Depolarization and cAMP Elevation Rapidly Recruit TrkB to the Plasma Membrane of CNS Neurons (original) (raw)
. Author manuscript; available in PMC: 2009 Jun 8.
Summary
Here, we describe a novel mechanism for the rapid regulation of surface levels of the neurotrophin receptor TrkB. Unlike nodose ganglion neurons, both retinal ganglion cells (RGCs) and spinal motor neurons (SMNs) in culture display only low levels of surface TrkB, though high levels are present intracellularly. Within minutes of depolarization or cAMP elevation, surface TrkB levels increase by nearly 4-fold, and this increase is not blocked by cycloheximide. These findings suggest that activity and cAMP elevation rapidly recruit TrkB to the plasma membrane by translocation from intracellular stores. We propose that a fundamental difference between peripheral nervous system (PNS) and central nervous system (CNS) neurons is the activity dependence of CNS neurons for responsiveness to their peptide trophic factors and that differences in membrane compartmentalization of the receptors underlie this difference.
Introduction
The signaling processes that control the survival and growth of central nervous system (CNS) neurons involve complex cell-cell interactions and are poorly understood. In order to simplify the problem, we have begun by studying the signals necessary to promote the survival of developing retinal ganglion cells (RGCs), which can be highly purified by immunopanning and maintained in long-term culture in serum-free medium containing only three peptide trophic factors that are normally produced by cells along the developing visual pathway: brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF) and insulin-like growth factor-1 (IGF-1) (Barres et al., 1988; Meyer-Franke et al., 1995). The survival of the purified RGCs is only promoted by these peptide trophic factors, however, if their intracellular levels of cAMP are simultaneously elevated. cAMP elevation by itself does not sifnificantly promote their survival. The effects of cAMP elevation are mimicked by depolarization induced either by K+ or by glutamate receptor activation, both of which elevate cAMP levels in the RGCs by activating a calcium-dependent type-1 adenylyl cyclase (Xia et al., 1991; Meyer-Franke et al., 1995; S. Shen and B. A. B., unpublished data).
In contrast, previous experiments have shown that purified postnatal peripheral nervous system (PNS) neurons respond well to peptide trophic factors such as neurotrophins without a requirement for cAMP elevation or other costimuli. cAMP elevation and depolarization, however, are sufficient by themselves to promote the survival of many PNS neurons in the absence of signaling by neurotrophic factors (Wakada et al., 1983; Rydell and Greene, 1988; Koike and Tanaka, 1991; Franklin and Johnson, 1992; Franklin et al., 1995). Thus, cAMP elevation and depolarization induce RGCs to be responsive to their peptide trophic factors, whereas they induce PNS neurons to be independent of growth factors. Like RGCs, depolarization also promotes the growth of cerebral cortical neurons in response to neurotrophins (McAllister et al., 1996), suggesting the possibility that CNS neurons in general may not optimally respond to their trophic peptides unless appropriate intracellular signaling pathways are activated.
Together, these findings have lead us to the hypothesis that the signaling mechanisms that promote the survival of PNS neurons and some or all types of CNS neurons may be fundamentally different (Meyer-Franke et al., 1995). To further test this hypothesis, we have addressed two questions in the present manuscript. First, are the apparent differences in the responsiveness of RGCs and PNS neurons to neurotrophins the result of genuine intrinsic differences or are they instead an artifactual result of differences in purification and culture methods? Whereas we have used an immunopanning method to purify and study acutely isolated RGCs cultured in serum-free medium, previous studies of PNS neurons have tended to use less pure cultures, different purification procedures, and different culture conditions. Thus, to determine whether there were genuine differences between cell types, it was crucial to examine the properties of acutely isolated PNS neurons purified by the same immunopanning method and cultured in the same serum-free medium used for the RGCs.
Second, we have asked how cAMP elevation enhances the responsiveness of RGCs to peptide trophic factors. cAMP could act simply by increasing the expression or density of trophic factor receptors on the plasma membrane. For instance, depolarization and cAMP elevation promote TrkA transcription and nerve growth factor (NGF) responsiveness of the adrenal medullary-derived neuronal cell line (Birren et al., 1992). Alternatively, cAMP might potentiate intracellular signal transduction pathways activated by these trophic factor receptors or activate a parallel pathway that synergizes with them (Sheng et al., 1990; Ghosh and Greenberg, 1995).
We show that two types of acutely isolated PNS neurons purified by immunopanning, superior cervical ganglion (SCG) neurons and nodose ganglion neurons, respond optimally to NGF and BDNF, respectively, in the absence of cAMP elevation or other costimuli. We further show that cAMP elevation induces a rapid increase in surface levels of full length TrkB in both RGCs and spinal motor neurons (SMNs) in culture. These results show that whereas PNS neurons are intrinsically responsive to neurotrophins, RGCs and SMNs are not and require signals from other cells to recruit trophic factor receptors to their surface.
Results
Responsiveness of Purified PNS Neurons to Neurotrophins
To determine whether acutely isolated, highly purified types of PNS neurons were responsive to neurotrophins when cultured in serum-free medium, we used immunopanning to obtain postnatal SCG- sympathetic and nodose ganglion sensory neurons (see Experimental Procedures). As in the RGC purification procedure, the cell suspensions were obtained enzymatically with papain, potentially contaminating cells were depleted from the cell suspension by panning, and the neurons of interest were selected by using a final Thy-1 antibody-coated panning dish. Thus, all cell types were treated nearly identically. These procedures resulted in purified populations of SCG and nodose ganglion neurons that were >95% pure, with a yield of at least 75% of the total neurons (see Experimental Procedures). The purified PNS neurons were cultured in serium-free medium in 96-well tissue culture plates (see Experimental Procedures). After 3 and 7 days of culture, we assessed their survival by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Mosmann, 1983; Meyer-Franke et al., 1995) (see Experimental Procedures).
As expected, in the absence of added growth factors, nearly all of the purified SCG (Figures 1A and 2A) and nodose ganglion (Figure 2B) neurons died within 3 days, with morphologies characteristic of apoptosis. In the presence of plateau levels of single neurotrophins, the majority of purified SCG (Figures 1B and 2A) and nodose ganglion neurons (Figure 2B) survived for 3 days in response to NGF (10 ng/ml) and BDNF (50 ng/ml), respectively. Similar results were obtained when SCG neurons were purified from postnatal day 8 (P8) ganglia (data not shown). As expected, elevation of cAMP by forskolin (10 μM), a specific activator of adenylyl cyclase, together with 3-isobutyl-1-methylxanthine (IBMX, 1 mM), a specific inhibitor of phosphodiesterases, was also sufficient by itself to promote significant survival of SCG and nodose ganglion neurons (Figure 2). Interestingly, while forskolin alone is sufficient to greatly increase cAMP levels in the RGCs (S. Shen and B. A. B., unpublished data), forskolin alone was insufficient to promote the survival of the PNS neurons, suggesting that unlike RGCs, these PNS neurons may have high resting levels of cyclic nucleotide phosphodiesterases (Virdee and Tolkovsky, 1996). Elevation of cAMP did not, however, increase the survival of the sympathetic or nodose ganglion neurons promoted by saturating levels of NGF or BDNF alone. Thus, single neurotrophins were sufficient to promote the survival of the majority of acutely isolated, purified PNS neurons in serum-free medium for 3 days (Figure 2) and even for as long as 7 days (data not shown).
Figure 1.
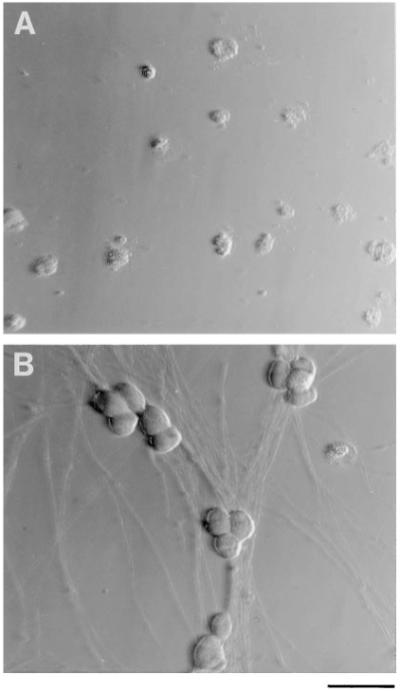
Hoffman Interference Contrast Micrographs of Purified SCG Neurons in Culture
The cells were cultured for 3 days in serum-free medium lacking NGF (A) or containing NGF (20 ng/ml) (B). Scale bar, 50 μm.
Figure 2.
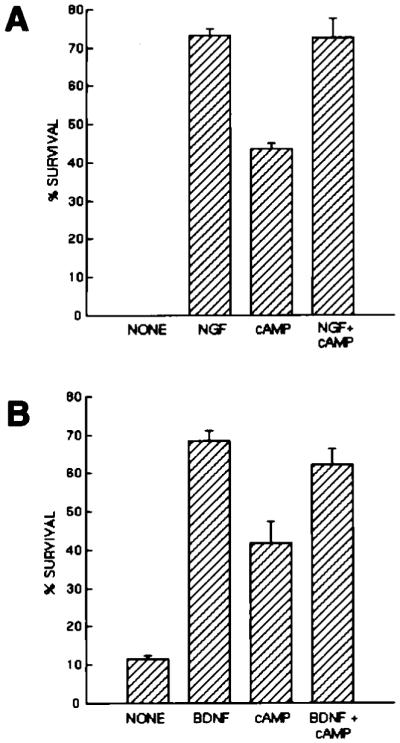
Effects of Neurotrophins and cAMP Elevation on the Survival of Purified PNS Neurons
Purified P2 sympathetic neurons (A) or nodose ganglion neurons (B) were plated at 5,000 cells per well in poly-l-lysine-coated 96-well tissue culture plates in serum-free medium containing the indicated substances. Plateau concentrations of neurotrophins were used (NGF was used at 20 ng/ml; BDNF was used at 50 ng/ml). cAMP was elevated by a combination of forskolin (5 μM) and IBMX (0.1 mM). Survival was measured after 3 days by the MTT assay. All values are means ± SEM (n = 3).
Effects of BDNF and cAMP on MAP Kinase Activation in Purified RGCs
The ability of the PNS neurons to survive in response to neurotrophins alone highlights the failure of RGCs to respond under the same conditions. As a first step in determining why RGCs do not respond to BDNF, we asked whether BDNF stimulation was sufficient to activate the mitogen-activated protein (MAP) kinases p44 ERK1 and p42 ERK2. These MAP kinases are activated in response to activation of many tyrosine kinase receptors. For instance, they are rapidly activated in response to NGF in PC12 cells, where they help to mediate NGF-promoted survival (Traverse et al., 1992; Xia et al., 1995; Vossler et al., 1997). If BDNF is sufficient to activate MAP kinase, then cAMP may act distally in the signal transduction pathway such as by enhancing activation of cAMP response element-binding protein (CREB), whereas an absence of MAP kinase activation in response to BDNF would be consistent with a more proximal site for cAMP action such as on the number of surface TrkB receptors.
We followed MAP kinase activation in two ways. First, we assessed the percentage of purified RGCs in culture that translocated MAP kinase from cytoplasm to nucleus in response to BDNF or cAMP elevation. Nuclear translocation has been shown to be closely correlated with phosphorylation and activation of MAP kinase and can be assessed by immunostaining with a specific anti-MAP kinase antibody (Martin et al., 1997) (see Experimental Procedures). For these experiments, purified P8 RGCs were incubated at 37°C for 1 hr to allow the reappearance on the plasma membrane of any constitutively secreted Trk receptors that had been enzymatically digested during the cell purification procedure. After 1 hr, the RGC cultures were incubated in either nothing, BDNF (50 ng/ml), forskolin (10 μM), or BDNF together with forskolin. The percentage of cells with nuclear MAP kinase immunostaining was determined after 30 min, 1 hr, and 3 hr. As shown in Figures 3 and 4, few cells exhibited MAP kinase translocation at 3 hr in control cells or in cells treated with forskolin or BDNF alone (Figures 3A and 4A), although BDNF did produce a transient response in some of the cells, particularly at 0.5 and 1 hr (Figure 4A). Sustained translocation was elicted in the majority of cells only when the RGCs were treated with BDNF and the elevation of cAMP together (Figures 3B and 4A). In contrast to the RGCs, NGF alone was sufficient to produce a rapid and sustained translocation of MAP kinase in purified SCG neurons in culture under the same conditions (Figure 4B).
Figure 3.

Immunofluoresence Micrographs of RGCs Stained by an Anti-MAP Kinase Antibody
Most cells incubated for 3 hr in serum-free medium lacking enhancers of cAMP levels, regardless of the presence of neurotrophins, have predominantly cytoplasmic staining (A). In contrast, most cells incubated for 3 hr in medium containing neurotrophins together with elevators of cAMP have both cytoplasmic and nuclear staining (B). Scale bar, 20 μm.
Figure 4.

Effects of Neurotrophins and cAMP Elevation on MAP Kinase Translocation into the Nucleus
Acutely isolated, purified P8 RGCs or P2 SCG neurons were plated onto PDL-coated coverslips for 1 hr and then treated with the indicated substances. Plateau concentrations of neurotrophins were used (NGF was used at 20 ng/ml; BDNF was used at 50 ng/ml). cAMP was elevated by a combination of forskolin (5 μM); in addition, IBMX (0.1 mM) was added to the SCG neurons. RGCs (A) or SCGs (B) were cultured for various times in the indicated substances and then labeled to determine the percentage of cells with MAP kinase staining in their nucleus. All values are means ± SEM. (n = 3).
To directly assess the amount of MAP kinase activation, we next measured the amount of MAP kinase phosphorylation (see Experimental Procedures) that was induced in the RGCs in response to treatment with BDNF and/or cAMP elevation. The above experiments were repeated except that the extent of MAP kinase activation in the RGCs in response to treatment with BDNF and/or cAMP elevation was directly assessed by Western blotting with a specific antibody for a phosphorylation-specific epitope on MAP kinase (see Experimental Procedures). As shown in Figure 5A, virtually no MAP kinase phosphorylation was observed in response to control treatment (nothing) or cAMP elevation. A transient activation at 0.5 and 1 hr, but not at 2 hr, was observed in response to BDNF alone, and a strong sustained response at all time points was observed in response to treatment with BDNF and cAMP elevation together. After a 2 hr treatment with BDNF and cAMP elevation together, the amount of activated MAP kinase was about 10-fold higher than that induced by BDNF treatment alone. Under all conditions, the total amount of MAP kinase protein was comparable, as shown by Western blotting with an antibody directed against an epitope present on both inactive and active MAP kinase (Figure 5A). In contrast to the cultures of RGC neurons, the neurotrophin NGF was sufficient alone to produce a sustained activation of MAP kinase in acutely isolated purified SCG neurons (Figure 5B). cAMP elevation did not potentiate this effect and interestingly, by itself was not sufficient to activate MAP kinase. Taken together, these results show that a single neurotrophin is sufficient to sustainedly activate MAP kinase in SCGs, but not RGCs, and suggest that cAMP may enhance RGC responsiveness to BDNF by acting proximally to MAP kinase.
Figure 5.
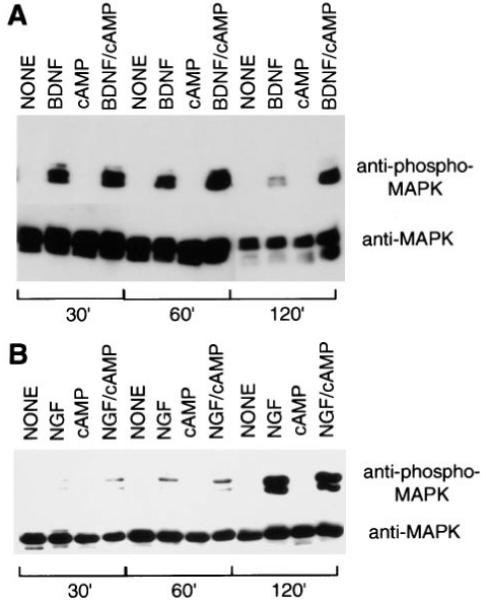
Effects of Neurotrophins and cAMP Elevation on MAP Kinase Activation in Purified RGCs and SCG Neurons
Acutely isolated RGCs (A) or SCGs (B) were treated with neurotrophins and/or cAMP elevation as described in the legend of Figure 2. After various time perods, the cells were lysed and a Western blot analysis was performed to detect the amount of phosphorylated MAP kinase (upper half of each panel) as well as the total amount of MAP kinase protein (bottom half of each panel).
Effects of MAP Kinase Inhibition on Survival of Purified Neurons
Purified RGCs do not respond to BDNF alone either by surviving or by sustainedly activating MAP kinase. To begin to address whether the failure of RGCs to survive in response to BDNF is at least in part caused by the failure to activate MAP kinase, we next determined whether RGC survival in response to BDNF and cAMP elevation is decreased by MAP kinase inhibition. Purified P8 RGCs were cultured for 3 days in the presence or absence of the specific MAP kinase kinase (MEK) inhibitor PD98059 (20 μM), which blocks the ability of MEK to activate MAP kinase (Alessi et al., 1995). The survival of the RGCs was decreased about 5-fold in the presence of PD98059 (Figure 6A). However, two different inhibitors of PI3 kinase (wortmannin, 1 μM; LY294002, 10 μM; Kimura et al., 1994; Sanchez-Margalet et al., 1994), another enzyme activated by neurotrophins that has been implicated in the promotion of cerebellar granule neuron survival (Dudek et al., 1997), did not significantly decrease BDNF-stimulated survival of RGCs. In contrast, inhibitors of MEK and PI-3 kinase each signficantly decreased NGF-stimulated survival of purified SCGs cultured under the same conditions and together produced a larger decrease of >50% in their survival (Figure 6B). These results suggest that activation of MAP kinase is necessary for the promotion of RGC survival mediated by the combination of BDNF and cAMP elevation.
Figure 6.
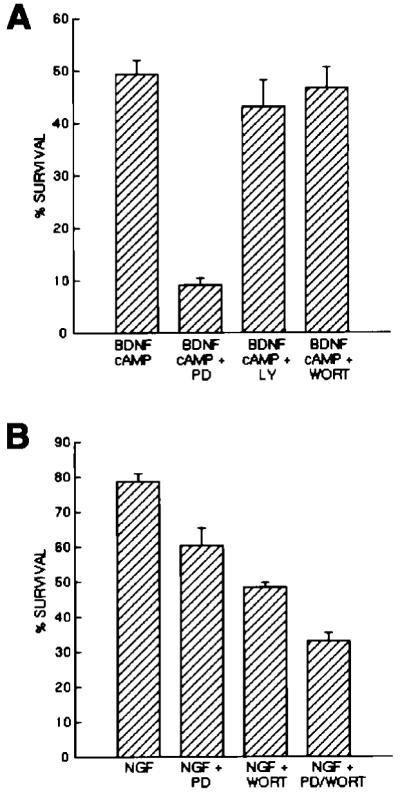
Effects of Pharmacological Inhibition of MAP Kinase and Pl3 Kinase on RGC and SCG Neuron Survival
Approximately 5,000 purified P8 RGCs (A) or P2 SCGs (B) were plated into each well of a 96-well tissue culture plate into serum-free medium containing the indicated substances. Drugs were used at concentrations previously determined to specifically inhibit MEK (PD98059, 20 μM) and Pl3 kinase (Ly294002, 10 μM; wortmannin, 1 μM). After 3 days of culture, the percentage of cells surviving in each condition was determined by the MTT assay. All values are means ± SEM (n = 3).
Effects of Depolarization and cAMP on Surface TrkB Levels of Retinal Ganglion Cells
One simple explanation for the inability of RGCs to respond to BDNF upstream of MAP kinase is that they may lack sufficient levels of surface TrkB; if so, then cAMP elevation may promote BDNF responsiveness by increasing the level of surface TrkB. To test this possibility, we assessed the amount of surface TrkB by biotinylating cell surface proteins. Purified P8 RGCs were cultured in Neurobasal (NB) containing 0.02% bovine serum albumin (BSA) for 1 hr after plating, and then equal numbers of cells were treated with or without forskolin. After 1 hr, the cell surfaces were biotinylated and the biotinylated proteins were examined by Western blotting with affinity-purified polyclonal antibodies directed against the extracellular domain of TrkB (see Experimental Procedures). As shown in Figure 7, only one band was detected by the anti-TrkB antibody, and it had the expected molecular weight of full length TrkB containing a tyrosine kinase domain (145kD) (Figure 7). No band was detectable in the molecular weight range of ∼95 kD, the expected size of truncated TrkB variants lacking most of the cytoplasmic domain (data not shown). The amount of TrkB on the surfaces of RGCs that had been treated with forskolin, however, was increased by 3.6 ± 0.2-fold (mean ± SEM, average of three separate experiments) (see Experimental Procedures) (Figure 7). If the cells were pretreated with BDNF (10 ng/ml) during the 2 hr incubation period to biotinylation, then the amount of surface biotinylated TrkB fell by 2-fold (Figure 7), suggesting that internalization of surface TrkB was elicited by the BDNF. Neurotrophin-stimulated internalization of Trk receptors has been reported previously (e.g., Grimes et al., 1997). Similar results were obtained when the amount of surface TrkB was assessed by the ability of iodinated NT4/5 to be cross-linked to surface binding sites on RGCs (data not shown; P. DeStefano and A. M.-F., unpublished data). In contrast, the amount of surface cadherins that could be biotinylated, as assessed by Western blotting with a pan-cadherin antibody, was not affected by cAMP elevation (data not shown). These findings demonstrate that cAMP elevation greatly increases the number of TrkB receptors on the surfaces of RGCs.
Figure 7.

Western Blot Analysis of Cell Surface TrkB on Purified RGCs
Acutely isolated, purified P8 RGCs were plated at about 700,000 cells per 10 cm tissue culture dish, allowed to recover for 1 hr, and then treated for 1 hr with either forskolin (5 μM), in order to elevate cAMP, or BDNF (50 ng/ml) as indicated. Surface proteins were biotinylated, and the biotinylated proteins were analyzed by Western blotting with an affinity-purified anti-TrkB antibody. Only bands corresponding to full length TrkB (∼145 kDa) were detected.
To further explore the effects of cAMP elevation on surface TrkB levels, we immunostained RGCs that had or had not been treated with forskolin with the same TrkB antibodies, which bind to an extracellular epitope of TrkB (see Experimental Procedures). Prior to staining, the cells were lightly fixed with 4% paraformaldehyde to prevent internalization of surface receptors after antibody cross-linking. Bright, ring-like staining of the plasma membrane of nearly 50% of the RGCs was observed after 1 hr of cAMP elevation but was seen in only 12% of cells that had not been treated with cAMP elevation (Figures 8A, 8B, and 10A). A large increase in surface TrkB immunoreactivity after 1 hr of cAMP treatment was also observed by confocal microscopy (Figures 9A and 9B). Similarly, depolarization with K+ or glutamate agonists significantly increased the percentage of cells that had bright surface TrkB immunoreactivity (Figure 10A). cAMP elevation did not increase immunoreactivity for all surface proteins on the RGCs; for instance, nearly all RGCs stained brightly on their surfaces with an anti-integrin-β 1 antibody (de Curtis et al., 1991) (see Experimental Procedures) regardless of whether or not their cAMP levels were increased (Figures 8E and 8F). In contrast to the RGCs, nearly all of the purified nodose ganglion neurons stained brightly on their surfaces with anti-TrkB antibodies regardless of whether or not they had been treated to elevate cAMP (Figures 8G and 8H).
Figure 8.

Immunofluorescence Micrographs of Purified RGCs and Nodose Ganglion Neurons Stained for Various Surface Proteins
Approximately 5,000 RGCs (A-F) or nodose ganglion neurons (G and H) were plated onto PDL-coated glass coverslips for 1 hr prior to treatment for 1 hr with (B, D, F, and H) or without (A, C, E, and G) forskolin (5 μM) to elevate cAMP levels. For nodose ganglion neurons, IBMX (0.1 mM) was also added to elevate cAMP. After incubation, the coverslips were fixed lightly with paraformaldehyde and in some cases permeabilized with 0.4% Triton X-100 (C and D); in most cases, the cells were not permeabilized (A, B, and E-H). Cells were then immunostained with the indicated primary antibody, which was detected by using a FITC-conjugated secondary antibody. Cells were stained with either an antibody that detects an extracellular epitope of TrkB (A-D, G, and H) or the integrin-β1 subunit (E and F). Scale bar, 25 μm.
Figure 10.

Effects of cAMP Elevation, Depolarization, and Cycloheximide on Surface TrkB Immunoreactivity
Approximately 10,000 acutely isolated, purified P8 RGCs were plated onto PDL-coated glass coverslips for 1 hr to allow recovery. In (A), the cells were then treated with the indicated substances for 1 hr prior to immunostaining the unpermeabilized cells with the anti-TrkB antibodies. cAMP elevation was induced by forskolin (5 μM), and depolarization was elicited either by KCl (50 mM) or by glutamate receptor activation (a combination of NMDA, 200 μM; kainate, 200 μM; and quisqualate, 200 μM). In (B), the cells were incubated in the indicated substances for various time periods prior to immunostaining the unpermeabilized cells with the anti-TrkB antibodies. Cycloheximide was used at 100 μM.
Figure 9.

Confocal Micrographs of Retinal Ganglion Cells Stained with Anti-TrkB Antibodies
Approximately 10,000 acutely isolated, purified P8 RGCs were plated onto PDL-coated glass coverslips for 1 hr to allow recovery and then treated with medium lacking (A) or containing (B) forskolin for 1 hr. The cells were then permeabilized and stained with the anti-TrkB antibodies prior to examination in a Multiprobe 2010 laser scanning confocal microscope (Molecular Dynamics). In (A), little surface immunoreactivity is detected; after cAMP treatment in (B), strong surface immunoreactivity is apparent. Scale bar, 10 μm.
cAMP elevation could increase surface TrkB levels either by increasing the rate of production of TrkB (for instance, by increasing transcription rate) or by increasing the rate of net addition of TrkB protein to the membrane (for instance, by inducing recruitment of TrkB from intracellular stores). Consistent with the latter possibility, we observed bright intracellular TrkB staining in the RGCs regardless of whether they were treated with cAMP elevation (Figures 8C and 8D). To further distinguish between these possibilities, we examined whether cycloheximide, an inhibitor of protein synthesis, would block the ability of cAMP elevation to increase surface TrkB immunoreactivity. One hundred micromolars cycloheximide, a concentration that is sufficient to block protein synthesis in PC12 cells (Twiss and Shooter, 1995) and to block apoptosis of RGCs cultured in the absence of peptide trophic factors (data not shown) did not decrease the ability of cAMP to increase surface TrkB immunoreactivity (Figure 10B), indicating that cAMP does not act by increasing the rate of transcription or protein synthesis. To determine whether cAMP acts by recruiting TrkB to the plasma membrane from intracellular stores, we measured the time course of the cAMP-mediated increase in surface TrkB immunoreactivity. By 30 min, the percentage of cells with surface TrkB immunoreactivity was significantly greater than in controls (no cAMP elevation) (Figure 10B), indicating that cAMP increases surface TrkB too rapidly to be accounted for by an increase in transcription or translation of TrkB mRNA.
Effects of cAMP Elevation on Surface TrkB Immunoreactivity of Spinal Motor Neurons
To explore whether TrkB on the surface of other types of CNS neurons is also regulated by cAMP elevation, we next examined the behavior of purified cultures of embryonic day 15 (E15) SMNs. We recently found that cAMP elevation on its own is sufficient to promote SMN survival (Hanson et al., 1998). Low concentrations of the protein kinase A inhibitor H89 (10 μM), however, blocked the survival induced by growth factors, suggesting that basal levels of cAMP might normally enhance growth factor responsiveness. To determine whether cAMP elevation increases surface TrkB immunoreactivity on the SMNs, we cultured the SMNs in NB containing 0.02% BSA and either nothing or forskolin together with IBMX for 1 hr and then immunostained the cultures with the anti-TrkB antibodies that bind to an extracellular epitope of rat TrkB. Prior to staining, the cells were lightly fixed with 4% paraformaldehyde to prevent internalization of surface receptors after antibody cross-linking. Bright, ring-like staining of the plasma membrane of ∼80% of the SMNs as well as bright staining along the processes was observed after cAMP elevation but was seen in only ∼20% of the cells that had not been treated with agents that elevate cAMP (Figures 11A, 11C, and 12). When the cells were permeabilized with Triton X-100 prior to immunostaining, however, bright intracellular TrkB immunoreactivity was present in the SMNs regardless of whether cAMP was elevated (Figure 11B).
Figure 11.
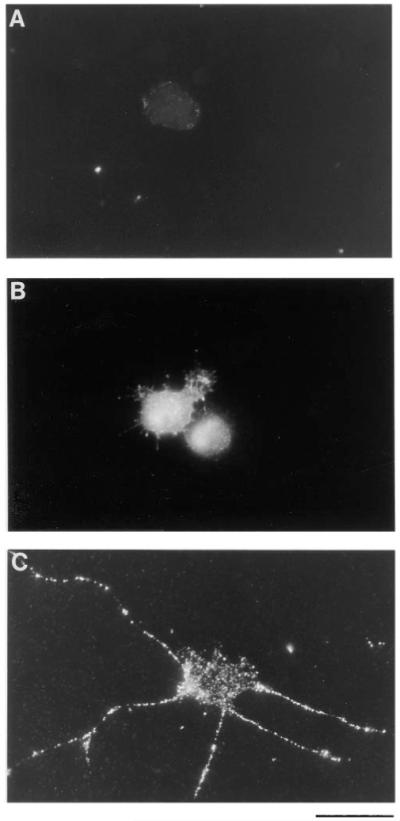
Immunofluorescence Micrographs of TrkB Immunoreactivity on Purified SMNs in Culture
Approximately 5,000 SMNs were plated onto PDL-coated glass coverslips for 1 hr without (A and B) or with (C) forskolin and IBMX to elevate their cAMP levels. After incubation, the coverslips were fixed lightly with paraformaldehyde and in some cases permeabilized with 0.4% Triton X-100 (B); in most cases, the cells were not permeabilized (A and C). Cells were then immunostained with anti-TrkB anti-bodies that bind to an extracellular epitope of rat TrkB. The primary antibodies were detected by using a FITC-conjugated secondary antibody. Scale bar, 50 μm.
Figure 12.

Effects of cAMP Elevation on Surface TrkB Immunoreactivity
Approximately 5,000 purified SMNs were plated onto PDL-coated glass coverslips for 1 hr in serum-free medium containing the indicated substances prior to immunostaining the unpermeabilized cells with the anti-TrkB antibodies. The percentage of cells that were brightly surface immunoreactive for TrkB was counted in each condition. Results are means ± SEM (n = 3) of a representative experiment; this experiment was repeated twice.
Discussion
Sympathetic and Sensory Ganglion Neurons Are Intrinsically Responsive to Neurotrophins but Retinal Ganglion Cells Need Signals from Other Cells to Be Responsive
The possibility that neuronal responsiveness to neurotrophins could be rapidly modified has received comparatively little previous attention. Results in this paper demonstrate a remarkable difference between the abilities of PNS neurons and RGCs to respond to neurotrophins. PNS neuron types, including SCG sympathetic neurons and nodose ganglion sensory neurons, respond well to single neurotrophins such as NGF or BDNF, measured both by survival of the majority of cells and by persistent activation of the MAP kinase signaling pathway. In contrast, RGCs do not survive or persistently activate MAP kinase in response to neurotrophins such as BDNF unless they are stimulated by depolarization or cAMP elevation. These differences cannot be attributed to differences in preparation or cell culture, as all of the cells were purified and cultured by the same stringent procedures. They also do not simply reflect a difference in signaling by different neurotrophins, as nodose ganglion neurons are able to respond to BDNF without a requirement for a costimulus such as cAMP elevation. Thus, the present results demonstrate unequivocally that PNS neurons but not RGCs are intrinsically programmed to respond to neurotrophins in the absence of inducing stimuli.
Although we have focused on responsiveness to neurotrophins, this difference is likely to be true of responsiveness to peptide trophic factors in general. Purified PNS neurons cultured in serum-free medium, such as parasympathetic ciliary neurons and SCG sympathetic neurons, have previously been shown to respond well to a wide variety of single peptide trophic factors including CNTF and NGF, respectively (Levi-Montalcini, 1987; Barde, 1989, 1990). In contrast, purified RGCs cultured in serum-free medium so far have not been observed to survive in response to a wide variety of peptides including CNTF, leukemia inhibitory factor (LIF), IGF-1, fibroblast growth factor (FGF), and transforming growth factor α (TGFα), as well as BDNF, unless their cAMP levels are increased (Meyer-Franke et al., 1995). One plausible explanation for the difference between PNS neurons and RGCs is the possibility that PNS neurons might normally have higher basal levels of cAMP. However, the protein kinase A inhibitor Rp-cAMP (200 μM) does not significantly decrease the survival of purified SCG neurons cultured in NGF (our unpublished data), although it does drastically decrease the survival of RGCs in response to BDNF and forskolin (Meyer-Franke et al., 1995).
This lack of intrinsic responsiveness to peptides does not appear to be limited to retinal ganglion cells but is increasingly being observed with many other CNS neuron types; for instance, cerebral cortical neurons do not respond to neurotrophins in the absence of electrical activity (McAllister et al., 1996). Similarly, the survival of purified spinal motor neurons in culture is only weakly promoted by single peptide trophic factors, particularly when cultured at low density, but is significantly increased by cAMP elevation and is almost completely abolished by protein kinase A inhibition (Hanson et al., 1998).
The present results indicate that RGCs must receive signals from neighboring cells that enable them to respond to neurotrophic factors. RGCs in mixed retinal cultures in serum-free medium have been shown to be responsive to BDNF and other trophic factors and to lose this responsiveness only when purified away from other cell types (Meyer-Franke et al., 1995). The ability of depolarization to enhance responsiveness suggests that depolarizing synaptic contacts from other cells are likely to mediate this cell-cell interaction. Not only do purified RGCs lose synaptic inputs from other cell types, but they are unable to form efficient synaptic contacts upon themselves because of the absence of glia (Pfrieger and Barres, 1997). Thus, although RGCs in mixed retinal cultures are spontaneously active, RGCs in purified cultures are electrically quiescent, which can account for their loss of responsiveness to trophic factors such as BDNF.
Depolarization and cAMP Rapidly Increase Surface TrkB on CNS Neurons
We observed that cAMP elevation rapidly induces a large increase in the number of full length TrkB receptors on the plasma membrane of retinal ganglion cells. cAMP elevation increased surface full length TrkB by nearly 4-fold, as measured by the amount of TrkB accessible to surface biotinylation, and produced a similar increase in the percentage of cells with detectable surface TrkB immunoreactivity. Depolarization, which increases cAMP levels in RGCs by promoting calcium influx and thus activating a type-1 adenylyl cyclase expressed by RGCs (Xia et al., 1991; S. Shen and B. A. B., unpublished data) produced a similar increase in surface TrkB levels. cAMP elevation also rapidly increased the levels of TrkB on the surfaces of SMNs, but this effect was not mimicked by depolarization, which does not increase cAMP in SMNs (Hanson et al., 1998).
How does cAMP elevation increase surface levels of TrkB? The presence of high levels of intracellular levels of TrkB immunoreactivity regardless of cAMP elevation, the inability of cycloheximide to block the effects of cAMP, and the comparatively rapid time course of increase in surface TrkB that is too rapid to be accounted for by a transcriptional or translational increase all provide strong evidence for a recruitment of TrkB from intracellular stores. The characteristics and kinetics of this recruitment are remarkably similar to an already well-described mechanism for the transient modification of cell surface membranes. To facilitate uptake of glucose into adipocytes and muscle cells, insulin induces the translocation of intracellular vesicles containing the glucose transporter GLUT4 to the cell surface, where they fuse (Cushman and Wardzala, 1980; Suzuki and Kono, 1980). A similar class of recycling vesicles has recently been described in many cell types including a neuronal cell line (Herman et al., 1994). Although insulin does not recruit these glucose transporters by elevating cAMP, cAMP elevation induced by hormones has previously been shown to induce a similar recruitment from intracellular stores of other membrane proteins, such as ion pumps and transporters, in a variety of nonneural cell types (Lewis and de Moura, 1984; Schwartz and Al-Awqati, 1986; Wade, 1986; Barres et al., 1989; Lencer et al., 1990; Murer and Biber, 1996; Yao et al., 1996). Thus, the simplest explanation for our findings is that cAMP induces the rapid translocation of TrkB stored in an intracellular synaptic vesicle-like organelle to the plasma membrane of CNS neurons. Our observation that cAMP elevation does not regulate the levels of all membrane proteins on the RGCs, such as integrin-β1 receptors and cadherins, is consistent with this possibility. Because we did not observe a cAMP-induced increase in TrkB on nodose ganglion neurons, we hypothesize that TrkB (as well as other receptors for trophic peptides) may be sorted to a regulated secretory pathway in CNS neurons but secreted constitutively in PNS neurons. A crucial future test of this hypothesis will be to examine and compare the intracellular ultrastructural distribution of TrkB in CNS and PNS neurons.
Our results do not rule out the additional possibility that depolarization or cAMP elevation could also transcriptionally regulate TrkB levels over much longer time periods. For instance, Birren et al. (1992) demonstrated that depolarization and cAMP elevation increase TrkA expression in the neuronal cell line over a period of several days in culture.
Does the cAMP-Mediated Increase in Surface TrkB on RGCs Account for the Enhancement of Responsiveness to BDNF?
Our data strongly suggest but do not yet conclusively prove that the rapid increase in surface TrkB in response to cAMP elevation is the explanation for how cAMP elevation enhances responsiveness of RGCs to BDNF. Future experiments should examine whether overexpression of TrkB is sufficient to restore responsiveness of RGCs to BDNF in the absence of cAMP elevation and find out whether blockade of the cAMP-induced TrkB recruitment to the membrane prevents the enhanced responsiveness.
Several arguments, however, point strongly toward the increase in surface receptor levels as being causally associated with the enhancement of responsiveness. Receptor number is clearly the simplest possible regulator of responsiveness, and changes in receptor levels correlated closely with the changes in RGC responsiveness to BDNF. Only the CNS neurons and not the PNS neurons had the large cAMP-mediated increase in surface TrkB, and only the CNS neurons had increased responsiveness to BDNF. Control of receptor levels in suggested by the requirement for cAMP elevation in order for BDNF to elicit MAP kinase activation and nuclear translocation, which places the site of action of cAMP as proximal to MAP kinase. Interestingly, we found that in the absence of cAMP elevation or depolarization, many RGCs displayed a transient responsiveness to BDNF as measured by MAP kinase activation and translocation, corresponding to the low levels of surface TrkB that were initially present under these conditions. The low level of TrkB on the surface of RGCs in the absence of forskolin could be due to low levels of constitutive secretion of TrkB to the plasma membrane or could instead be the result of recruitment induced by basal levels of cAMP. A simple explanation for this transient response is that BDNF induces internalization of TrkB, thus depleting surface levels, unless cAMP levels are sufficient to induce recruitment to the surface of additional TrkB from intracellular stores.
Implications of Activity-Dependent Recruitment of TrkB for Activity-Dependent Plasticity and Disease Processes
The present results raise the question of whether the responsiveness of CNS neurons in vivo to peptide trophic factors is normally dynamically regulated by electrical activity and cAMP elevation. Consistent with this possibility, experiments in progress in our lab demonstrate that physiological levels of activity normally play a crucial role in enhancing the responsiveness of RGCs in intact postnatal retina (S. Shen and B. A. B., unpublished data.).
Why regulate trophic responsiveness? One provocative possibility is that activity-dependent control of trophic responsiveness may be part of a mechanism that controls the activity-dependent structural plasticity of CNS neurons during development. Intracellular and surface TrkB immunoreactivity extends along RGC and SMN somas, dendrites, axons, and growth cones in vitro (our unpublished data). In the RGCs, depolarization rapidly elevates cAMP in the dendrites, axons, and growth cones as well as in the soma (S. Shen and B. A. B., unpublished data). We have observed large cAMP-mediated increases in surface TrkB along dendrites, somas, axons, and growth cones (e.g., Figure 11), raising the question of whether activity-dependent changes in surface TrkB levels could play an important role in activity-dependent plasticity. Such a mechanism would simply account for activity-dependent growth of dendrites of developing cerebral cortical neurons in ferret brain slices in response to neurotrophins (McAllister et al., 1996). If the surfaces of active processes have more TrkB, then active processes would be better able to compete for and respond to neurotrophins. In addition, as TrkB-bound BDNF is rapidly internalized and cleared from the extracellular fluid, active neurons may be better able to deprive nearby, inactive neurons of BDNF. Active processes may be better able to respond to glutamate, as TrkB activation potentiates NMDA responsiveness (Levine et al., 1997, Soc. Neurosci., abstract). Lastly, activity-dependent, local recruitment of trophic receptors such as TrkB onto the surface of a potentiated synapse could constitute the “synaptic tags” postulated to help maintain late phase, long-term potentiation (Frey and Morris, 1997).
Most importantly, our findings raise the question of whether the intrinsic differences in trophic responsiveness between CNS and PNS neurons help to explain the lesser ability of the CNS, compared to the PNS, to survive and regenerate after injury. After CNS injury, electrical activity or intracellular cAMP levels may decrease, for instance as a consequence of axotomy, inflammation, or synaptic stripping by microglia. This loss of activity would be expected to produce loss of responsiveness to endogenous trophic factors and thus death of cells and/or inability of their processes to regenerate in response to these factors. If so, treatment aimed at promoting the survival of injured neurons by delivery of exogenous survival factors will not be effective unless responsiveness to these signals is simultaneously enhanced.
Experimental Procedures
Detailed step-by-step protocols for all procedures are available upon request (barres@stanford.edu).
Reagents
Several recombinant trophic factors were generously provided by Regeneron (human BDNF, NT-4/5, and CNTF). Other recombinant peptide trophic factors were obtained from Peprotech (NJ). Insulin was obtained from Sigma.
Preparation of Retinal Suspensions
P8 retinas were obtained from Sprague-Dawley rats (Simonsen Labs, CA) and dissociated enzymatically to make a suspension of single cells essentially as described by Huettner and Baughman (1986). Briefly, the tissue was incubated at 37°C for 30 min in a papain solution (15 units/ml for retina; Worthington) in an Earle’s Balanced Salt Solution (EBSS; Gibco) containing l-cysteine. The tissue was then triturated sequentially with a 1 ml pipette in a solution containing ovomucoid (2 mg/ml; Boehringer-Mannheim), DNAse (0.004%; Sigma), and BSA (1 mg/ml; Sigma) to yield a suspension of single cells. After centrifugation at 200 × g, the cells were rewashed in another ovomucoid/BSA solution (10 mg/ml each).
Purification of Retinal Ganglion Cells
Retinal ganglion cells from P8 Sprague-Dawley rats were purified essentially as previously described (Barres et al., 1988). By using sequential immunopanning, RGCs were isolated to a >99.5% purity. Typically, about 20% of the RGCs were isolated, which is ∼40,000 RGCs per P8 animal. A brief description of the procedure follows.
Preparation of Panning Plates
Secondary antibodies included affinity-purified goat anti-mouse IgM (μ-chain specific; Jackson) and affinity-purified goat anti-rabbit IgG (H + L; Jackson). Primary antibodies included monoclonal supernatant IgM antibody against mouse Thy1.1 (T11D7e2, American Type Culture Collection, T. I. B. 103). Petri dishes (two 150 × 15 mm and one 100 × 15 mm; Fisher) were incubated with 5-15 ml of Tris buffer solution (pH 9.5) with 10 μg/ml of secondary antibody (anti-mouse IgM on the small dish or anti-rabbit IgG on the large dishes) for 12 hr at 4°C. The dishes were washed 3 times with 8 ml of phosphate buffered saline (PBS), and the small dish was further incubated with 5 ml of Thy 1.1 IgM monoclonal supernatant for 2 hr at room temperature. The supernatant was removed and the plate was washed 3 times with PBS. To prevent nonspecific binding of cells to the panning dish, 5-15 ml of PBS with 2 mg/ml BSA was placed on the plates for 20 min.
Panning Procedure
The retinal suspension was incubated in anti-rat macrophage antiserum (Axell; 1:100) for 20 min, centrifuged, resuspended in PBS, and incubated on a 150 mm anti-rabbit IgG panning plate at room temperature for 45 min. The plate was gently swirled every 15 min to ensure access of all cells to the surface of the plate. If cells from greater than eight retinas were panned, the nonadherent cells were transferred to another 150 mm anti-rabbit IgG panning plate for another 30 min. The nonadherent cells were removed with the suspension, filtered through 15 μm Nitex mesh (Tetko), and placed on the T11D7 panning plate. The cells were incubated on the plate as described above. After 45 min, plates were washed 8 times with 10 ml of PBS and swirled moderately vigorously to dislodge nonadherent cells. Progress of nonadherent cell removal was monitored under the microscope. The washing was terminated when only adherent cells remained.
Removing Adherent Cells from the Plate
Four mililiters of a trypsin solution (0.125%) was prepared by diluting a trypsin 20× stock (Sigma) into EBSS. Cells on the panning dish were incubated with this solution for 10 min in a 10% CO2 incubator at 37°C. The cells were dislodged by gently pipetting trypsin solution around the plate. Ten mililiters of a 25% fetal calf serum (FCS) solution were added to inactivate the trypsin, and the cells were spun and collected as above.
Culture of Purified Retinal Ganglion Cells
Approximately 5,000 purified retinal ganglion cells were cultured in 96-well plates (Falcon) that had been coated with poly-d-lysine (PDL) (70 kD, 10 μg/ml; Sigma) followed by merosin (2 μg/ml; Telios/Gibco) in 100 μl of a serum-free medium containing NB, a recently described medium that has been optimized for neuronal cell culture (Brewer et al., 1993). The use of NB Medium instead of Dulbecco’s Modified Eagle’s Medium (DMEM) was essential for good survival of the purified RGCs. The serum-free additives included BSA, selenium, putrescine, thryoxine, tri-iodothyronine, transferrin, and progesterone (modified from Bottenstein and Sato, 1979 as previously described in Lillien and Raff, 1990) (B-S medium), sodium pyruvate (1 mM), glutamine (1 mM), the B27 serum-free supplement (1:50; Gibco), and the indicated trophic factors.
Purification of SCG Sympathetic and Nodose Ganglion Sensory Neurons
The procedure was similar to that used to isolate and purify RGCs. Briefly, SCGs or nodose ganglia were obtained from P2 rats and digested in papain (33 U/ml in DPBS; Worthington) for 45 min at 37°C. The tissue was then triturated in a solution containing ovomucoid and BSA through 21 and 23 gauge syringe needles, filtered through nitex mesh, spun down and resuspended in an ovomucoid/BSA solution, and centrifuged. The pellet was resuspended in DPBS containing 0.04% BSA and insulin (5 μg/ml).
The cell suspension was immunopanned by incubating sequentially on a Petri dish coated with the monoclonal RAN-2 antibody (Bartlett et al., 1981) for 30 min to remove some satellite cells, on a Petri dish coated with the monoclonal O4 antibody (Sommer and Schachner, 1981, 1982) for 30 min to remove Schwann cells, and finally on a Petri dish coated with the anti-Thy1.1 monoclonal T11D7 antibody for 1 hr to select for the SCG neurons or nodose ganglion neurons. A rat SCG ganglion contains ∼25,000 neurons; this procedure yielded 20,000 SCG neurons per ganglion, a yield of about 75% of the SCG neurons, with greater than 95% purity. A rat nodose ganglion contains ∼6,000 neurons per ganglion; our purification procedure yielded ∼5,000 nodose ganglion neurons per ganglion, a yield of ∼80% of the nodose neurons, with >95% purity.
E15 spinal motor neurons were purified and cultured as previously described (Hanson et al., 1998).
MTT Survival Assay
The MTT survival assay was performed as described by Mosmann (1983). MTT (Sigma) was dissolved in PBS at 5 mg/ml and sterilized by passage through a Millipore filter (0.22 μm). This stock solution was added to the culture well (1:9) and incubated at 37°C for 1 hr. Viable cells with active mitochondria cleaved the tetrazolium ring into a visible dark blue formazan reaction product. The viable and dead cells in each well were counted by bright field microscopy. In all cases, the percentage of survival determined by the MTT assay was nearly identical to the values determined by morphology alone. All values are given as means ± SEM of at least three cultures. The results of representative experiments are shown.
Measurement of Nuclear Translocation of MAP Kinase
Approximately 20,000 purified RGCs or SCG neurons were plated onto PDL-coated glass coverslips. Cells were incubated in 500 μl NB containing 0.02% crystalline BSA (Sigma) for 1 hr to recover. After 1 hr, 500 μl of NB/0.02% BSA was added with either no factors (control), 100 ng/ml BDNF (RGCs and nodose ganglion neurons) or 20 ng/ml NGF (SCG neurons), and/or forskolin (5 μM). Cells were fixed for 30 min in 4% paraformaldehyde after various incubation time points, permeabilized in blocking buffer containing 0.4% Triton X-100 for 30 min, and then incubated for 1 hr with a polyclonal antiserum directed against MAP Kinase (10 μg/ml; Santa Cruz; cat#sc-93-G, reactive with ERK1/ERK2). The primary antibody was detected with a FITC-coupled anti-rabbit IgG (10 μg/ml; Jackson Immunoresearch) for 1 hr. Cells with bright nuclear staining were counted as positive. Two hundred cells were evaluated per cover slip and three coverslips were assayed per condition.
Western Blot Analysis for Active MAPK
Approximately 50,000 purified RGCs or SCG neurons were plated onto PDL-coated wells in a 48-well tissue culture plate (Falcon). Cells were allowed to recover for 1 hr in a solution containing NB/0.02% BSA at 37°C. After 1 hr, either NB/0.02% BSA alone (control) or containing 50 ng/ml BDNF (RGCs), 20 ng/ml NGF (SCGs), and/or forskolin (5 μM) was added. After different time points, cells were washed twice with cold calcium and magnesiusm-free Earle’s balanced salt solution (EBSS; Gibco/BRL). They were then lysed in sample buffer, boiled for 5 min, and the lysates were run on 8% polyacrylamide gels. The separated proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Qiagen) and incubated in a blocking buffer (PBS containing 5% nonfat baby milk, 1% BSA, 0.01% gelatine, and 0.1% Nonidet P-40) for 1 hr. The membranes were incubated in the blocking buffer containing a rabbit anti-phosphorylated MAP kinase antiserum (anti-dually phosphorylated MAPK; 10 μg/ml; Promega; cat# V6671, recognizes p44 ERK1 and p42 ERK2 phosphorylated amino acid residues Thr-202 and Tyr-204) overnight at 4°C. Subsequently, the labeled proteins were visualized by incubation with an horseradish peroxidase- (HRP-) conjugated anti-rabbit IgG (10 μg/ml; Jackson Immunoresearch) followed by development with a chemiluminescence substrate for HRP (Pierce). To determine the amounts of MAPK present in each lane, the PVDF membranes were stripped of the antibodies in 62.5 mM Tris/HCl (pH 6.7) containing 100 μM 2-Mercaptoethanol and 2% SDS for 30 min at 60°C and incubated with a rabbit anti-MAPK antiserum (Santa Cruz; cat# sc-93-G) and then visualized as described above.
Generation of a Polyclonal Antiserum Directed against the Extracellular Domain of Rat TrkB
An affinity-purified polyclonal antiserum directed against the extracellular domain of rat TrkB that did not cross-react with trkA or TrkB was raised by G. Wilkinson and L. Reichardt as previously described (Wilkinson, 1997; Farinas et al., 1998). In brief, a cDNA segment encoding the extracellular domain of rat TrkB (Rtb-ex) was obtained by PCR to amplify that portion of the cDNA from a full length cDNA clone. The olignucleotide sequences used to amplify rTrkB-ex were (5′-3′ direction) CCC GAA TTC GCC ACC ATG TCG CCC TGG CCG AGG TGG CAT TGG and (3′-5′ direction) GTT GCT GAC CAA ACC AAT CGG GAG CAT CAC CAT CAC CAT CAC GGC GAG CAG AAG CTG ATC TCC GAG GAG GAC CTG TAG CTC GAG AAG G. To purify the encoded peptide, the amplified PCR fragments were cloned into a plasmid expression vector, and the constructs were transfected by using lipofectamine into large scale cultures of Cos cells. Supernatants were harvested and passed over a wheat germ lectin column. Lectin-binding proteins were eluted with 500 mM N-acetyl-d-glucosamine (Sigma), concentrated, and the purified rTrkB-ex was used to immunize a rabbit. The anti-TrkB antiserum was then affinity purified on a column of purified rTrkB-ex. The specificity of the antibody for TrkB was confirmed by Western blot analyses of extracts of Cos cells transfected with rat TrkA, rat TrkB, or rat TrkC expression vectors.
TrkB Immunofluorescence Staining
Approximately 20,000 RGCs or nodose ganglion neurons were plated onto PDL-coated coverslips. Cells were incubated in various solutions for several hours as described in the text. To immunostain for TrkB, the coverslips were fixed with 4% paraformaldehyde for 30 min at room temperature and then incubated for 30 min in a 50% goat serum solution containing 1% BSA and 100 mM l-lysine to block nonspecific binding. In some cases, when indicated, coverslips were incubated in Triton X-100 (0.4%) for 5 min in order to permeabilize the membrane. The cells were then incubated in the affinity-purified anti-TrkB antibody ∼5 μg/ml for 1 hr), which was detected by a fluorescein-coupled goat anti-rabbit lgG (10 μg/ml; Jackson). Similar results were obtained with an anti-TrkB antiserum obtained from Transduction Laboratories. Some cells were also stained with a rabbit anti-integrin-β1 subunit antiserum (Chemicon, AB1937). The coverslips were mounted in Citifluor on glass slides, sealed with nail varnish, and examined in a Nikon fluoresence microscope.
Surface Biotinylation and Western Blot Analysis
Acutely isolated, purified P8 RGCs were plated at ∼700,000 cells per 10 cm tissue culture dish, allowed to recover for 1 hr in NB containing 0.02% BSA, and then treated for 1 hr with either forskolin (5 μM) in order to elevate cAMP or BDNF (50 ng/ml)as indicated. The cell surfaces were then biotinylated, and the biotinylated proteins were examined by Western blotting with the affinity-purified anti-TrkB antibody as follows. The cells were washed with ice-cold PBS containing calcium and magnesium (pH 7.4) in order to stop cellular protein trafficking. Surface biotinylation was performed by adding Sulfo-NHS-LC-biotin (Pierce) at 250 μg/ml in PBS at 4°C for 30 min (Sargiacomo et al., 1989). This solution was removed, and the reaction was quenched by the addition of 10 mM ice-cold glycine in PBS at 4°C for 20 min. Subsequently, cells were washed 3 times with ice-cold PBS and lysed by addition of RIPA buffer (Barker and Shooter, 1994). Biotinylated proteins were immunoprecipitated with streptavidin-agarose (Pierce) and subjected to SDS-PAGE on 7.5% gels (Laemmli, 1970). After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose membranes (Schleicher and Schuell) and blocked by overnight incubation at 4°C in 5% nonfat dry milk in PBST (PBS containing 0.1% Tween 20). Western blot analysis of TrkB was performed by using an affinity-purified rabbit antibody raised against an extracellular domain of rat TrkB (see above). After incubation with HRP-conjugated anti-rabbit IgG (Sigma) to detect the primary antibody, bands were visualized by using a “Renaissance” chemoluminescence kit (DuPont) and XAR films (Kodak). The blots were also probed with a rabbbit anti-pan cadherin antibody (Sigma, C-3678). Stripping and reprobing were performed according to standard protocols (Canossa et al., 1996). Blots were scanned with a P. D. I. imaging system and analyzed with Quantity One Discovery Series software.
Acknowledgments
This research was possible thanks to the support of the National Eye Institute (RO1 EY11030) to B. A. B., the National Institutes of Health (MH48200) and the Howard Hughes Medical Institute to L. F. R., an individual predoctoral fellowship from the National Science Foundation and support from the National Institutes of Health (GM 07449) to G. A. W., and a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft to A. M.-F. We thank Axel Franke for help with the preparation of the figures.
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel A. PD098059 is a specific inhibitor of the activation of mitogen activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- Barde YA. The nerve growth family. Prog. Growth Factor Res. 1990;2:237–248. doi: 10.1016/0955-2235(90)90021-b. [DOI] [PubMed] [Google Scholar]
- Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75NGFR reduces NGF binding to TrkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LLY. Immunological, morphological and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Barres BA, Chun L, Corey DP. Calcium current in cortical astrocytes: induction by cAMP and neurotransmitters. J. Neurosci. 1989;9:3169–3175. doi: 10.1523/JNEUROSCI.09-09-03169.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett PF, Noble MD, Pruss RM, Raff MC, Rattray S, Williams CA. Rat neural antigen-2 (RAN-2): a cell surface antigen on astrocytes, ependymal cells, Muller cells and leptomeninges defined by a monoclonal antibody. Brain Res. 1981;204:339–351. doi: 10.1016/0006-8993(81)90593-x. [DOI] [PubMed] [Google Scholar]
- Birren SJ, Verdi JM, Anderson DJ. Membrane depolarization induces p140trk and NGF responsiveness but not p75LNGFR in MAH cells. Science. 1992;257:395–397. doi: 10.1126/science.1321502. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. J. Neurosci. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer J, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Canossa M, Twiss JL, Verity AN, Shooter EM. p75NGFR and TrkA receptors collaborate to rapidly activate a p75NGFR associated protein kinase. EMBO J. 1996;15:3369–3376. [PMC free article] [PubMed] [Google Scholar]
- Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 1980;255:4758–4762. [PubMed] [Google Scholar]
- de Curtis I, Quaranta V, Tamura RN, Reichardt LF. Laminin receptors in the retina. J. Cell Biol. 1991;113:405–415. doi: 10.1083/jcb.113.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal R, Kaplan DR, Greenberg ME. Regulation of the neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Farinas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia. Neuron. 1998;21:325–334. doi: 10.1016/s0896-6273(00)80542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin JL, Johnson EM. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr. Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J. Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RGM. Synaptic tagging and long term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Gosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–246. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Grimes M, Zhou J, Beattie E, Yuen E, Hall D, Valletta J, Topp K, LaVail J, Bunnett N, Mobley W. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 1997;16:7950–7984. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Jr., Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J. Neurosci. 1998 doi: 10.1523/JNEUROSCI.18-18-07361.1998. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman GA, Bonzelius F, Cieutat AM, Kelly RB. A distinct class of intracellular storage vesicles, identified by expression of the glucose transporter GLUT4. Proc. Natl. Acad. Sci. USA. 1994;91:12750–12754. doi: 10.1073/pnas.91.26.12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J. Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Hattori S, Kabuyama Y, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of PI3 kinase. J. Biol. Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- Koike T, Tanaka S. Evidence that nerve growth factor dependence of sympathetic neurons for survival in vitro may be determined by levels of cytoplasmic free calcium. Proc. Natl. Acad. Sci. USA. 1991;88:3892–3896. doi: 10.1073/pnas.88.9.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lencer W, Verkman A, Arnaout M, Ausiello D, Brown D. Endocytotic vesicles from renal papilla which retrieve the vasopressin sensitive water channel do not contain a functional H+-ATPase. J. Cell Biol. 1990;111:379–389. doi: 10.1083/jcb.111.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor: 35 years later. EMBO J. 1987;6:1145–1154. doi: 10.1002/j.1460-2075.1987.tb02347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S, de Moura J. Apical membrane area of rabbit urinary bladder increases by fusion of intracellular vesicles. J. Membr. Biol. 1984;82:123–136. doi: 10.1007/BF01868937. [DOI] [PubMed] [Google Scholar]
- Lillien LE, Raff MC. Analysis of the cell-cell interactions that control type-2 astrocyte development in vitro. Neuron. 1990;4:525–534. doi: 10.1016/0896-6273(90)90110-2. [DOI] [PubMed] [Google Scholar]
- Martin K, Michael D, Rose J, Barad M, Casadio A, Zhu H, Kandel E. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Murer H, Biber J. Molecular mechanisms of renal apical Na/phophate contransport. Annu. Rev. Physiol. 1996;58:607–618. doi: 10.1146/annurev.ph.58.030196.003135. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Rydell RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of NGF. Proc. Natl. Acad. Sci. USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Margalet V, Goldfine ID, Vlahos CJ, Sung CK. Role of PI3 kinase in insulin receptor signaling: studies with inhibitor LY294002. Biochem. Biophys. Res. Commun. 1994;204:446–452. doi: 10.1006/bbrc.1994.2480. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M, Lisanti L, Graever L, Le Bivic A, Rodriquez-Boulan E. Integral and peripheral protein composition of the apical and basolateral membrane domains in MDCK cells. J. Membr. Biol. 1989;107:277–286. doi: 10.1007/BF01871942. [DOI] [PubMed] [Google Scholar]
- Schwartz G, Al-Awqati Q. Regulation of transepithelial H+ transport by exocytosis and endocytosis. Annu. Rev. Physiol. 1986;48:153–161. doi: 10.1146/annurev.ph.48.030186.001101. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of the transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytochemical study in the central nervous system. Dev. Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Cells that are O4-antigen positive and O1-negative differentiate into O1-positive oligodendrocytes. Neurosci. Lett. 1982;29:183–188. doi: 10.1016/0304-3940(82)90351-2. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. USA. 1980;77:2542–2545. doi: 10.1073/pnas.77.5.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein kinase cascade may be required for differentiation of PC12 cells. Biochem. J. 1992;288:351–355. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss JL, Shooter EM. Nerve growth factor promotes neurite regeneration in PC12 cells by translational control. J. Neurochem. 1995;64:550–557. doi: 10.1046/j.1471-4159.1995.64020550.x. [DOI] [PubMed] [Google Scholar]
- Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor induced survival of sympathetic neurons. J. Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf-and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Wade JB. Role of membrane fusion in hormonal regulation of epithelial transport. Annu. Rev. Physiol. 1986;48:213–223. doi: 10.1146/annurev.ph.48.030186.001241. [DOI] [PubMed] [Google Scholar]
- Wakada AR, Edgar D, Thoenen H. Both nerve growth factor and high K+ concentrations support the survival of chick embryo sympathetic neurons. Exp. Cell Res. 1983;144:377–384. doi: 10.1016/0014-4827(83)90417-2. [DOI] [PubMed] [Google Scholar]
- Wilkinson GA. Ph. D. Thesis. University of California; San Francisco, California: 1997. Neurotrophin-3 and Trk receptors in the developing mouse trigeminal ganglion. [Google Scholar]
- Xia Z, Refsdal CD, Merchant K, Dorsa D, Storm DR. Distribution of mRNA for the calmodulin sensitive adenylyl cyclase in rat brain. Neuron. 1991;6:431–443. doi: 10.1016/0896-6273(91)90251-t. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg M. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yao X, Karam S, Ramilo M, Rong Q, Thibodeau A, Forte JG. Stimulation of gastric acid secretion by cAMP in a novel toxin permeabilized gland model. Am. J. Physiol. 1996;271:C61–C73. doi: 10.1152/ajpcell.1996.271.1.C61. [DOI] [PubMed] [Google Scholar]