
Fig. 1.

Schematic of the experimental setup illustrating the breathing circuit with the 2 inspiratory pathways connected to pressurized ambient air (21% O2) or a hypoxic gas mixture (95% N2 and 5% O2).
IVGTT.
The intravenous line in the dominant arm was used for blood sampling. Glucose and insulin administration for the frequently sampled IVGTT occurred via the contralateral antecubital vein. Basal sampling occurred at −15, −10, −5, and −1 min before glucose administration. At time 0 min, a weight-adjusted dose of glucose (50% dextrose, 0.3 g/kg) was administered intravenously over 1 min followed by infusion of normal saline. Twenty minutes after the glucose dose, a weight-adjusted dose of regular insulin (0.03 U/kg) was administered. Blood samples were collected after the glucose infusion at the following times: 2, 3, 4, 5, 6, 8, 10, 12, 14, 16, 19, 22, 24, 25, 27, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, and 180 min. Glucose was measured enzymatically in duplicate using a Glucose Analyzer II (Beckman Instruments, Fullerton CA). Insulin concentrations were determined in duplicate by radioimmunoassay using standard commercial kits (Linco Research; St Charles, MO). The resulting glucose and insulin values during the intermittent hypoxia and normoxia conditions were subjected to the minimal model analysis (5, 7, 9) for determination of insulin sensitivity (SI), glucose effectiveness (SG), and glucose effectiveness at zero insulin (GEZI).
SI quantifies the effect of insulin in enhancing glucose disposal. SG quantifies the effects of glucose on its own disposal independent of any insulin response. SG is further divided into two components: the contribution of hyperglycemia per se to increase glucose disposal and the effect of basal insulin levels. The basal component of SG is referred to as the basal insulin effect (BIE) and is the product of basal insulin (Ib) and SI. The contribution of non-insulin-dependent glucose uptake (GEZI) to glucose disposal is the difference between total SG and the BIE: GEZI = SG − (Ib × SI). In addition to estimating SI, SG, and GEZI, the acute insulin response to glucose (AIRg), an index of pancreatic β-cell response, was also determined as the area under the insulin curve between 0 and 10 min. Finally, the disposition index (DI), which is an integrated measure of pancreatic β-cell function, was calculated as the product of SI and AIRg.
Heart rate variability analysis.
A single-lead electrocardiogram (ECG) was continuously recorded during each of the two experimental periods. Heart rate variability analysis was conducted according to standard guidelines (36) to derive time and frequency domain measures of sympathovagal balance. The heart rate trace was initially reviewed and regions of artifact were visually removed and the remaining normal-to-normal R-R intervals were used for further analysis. Frequency domain measures were computed in 5-min epochs of the ECG using a computerized algorithm. Power spectral analysis of the R-R intervals provided the following frequency domain measures: very-low-frequency power (VLF: 0.003–0.04 Hz), low-frequency power (LF: 0.04–0.15 Hz), high-frequency power (HF: 0.15–0.4 Hz), and the normalized low-frequency power [LF/(LF + HF)]. HF power reflects the activity of parasympathetic nervous system activity, whereas LF power reflects a combination of sympathetic and parasympathetic activity.
Statistical analysis.
Data were analyzed with the SAS 9.0 software package (SAS Institute, Cary NC). All results are presented as means along with the corresponding standard error of the mean (SE). The _t_-test and Wilcoxon's rank-sum test were used to compare the primary outcome measures (SI, SG, GEZI, and AIRg) between the intermittent hypoxia and normoxia conditions. A P value of <0.05 was used as a threshold for statistical significance.
RESULTS
The study sample consisted of 13 healthy volunteers with a mean age of 24.3 yr (range 18–35 yr). The average body mass index and percent body fat were 25.8 kg/m2 (SE 0.8) and 20.8 (SE 1.7), respectively. Sleep duration by actigraphy was, on average, 7.3 h (SE 0.2) before enrollment, 7.6 h (SE 0.2) before the intermittent hypoxia condition, and 7.3 h (SE 0.2) before the normoxia condition (P = 0.12). Thus the study sample had in excess of 7 h of average sleep before both experimental conditions. Body weight before the intermittent hypoxia and normoxia condition was 86.0 kg (SE 3.0) and 86.3 kg (SE 3.0), respectively (P = 0.45). No adverse effects were observed with exposure to intermittent hypoxia. The subjects' rating of whether they had been exposed to hypoxic or normoxic gas mixture on a particular day was no better than by chance, indicating the success of subject blinding.
Figure 2 displays a recording of the oxyhemoglobin saturation (SpO2) in a representative subject for the 8-h exposure to intermittent hypoxia along with a 5-min expanded view of the fractional inspired oxygen and the single-lead electrocardiogram. Across the 13 volunteers, a total of 2,564 drops in oxyhemoglobin saturation were elicited at an average rate of 24.3 events/h (SE 0.9). The average duration of exposure to the hypoxic gas mixture per event was 70.8 s (SE 0.66). Heart rate was temporally correlated with changes in oxyhemoglobin saturation. It progressively increased during the exposure to the hypoxic gas, reaching a maximum at the nadir of oxyhemoglobin saturation. With cessation of the hypoxic exposure, heart rate returned to baseline values as oxyhemoglobin saturation recovered to preexposure levels. Table 1 shows frequency of hypoxic events along with other measures of oxyhemoglobin saturation for different segments of the experimental period. As expected, the average and minimum oxyhemoglobin saturation were lower with the intermittent hypoxia than with the normoxia condition.
Fig. 2.

Oxyhemoglobin saturation (SpO2) profile from 1 subject along with a 5-min expanded view of the fractional inspired oxygen concentration (FiO2). The electrocardiogram during 1 hypoxic event is also shown. IVGTT, intravenous glucose tolerance test.
Table 1.
Summary statistics of oxyhemoglobin saturation under conditions of intermittent hypoxia and normoxia by experimental segment
| Parameter | Before IVGTT (0–5 h) | During IVGTT (5–8 h) | Experimental Duration (0–8 h) | |||
|---|---|---|---|---|---|---|
| Normoxia | Intermittent hypoxia | Normoxia | Intermittent hypoxia | Normoxia | Intermittent hypoxia | |
| Exposure time, min | 297.8 (3.6) | 308.2 (4.6)* | 182.3 (1.8) | 182.2 (1.0) | 480.0 (2.5) | 486.3 (2.2) |
| Event frequency, no./h | 0.0 (0.0) | 24.3 (0.9)* | 0.0 (0.0) | 24.0 (1.0)* | 0.0 (0.0)* | 24.3 (0.8)* |
| SpO2, % | ||||||
| Average | 97.1 (0.2) | 90.6 (0.1)* | 97.1 (0.2) | 91.3 (0.2)* | 97.1 (0.2) | 90.9 (0.1)* |
| Maximum | 98.0 (0.0) | 98.0 (0.0)* | 98.0 (0.0) | 98.0 (0.0)* | 98.0 (0.0) | 98.0 (0.0)* |
| Minimum | 91.5 (1.1) | 75.4 (0.9)* | 93.1 (0.5) | 77.2 (0.9)* | 91.2 (1.1) | 74.8 (0.9)* |
| Time SpO2 <90%, min | 0.1 (0.0) | 114.9 (3.5)* | 0.00 (0.0) | 63.5 (2.6)* | 0.1 (0.1) | 178.3 (5.4)* |
| Time SpO2 <85%, min | 0.0 (0.0) | 47.0 (3.5)* | 0.00 (0.0) | 21.2 (3.2)* | 0.0 (0.0) | 68.3 (6.0)* |
| Time SpO2 <80%, min | 0.0 (0.0) | 9.6 (2.4)* | 0.00 (0.0) | 4.6 (1.4)* | 0.0 (0.0) | 14.2 (3.2)* |
Minimal-model analysis of the IVGTT data showed that SI, SG, and GEZI were lower during the intermittent hypoxia condition compared with the normoxia condition (Fig. 2). The AIRg was, however, similar between the two groups (398.9 ± 85.9 (mU/l)·min during normoxia vs. 388.6 ± 95.2 (mU/l)·min during intermittent hypoxia; P = 0.85), indicating that, despite a decrease in insulin sensitivity with intermittent hypoxia, the expected compensatory increase in insulin secretion was absent. As a result, the disposition index (DI = SI × AIRg), which expresses the insulin response for the degree of insulin resistance present, was lower during the intermittent hypoxia than the normoxia condition (Fig. 3; P = 0.0015). Finally, glucose effectiveness, the ability of glucose to mobilize itself independent of an insulin response, was also lower during the intermittent hypoxia condition, as was the GEZI.
Fig. 3.
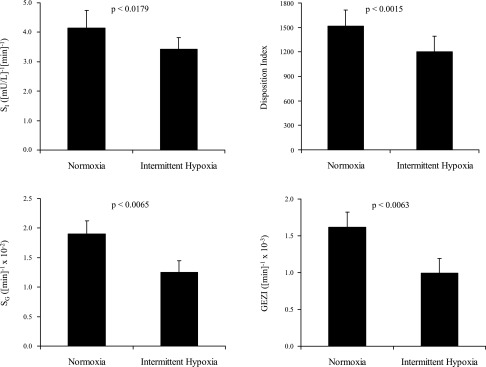
Values of insulin sensitivity (SI), the disposition index (DI = SI × AIRg, where AIRg is acute insulin response to glucose), glucose effectiveness (SG), and glucose effectiveness at zero insulin (GEZI) with intermittent hypoxia and normoxia. Values are means and SEs of the mean.
Time domain and frequency domain analysis of the heart rate data (Table 2) showed that intermittent hypoxia was associated with a shift in sympathovagal balance toward an increase in sympathetic nervous system activity. The average R-R interval decreased from 919.9 ms (SE 41.0) during the normoxia condition to 874.4 ms (SE 26.5) during the intermittent hypoxia condition (P = 0.04). The standard deviation of all normal R-R intervals (SDNN) was also higher with intermittent hypoxia than with normoxia (107.8 vs. 93.3; P = 0.0085), suggesting a greater degree of heart rate modulation with exposure to intermittent hypoxia. Frequency domain analysis confirmed a shift in sympathovagal balance toward an increase in sympathetic nervous system activity. Intermittent hypoxia was associated with a significant increase in average VLF and LF power, but not HF power, compared with the normoxia condition. Although the normalized LF power [LF/(HF + LF)], a measure of sympathovagal balance, was higher during intermittent hypoxia than normoxia, it did not reach statistical significance. However, sensitivity analyses showed that exclusion of one outlying subject strengthened the differences in the normalized LF power between the two conditions (P < 0.07). Finally, serum cortisol values at the end of the experiment were similar under the two conditions. During intermittent hypoxia condition, the preexposure (8 AM) and postexposure (4 PM) cortisol values were 13.2 μg/dl (SE 1.0) and 8.3 μg/dl (SE 0.8), respectively. The corresponding values during the normoxia condition were 12.8 μg/dl (SE 1.1) and 7.5 μg/dl (SE 0.9), respectively.
Table 2.
HRV parameters under conditions of intermittent hypoxia and normoxia
| HRV Parameter | Normoxia | Intermittent Hypoxia | P Value |
|---|---|---|---|
| Average R-R interval, ms | 919.9 (41.0) | 874.4 (26.5) | 0.04 |
| SDNN, ms | 93.3 (6.5) | 107.8 (5.1) | 0.009 |
| VLF power, ms2 | 4,460.2 (729.3) | 9,284.8 (1,408.3) | <0.0001 |
| LF power, ms2 | 3,534.2 (332.1) | 4,450.2 (620.1) | 0.047 |
| HF power, ms2 | 1,968.8 (314.3) | 1,839.7 (228.5) | 0.53 |
| LF/(LF + HF) | 0.64 (0.04) | 0.69 (0.04) | 0.07 |
DISCUSSION
The primary objective of this study was to examine the effects of intermittent hypoxia on glucose metabolism in healthy volunteers. Recurrent transient drops in oxyhemoglobin saturation were induced using brief periodic exposures to a hypoxic gas mixture to mimic the oxygen saturation profile commonly seen in patients with moderate obstructive sleep apnea. In a randomized controlled study, intermittent hypoxia caused alterations in insulin-dependent and insulin-independent mediated glucose disposal. Compared with the normoxic condition, intermittent hypoxia led to a decrease in insulin sensitivity that was not accompanied by a commensurate increase in insulin secretion. Intermittent hypoxia also resulted in a notable decrease in glucose effectiveness, which measures the ability of glucose to enhance its own disposal independent of an insulin response. Finally, although intermittent hypoxia had no effect on serum cortisol levels, it was associated with a shift in sympathovagal balance toward heightened sympathetic nervous system activity. The major implication of these findings is that intermittent hypoxemia, in the absence of other pathophysiological changes, may be an important intermediate in the genesis of metabolic dysfunction in obstructive sleep apnea.
The acute and chronic effects of sustained hypoxia on glucose metabolism are well documented. Experimental evidence from animal models indicates that exposure to sustained hypoxia for as little as 2 h can induce insulin resistance in newborn calves (11). With chronic exposure (∼7 days), serum glucose and insulin levels in rats remain elevated despite a decrease in body weight (30, 31). Additional support for the adverse metabolic effects of sustained hypoxia can also be found in several human studies that show that altitude hypoxia in healthy men and women can markedly reduce insulin sensitivity (10, 21). Moreover, exposure to even brief periods of sustained hypoxia (∼30 min) in humans can decrease insulin sensitivity and increase sympathetic nervous system activity (25). Collectively, the available body of experimental and observational evidence indicates that sustained hypoxia has a negative impact on glucose metabolism.
The effects of intermittent hypoxia on glucose metabolism have also been explored but only in animal models. Polotsky et al. (26) have shown that leptin-deficient C57BL/6J obese mice exposed to intermittent hypoxia for 12 wk experience a time-dependent worsening of glucose tolerance with a concomitant increase in fasting serum levels. Recent work by Iyori et al. (16) confirmed that intermittent hypoxia (∼9 h) can cause insulin resistance even in lean, otherwise healthy, mice and that this reduction in insulin sensitivity occurs despite hexamethonium-induced blockade of autonomic activity. As the exposure duration to intermittent hypoxia is extended further (∼80 h), insulin resistance persists and is associated with an increase in pancreatic β-cell replication, a response to perhaps help compensate for the decrease in insulin sensitivity (37). The results of our study, the only one to date on the metabolic consequences of intermittent hypoxia in humans, corroborate the notion that intermittent hypoxia can alter glucose disposal by worsening insulin-dependent and insulin-independent mechanisms of glucose disposal.
It is important to note that the decrease in insulin sensitivity with intermittent hypoxia in the present study was not accompanied by an increase in insulin secretion. It is well established that insulin sensitivity and insulin secretion are inextricably linked such that changes in insulin sensitivity are compensated by an increase in insulin secretion (17). In fact, mathematical modeling of insulin sensitivity and insulin secretion in healthy individuals reveals a hyperbolic relationship (18), such that the product of the two—the disposition index—remains constant. As the disposition index decreases, the risk for developing glucose intolerance and Type 2 diabetes increases (17). Whether the short-term exposure to intermittent hypoxia in the present study was insufficient to evoke an adaptive response by the pancreatic β-cell or whether it inhibited the expected pancreatic compensation remains to be determined. Experimental evidence from human studies indicates that an acute adaptation in insulin secretion is possible with short-term changes in insulin sensitivity (2). For example, suppression of insulin sensitivity with oral dexamethasone for 2 days in healthy volunteers has been shown to increase insulin secretion (4, 22). Thus we speculate that intermittent hypoxemia may, in fact, have inhibited pancreatic ability to compensate for the hypoxia-associated reduction in insulin sensitivity.
There are several causal mechanisms that could explain the observed effects of intermittent hypoxia on glucose metabolism. One candidate mechanism is the activation of sympathetic nervous system with intermittent hypoxia. Catecholamines are known to decrease insulin sensitivity and reduce insulin-mediated glucose uptake (13). Indeed, administration of epinephrine to healthy volunteers decreases glycogenesis, increases glycolysis, and diminishes the ability of glucose to stimulate its own disposal (13, 23). Despite strong evidence for a potential role for the sympathetic nervous system in glucose metabolism, Iyori et al. (16) have shown that blockade of autonomic activity does not mitigate the negative effects of intermittent hypoxia on insulin sensitivity, suggesting that alternative pathways may be involved. A second candidate mechanism is alterations in corticotropic function induced by intermittent hypoxia. Studies at high altitude or under hypobaric conditions show that hypoxia modifies hypothalamic-pituitary-adrenal (HPA) function and leads to higher levels of circulating cortisol (12, 15). It is well known that cortisol increases hepatic gluconeogenesis, inhibits pancreatic β-cell insulin secretion, and induces insulin resistance (3, 20). Interestingly, cortisol levels in the present study were not altered with intermittent hypoxia. This finding suggests that perhaps intermittent hypoxia has no effect on the HPA axis or the duration of exposure in the present study was insufficient for a detectable response. A third candidate mechanism for metabolic dysfunction with intermittent hypoxia is the formation of reactive oxygen species, which can suppress insulin secretion and worsen insulin sensitivity (8, 33). Finally, it is also possible that release of proinflammatory cytokines, such as interleukin-6 and tumor necrosis factor-α, is responsible for metabolic dysfunction with intermittent hypoxia as both of these cytokines have been causally implicated in pathogenesis of insulin resistance and Type 2 diabetes (14, 24, 27). Although the above candidate mechanisms may act synergistically to alter glucose metabolism with intermittent hypoxia or in obstructive sleep apnea, it is abundantly clear that our understanding of intermediate pathways is in its infancy, and future research efforts using animal and human studies are desperately needed.
There are several limitations in our study that merit discussion. First, the duration of intermittent hypoxia was brief and limited to only 8 h. Additional exposure (i.e., days), however, would place substantial participant burden and could have untoward effects. Second, intermediate mechanisms were crudely assessed using heart rate variability as a marker of sympathetic activity and serum cortisol levels as a surrogate of corticotropic function. Microneurography for assessment of sympathetic activity and the collection of additional blood samples for cortisol would have precluded execution of an already cumbersome experimental paradigm. Third, intermittent hypoxia was induced during wakefulness and not during sleep. The decision to characterize the metabolic effects during wakefulness was based on the awareness that hypoxic exposure during sleep would induce a range of pathophysiological abnormalities, including periodic breathing and sleep disruption. Thus, to characterize the isolated effects of intermittent hypoxia, the experiment was undertaken during wakefulness. Fourth, the experimental paradigm focused strictly on the effects of intermittent hypoxia and did not include other pathophysiological concomitants of obstructive sleep apnea, including hypercapnia or asphyxia. Finally, although the IVGTT results show a difference in insulin sensitivity comparing the intermittent hypoxia and normoxia conditions, the locus of insulin resistance (hepatic or extrahepatic) was not assessed. These limitations notwithstanding, the present study also several strengths. These include a critical assessment of the independent effects of intermittent hypoxemia on glucose metabolism, the concurrent assessment of insulin sensitivity and insulin secretion using the frequently sampled IVGTT, the inclusion of a control condition, and the exclusion of potentially confounding factors with the use of healthy volunteers.
In conclusion, short-term episodic hypoxia during the daytime alters glucose disposal in healthy volunteers by decreasing insulin sensitivity and glucose effectiveness. The decrease in insulin action was not accompanied by a commensurate increase in insulin secretion. Intermittent hypoxia was also associated with a shift in sympathovagal balance toward an increase in sympathetic nervous system activity. Thus we speculate that intermittent hypoxemia is a central mechanism responsible for metabolic dysfunction in patients with obstructive sleep apnea and that the sympathetic nervous system is a putative mediator.
GRANTS
This publication was made possible by National Center for Research Resources Grant UL1-RR-025005 and by National Institutes of Health Roadmap for Medical Research. Additional support was provided by National Heart, Lung, and Blood Institute Grant HL-07578.
Acknowledgments
We express our appreciation to the research volunteers who participated in this study and to Kelly Devine and Melissa Minotti for their efforts with implementation and execution of the study protocol.
REFERENCES
- 1.Adeghate E, Schattner P, Dunn E. An update on the etiology and epidemiology of diabetes mellitus. Ann NY Acad Sci 1084: 1–29, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Ahren B, Pacini G. Islet adaptation to insulin resistance: mechanisms and implications for intervention. Diabetes Obes Metab 7: 2–8, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci (Lond) 96: 513–523, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Beard JC, Halter JB, Best JD, Pfeifer MA, Porte D Jr. Dexamethasone-induced insulin resistance enhances B cell responsiveness to glucose level in normal men. Am J Physiol Endocrinol Metab 247: E592–E596, 1984. [DOI] [PubMed] [Google Scholar]
- 5.Bergman RN, Finegood DT, Ader M. Assessment of insulin sensitivity in vivo. Endocr Rev 6: 45–86, 1985. [DOI] [PubMed] [Google Scholar]
- 6.Bergman RN Minimal model: perspective from 2005. Horm Res 64, suppl 3: 8–15, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol Endocrinol Metab Gastrointest Physiol 236: E667–E677, 1979. [DOI] [PubMed] [Google Scholar]
- 8.Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal 7: 1553–1567, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther 5: 1003–1015, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Braun B, Rock PB, Zamudio S, Wolfel GE, Mazzeo RS, Muza SR, Fulco CS, Moore LG, Butterfield GE. Women at altitude: short-term exposure to hypoxia and/or α1-adrenergic blockade reduces insulin sensitivity. J Appl Physiol 91: 623–631, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Cheng N, Cai W, Jiang M, Wu S. Effect of hypoxia on blood glucose, hormones, and insulin receptor functions in newborn calves. Pediatr Res 41: 852–856, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Coste O, Beers PV, Bogdan A, Charbuy H, Touitou Y. Hypoxic alterations of cortisol circadian rhythm in man after simulation of a long duration flight. Steroids 70: 803–810, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest 65: 717–721, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, Hoogeveen R, Folsom AR, Heiss G. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 52: 1799–1805, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Humpeler E, Skrabal F, Bartsch G. Influence of exposure to moderate altitude on the plasma concentration of cortisol, aldosterone, renin, testosterone, and gonadotropins. Eur J Appl Physiol Occup Physiol 45: 167–176, 1980. [DOI] [PubMed] [Google Scholar]
- 16.Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O'Doherty RM, Polotsky VY, O'Donnell CP. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med 175: 851–857, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kahn SE The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46: 3–19, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42: 1663–1672, 1993. [DOI] [PubMed] [Google Scholar]
- 19.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 21: 1414–1431, 1998. [DOI] [PubMed] [Google Scholar]
- 20.Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest 99: 414–423, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen JJ, Hansen JM, Olsen NV, Galbo H, Dela F. The effect of altitude hypoxia on glucose homeostasis in men. J Physiol 504: 241–249, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsson H, Ahren B. Insulin resistant subjects lack islet adaptation to short-term dexamethasone-induced reduction in insulin sensitivity. Diabetologia 42: 936–943, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Lembo G, Capaldo B, Rendina V, Iaccarino G, Napoli R, Guida R, Trimarco B, Sacca L. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am J Physiol Endocrinol Metab 266: E242–E247, 1994. [DOI] [PubMed] [Google Scholar]
- 24.Moller DE Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11: 212–217, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Oltmanns KM, Gehring H, Rudolf S, Schultes B, Rook S, Schweiger U, Born J, Fehm HL, Peters A. Hypoxia causes glucose intolerance in humans. Am J Respir Crit Care Med 169: 1231–1237, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol 552: 253–264, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286: 327–334, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Punjabi NM, Polotsky VY. Disorders of glucose metabolism in sleep apnea. J Appl Physiol 99: 1998–2007, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 160: 521–530, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Raff H, Bruder ED, Jankowski BM. The effect of hypoxia on plasma leptin and insulin in newborn and juvenile rats. Endocrine 11: 37–39, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Raff H, Bruder ED, Jankowski BM, Colman RJ. Effect of neonatal hypoxia on leptin, insulin, growth hormone and body composition in the rat. Horm Metab Res 33: 151–155, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Reichmuth KJ, Austin D, Skatrud JB, Young T. Association of sleep apnea and type II diabetes: a population-based study. Am J Respir Crit Care Med 172: 1590–1595, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson RP Oxidative stress and impaired insulin secretion in type 2 diabetes. Curr Opin Pharmacol 6: 615–619, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365: 1333–1346, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 29: 777–783, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation, and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Circulation 93: 1043–1065, 1996. [PubMed] [Google Scholar]
- 37.Yokoe T, Alonso LC, Romano LC, Rosa TC, O'Doherty RM, Garcia-Ocana A, Minoguchi K, O'Donnell CP. Intermittent hypoxia reverses the diurnal glucose rhythm and causes pancreatic beta-cell replication in mice. J Physiol 586: 899–911, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]