Failure of neuronal homeostasis results in common neuropsychiatric phenotypes (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 16.
Published in final edited form as: Nature. 2008 Oct 16;455(7215):912–918. doi: 10.1038/nature07457
Abstract
Failure of normal brain development leads to mental retardation or autism in about 3% of children. Many genes integral to pathways by which synaptic modification and the remodelling of neuronal networks mediate cognitive and social development have been identified, usually through loss of function. Evidence is accumulating, however, that either loss or gain of molecular functions can be deleterious to the nervous system. Copy-number variation, regulation of gene expression by non-coding RNAs and epigenetic changes are all mechanisms by which altered gene dosage can cause the failure of neuronal homeostasis.
Homeostasis is the ability of a system to return to a set point following perturbation. Much is understood about neuronal homeostatic mechanisms at the level of physiological output (for example, the nerve action potential), but the underlying molecular processes are the subject of intense investigation. The developing and learning brain is composed of neurons that must continue to carry out routine functions even while neurogenesis is occurring and the synaptic connections between neurons are being strengthened, remodelled and pruned. Neurons within affected networks must therefore make homeostatic responses to reestablish a proper balance of excitation and inhibition. Recent clinical studies show that loss or gain in dosage of proteins or RNAs in different neurodevelopmental syndromes often results in similar or overlapping sets of neurological symptoms, suggesting that the molecules involved in cognitive and behavioural processes participate in highly regulated homeostatic mechanisms. This inability of neurons in vivo to compensate for seemingly minor alterations in protein or RNA dosage should teach us something about the nervous system. It may help us to answer questions such as: Why are there hundreds of known genes whose alteration causes mental retardation or autism, yet rarely can we predict genotype solely on the basis of neurophenotype? And why is autism a prominent feature of so many disorders affecting cognition?
Inherent in this discussion is the fact that single genes or proteins do not encode specific behaviours or neurological deficits but instead encode biological functions. When disruption of biological processes leads to neuronal dysfunction in a developing brain, numerous phenotypes may result, depending on the function(s) subserved by the affected cell type and other neurons within the network. In other words, neurodevelopmental disorders are not cell autonomous because even neuron-specific changes will always affect the neuronal network. Loss or gain of function of any protein that alters synaptic output may cause neurological or psychiatric phenotypes because changes in neuronal excitability demand compensation, which may, in turn, exhaust the homeostatic capacity of the network. In vitro and ex vivo model systems have demonstrated the exceptional ability of neurons to compensate for experimental perturbations by modulating ion channels, receptors, signalling pathways and neurotransmitters. At the molecular level, such homeostatic processes require chromatin remodelling, changes in gene expression and repression, changes in protein production and turnover, and cytoskeletal rearrangement.
The disorders discussed in this Review all represent the cumulative impact of inappropriate gene dosage on a neuronal system and variously involve abnormal regulation of gene expression by small non-coding RNA molecules, alteration of RNA metabolism leading to abnormal protein synthesis, abnormal protein turnover, abnormal chromatin modulation and gene expression, and altered cytoskeletal dynamics. Amazingly, these disorders show that the inability to maintain neuronal homeostasis at the level of a variety of molecular processes is sufficient to cause common end points such as mental retardation, epilepsy and autism (Fig. 1). There are almost no medical therapies for these conditions, yet we know from animal models of some of them — for example, Angelman syndrome, fragile X syndrome and Rett syndrome — that the therapeutic potential exists. Thus, never before has the promise for neuroscience to uncover the molecular underpinnings of cognition, social interaction and behaviour been greater, nor has the potential to discover treatments that restore homeostasis to a diseased brain been riper.
Figure 1. Loss of protein or RNA function causes neurodevelopmental disorders with phenotypes overlapping those caused by gain of protein or RNA function.

Each pair of ovals demonstrates loss and duplication of the same chromosomal region. Phenotypes unique to loss of function are shown in yellow; phenotypes unique to gain of function are shown in blue; phenotypes common to both loss of function and gain of function are shown in green. Each of these alterations in neuronal function converges on pathways that cause mental retardation and autism or abnormal behaviours. (Both Prader–Willi syndrome (PWS) and Angelman syndrome (bottom) involve deletion of 15q11–q13.) MECP2, gene encoding methyl-CpG-binding protein 2; PLS, Potocki–Lupski syndrome; SMS, Smith–Magenis syndrome; WBS, Williams–Beuren syndrome.
In this Review, we discuss the pathophysiology of six neurodevelopmental disorders that illustrate instances in which loss or gain of molecular function converge on the common phenotypes of mental retardation and autism spectrum behaviours. We put forward a hypothesis in which the failure of neuronal homeostasis or homeostatic compensation leads to neuronal networks with weakened synaptic flexibility and to common clinical end points.
Abnormal protein synthesis disrupts synaptic function
Levels of RNA and protein are regulated by transcription and translation, respectively. Whether a messenger RNA molecule will be translated into protein often depends on the presence of RNA-binding proteins that regulate the translation of specific mRNA molecules, thus providing a mechanism for regulating temporal and spatial patterns of protein expression. Fragile X syndrome, the disorder we discuss first, involves abnormal dosage of an RNA-binding protein that results in a devastating neurological outcome.
The discovery of a CGG repeat expansion in the 5′ untranslated region of the gene FMR1 led to insight into the pathophysiology of a common form of heritable mental retardation, fragile X syndrome1. Hypermethylation of the expanded CGG repeats and upstream CpG island leads to transcriptional silencing of FMR1 (ref. 2). Mental retardation, characteristic facies (facial appearance), macro-orchidism, autism and epilepsy occur with a full mutation of >200 repeats2. _Fmr1_-null mice have abnormal maturation of, and pruning of, dendritic spines, resulting in abnormal dendritic-spine morphology similar to that observed in the cerebral cortex of patients with fragile X syndrome3 (Fig. 2b). Transgenic mice that overexpress a human FMR1 transgene develop hypoactivity, anxiety and abnormal responses to sensory stimulation4 and predict a human gain-of-function disorder.
Figure 2. Loss or gain of protein or RNA function results in altered neuronal homeostasis or ‘imbalance’.
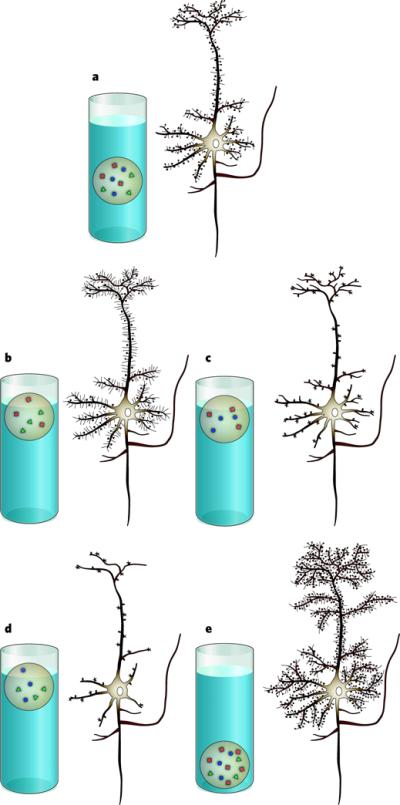
Homeostasis is represented by the balance of a weighted ball floating in water. Individual proteins involved in the following syndromes are each represented by a coloured shape. a, Representation of a healthy neuron, with proteins in balance. b, Representation of a neuron in fragile X syndrome, with normal dendrites but an increased density of longer, thinner, immature-appearing dendritic spines. c, Representation of a neuron in Angelman syndrome, with normal dendrites but a decreased density of, and abnormally shaped, dendritic spines. d, Representation of a neuron in Rett syndrome or _Mecp2_-null mice, with decreased density of dendrites and decreased density of, and abnormally shaped, synapses (represented by dendritic spines). e, Representation of a neuron from mice in which Mecp2 is duplicated, with increased density of synapses (represented by increased density of dendrites and dendritic spines).
The product of normal FMR1 is an RNA-binding protein, FMRP, that associates with polyribosomes and suppresses the translation of specific mRNA molecules2. Identification of mRNAs differentially associated with the FMRP ribonucleoprotein complex suggested that activity-dependent alterations in local protein synthesis at synapses leads to the neurological consequences of fragile X syndrome5,6. The fact that activation of group 1 metabotropic glutamate receptors (mGluR) stimulates synaptic protein synthesis in an mRNA-translation-dependent manner led to the mGluR theory of fragile X pathogenesis7,8. In the absence of FMRP, mGluR5 signalling stimulates increased internalization of the ionotropic AMPA receptors for glutamate9. Also, the mGluR-induced translation of proteins that are crucial for surface expression of the AMPA receptor, such as AMPA receptor subunits and postsynaptic-density protein 95 (PSD95), does not occur10. These data support a model in which loss of FMRP leads to an increase in baseline protein synthesis to the extent that synaptic activation does not induce the additional local protein synthesis necessary to regulate synaptic plast icity appropriately10.
Treatment with a non-competitive mGluR5 antagonist rescues the AMPA-trafficking defect in cultured hippocampal neurons9, the seizure phenotype of _Fmr1_-null mice11, and the synaptic plasticity, courtship behaviour and mushroom-body defects in a Drosophila model of fragile X syndrome12. Furthermore, a 50% reduction in mGluR5 expression in _Fmr1_-null mice rescued synaptic plasticity, dendritic-spine density, protein synthesis, and memory and epilepsy phenotypes8. This discovery, and the fact that a 50% reduction in mGluR5 does not lead to overt behavioural phenotypes in normal mice8, has generated much excitement over the potential of mGluR5 antagonists to improve the neurodevelopmental phenotypes of fragile X syndrome. In summary, FMRP and mGluR5 signalling seem to oppose each other functionally in the regulation of local synaptic protein synthesis in an activity-dependent, homeostatic manner. In addition to causing abnormal protein synthesis and dynamically weakened neurons (neurons with attenuated ability to respond to change), loss of FMRP probably also causes secondary homeostatic changes that further damage neuronal networks.
MicroRNAs and cognition
Small non-coding microRNAs (miRNAs) that repress the translation of target mRNAs are emerging as important second-tier regulators of protein levels. In our next example, that of the heterozygous 22q11.2 deletion and duplication syndromes, altered dosage of an RNA-binding protein important for the processing of miRNAs may underlie significant neurodevelopmental and psychiatric morbidity.
The 22q11.2 deletion syndrome is caused by the loss of the q11.2 region from one copy of chromosome 22. Individuals with this deletion syndrome have highly variable phenotypes, including the pheno typic constellations that are typically associated with DiGeorge syndrome and/or velocardiofacial syndrome. Developmental and neuropsychiatric phenotypes are prevalent in individuals with the 22q11.2 deletion syndrome and include developmental delay, learning difficulties, epilepsy, hyperactivity, anxiety, autism, obsessive–compulsive disorder, bipolar disorder and schizophrenia13–15. In fact, 22q11.2 deletion conveys the strongest DNA-based risk factor identified so far for developing schizophrenia14. Unexpectedly, the duplication of 22q11.2 (the reciprocal of the 22q11.2 deletion) was recently identified in individuals with developmental delay, learning disabilities, behavioural problems, cardiovascular abnormalities, velopharyngeal insufficiency, growth retardation and dysmorphic features — phenotypes similar to those of individuals with the 22q11.2 deletion syndrome16,17 (Fig. 1).
Cardiovascular anomalies and impaired sensorimotor gating (the abnormal modulation of sensory information transmitted to a motor system, resulting in abnormal behavioural output) are reproduced in a mouse model with deletion of the region homologous to human 22q11.2 (refs 18, 19). Mutation of a single gene in the critical region, TBX1 (which encodes a T-box-containing transcription factor), reproduces most physical features of DiGeorge syndrome and velocardio facial syndrome in human patients20. TBX1 mutation has been implicated in nonspecific cognitive dysfunction and in a boy with Asperger's syndrome18, and haploinsufficiency for Tbx1 or Gnb1l (which encodes a G-protein-β-subunit-like protein) is sufficient to cause abnormal sensorimotor gating in mice18. Transgenic mice that overexpressed a region of human 22q11.2 encompassing four genes, CDCREL (which encodes a substrate for the ubiquitin–protein ligase parkin), GP1B (which encodes a platelet-surface membrane protein), TBX1 and GNB1L, showed hyperactivity and a lack of habituation that could be ameliorated by treatment with anti psychotic medication21. Other genes in the 22q11.2 deletion region have been put forward as candidate genes for schizophrenia; however, none has been incontrovertibly linked to psychiatric or neurodevelopmental phenotypes22.
Transcriptional profiling in a mouse model lacking the 22q11.2-homologous region revealed consistent upregulation of miRNA-related transcripts in the hippocampus and prefrontal cortex23. One of the genes mapping to the 22q11.2 deletion region is DGCR8, which encodes a double-stranded RNA-binding protein that is part of a complex that processes primary miRNAs to mature miRNAs. Thus Kimberly Stark et al.23 proposed that DGCR8 might mediate this effect. Indeed, haploinsufficiency for Dgcr8 in mice resulted in abnormal miRNA biogenesis, smaller dendritic spines, a less complex dendritic tree and impaired sensorimotor gating and spatial working memory, suggesting the hypothesis that abnormal processing of miRNAs may lead to the neurological and behavioural phenotypes observed in the 22q11.2 deletion syndrome23, perhaps through the inability of neurons to regulate translational responses to stimuli. Like the loss of FMRP, the loss of DGCR8 may lead to dynamically weakened neurons and compensatory homeostatic mechanisms that damage neuronal networks. The study by Stark and colleagues23 put forward a new role for miRNAs in cognitive processes and predicted that other components of this pathway might contribute to neurological and psychiatric disease in humans.
Imprinting failure disrupts protein and RNA dynamics
A key mechanism for fine-tuning gene dosage is genomic imprinting, the phenomenon in which only the maternal or paternal copies of certain genes are expressed as a result of differential cytosine methylation on the maternally and paternally inherited homologous chromosomes. Biological processes that are extremely vulnerable to altered protein dosage must exist to explain why some gene dosages are so critical that imprinting is necessary. The disorders discussed next illustrate the extreme dosage sensitivity of certain genes required for proper neuronal function.
Chromosome region 15q11–q13 is the best-characterized imprinted region in the human genome24 because altered gene dosage resulting from uniparental disomy, imprinting-centre mutations or hemizygous deletion results in two distinct disorders: Angelman syndrome (which involves the loss of maternal genetic information) and Prader–Willi syndrome (PWS, which involves the loss of paternal genetic information)25. Individuals with Angelman syndrome typically show mental retard ation, autism, microcephaly, gait ataxia, tremor, epilepsy, and inappropriate hand flapping, laughing and excitability24. PWS is characterized by infantile hypotonia (abnormally low muscle tone) and failure to thrive, which progresses to excessive eating and obesity by early childhood, and mental retardation, hypogonadism and abnormal behaviours (including autism, psychosis and obsessive–compulsive behaviour)26. An overlapping clinical syndrome (Fig. 1) caused by increased maternal copy number of 15q11–q13 (the critical region for Angelman syndrome and PWS) also occurs, and includes hypotonia, mental retardation, autism, stereotypies, epilepsy and a spectrum of congenital anomalies25.
The gene responsible for Angelman syndrome has been identified as that encoding ubiquitin–protein ligase E3A (UBE3A), because point mutations in this gene are sufficient to cause Angelman syndrome24. UBE3A marks proteins for degradation by the proteasome; however, the target proteins that fail to be degraded in Angelman syndrome have not been identified24. UBE3A is expressed from the maternal allele in the brain but is expressed from both alleles in peripheral tissues, suggesting that, to maintain proper function, neurons in the brain have a dosage requirement that differs from that of other cell types24. Mice deficient in the maternal Ube3a allele have abnormal dendritic spines (Fig. 2c), microcephaly, impaired motor function, impaired spatial learning and epilepsy, closely mimicking the human disorder27. The recent identification of an individual with typical PWS and paternal deletion of the genes encoding small nucleolar RNAs (snoRNAs, which are non-coding RNA molecules generally involved in RNA processing) confirmed previous hypotheses that paternal deficiency of the HBII-85 snoRNA cluster is sufficient to cause most features of PWS26,28. Mouse models with deletion of the homologous snoRNA cluster reproduce the neonatal hypotonia, growth retardation, abnormal motor learning, increased anxiety and hyperphagia observed with larger deletions29.
It follows that the identification of UBE3A and HBII-85 targets is likely to demonstrate that failure of normal protein turnover in Angelman syndrome and failure of proper RNA processing in PWS also lead to dynamically weakened neurons and neuronal networks that are at the mercy of secondary compensatory mechanisms. The identification of these target proteins and RNAs will pinpoint novel pathways that could be targeted therapeutically.
A dosage-sensitive transcription factor
It is no surprise that improper dosage of a transcription factor essential for the regulation of gene expression can lead to detrimental consequences for neuronal networks in a developing brain. The disorders discussed in this section are caused by the altered dosage of such a protein.
Smith–Magenis syndrome (SMS), a constellation of infantile hypotonia and failure to thrive, congenital anomalies, mental retardation, behavioural problems (including self-mutilation, aggression, hyperactivity, stereotypies and often autism), obesity, sleep disturbance, decreased pain sensitivity and epilepsy30,31, is in most individuals due to heterozygous deletion of 3.7 Mb in chromosome 17p11.2 (refs 32, 33). A few patients are haploinsufficient for the retinoic-acid-induced 1 (RAI1) gene product as a result of truncating mutations, indicating that RAI1 is the primary dosage-sensitive culprit in SMS30,34.
In a mouse cell line (P19 embryonal carcinoma cells), Rai1 is upregulated subsequent to retinoic-acid-induced neuronal differentiation35. The RAI1 protein contains a bipartite nuclear-localization signal, polyserine and polymorphic polyglutamine tracts, and a carboxy-terminal PHD/zinc-finger domain36. The amino-terminal region of RAI1 is sufficient to activate transcription in heterologous reporter gene assays37. Therefore, RAI1 may function in chromatin remodelling and transcriptional processes31. Targeted disruption of Rai1 in mice led to embryonic or peri-natal lethality and severe craniofacial and skeletal abnormalities in the homozygous state37, although a small percentage of null animals survived the neonatal period and frequently developed epilepsy and motor and learning deficits38. _Rai1_-haploinsufficient mice experienced obesity, less severe craniofacial abnormalities37 and less frequent epilepsy38.
Duplications of 17p11.2 (the reciprocal of the SMS-causing microdeletion) cause Potocki–Lupski syndrome (PLS), which is characterized by infantile hypotonia and failure to thrive, mental retardation, expressive language deficits, autism, sleep-disordered breathing and cardiovascular abnormalities39,40. The duplication-critical region has been narrowed down to a 1.3-Mb interval containing the RAI1 locus; however, contributions of other genes or regulatory regions to the PLS phenotype cannot be excluded40. Mice with duplication of the region homologous to human chromosome 17p11.2 (located in mouse chromosome 11) are abnormally small and hyperactive and have deficits in learning and memory41. Transgenic mice that overexpress Rai1 show growth retardation, hyperactivity, anxiety and increased sensitivity to pain, as well as impaired gait, forelimb grip strength and sensorimotor activity; these phenotypes worsen as gene dosage increases31.
Altered RAI1 dosage may not explain all of the phenotypes associated with SMS and PLS, but RAI1 seems to be the most dosage-sensitive gene in the 17p11.2 deletion/duplication region. Gain or loss of RAI1 function leads to overlapping neurological syndromes (Fig. 1). Individuals with SMS can interact socially to some extent but have severely maladaptive and aggressive behaviour30, and individuals with PLS primarily develop autistic features40, suggesting that RAI1 dosage is crucial in pathways that modulate these behaviours. Because little is known about the function and targets of RAI1, the next logical steps to delineating the underlying molecular pathways are to carry out expression array studies and genome-wide binding-site analysis of brain tissue from animals with loss or gain of RAI1 protein dosage, as well as to identify the binding partners of RAI1. Such studies should also provide exciting insight into the role of RAI1 in cognitive and behavioural processes.
Chromatin modulation essential for synaptic homeostasis
Chromatin remodelling occurs through mechanisms such as modification of histones, methylation of cytosine, regulation of DNA-binding proteins and recruitment of transcriptional activator and repressor protein complexes. The disorders discussed in this section are the result of improper dosage of a prototypical chromatin-remodelling protein.
Rett syndrome is one of the most common causes of mental retardation in females42. After 6 to 18 months of apparently normal development, affected females lose speech, social-interaction skills and the ability to learn43. They develop characteristic hand stereotypies, anxiety, microcephaly, epileptic seizures and impaired motor and autonomic control43. Mutations in the X-linked gene encoding methyl-CpG-binding protein 2 (MECP2) cause Rett syndrome44. Recently, males with Xq28 duplications that include the MECP2 locus were found with infantile hypotonia, mental retardation, poor speech development, recurrent infections, epilepsy, progressive spasticity and autistic behaviours45–49, features that overlap with the Rett syndrome phenotype (Fig. 1). The smallest region of overlap among the duplications studied suggests that MECP2 is the primary dosage-sensitive gene in the duplicated region and is responsible for most of the observed phenotypes45–49.
Mouse models of altered Mecp2 gene dosage exist and were reviewed recently43. Briefly, male mice that produce no MeCP2 protein are normal at birth but develop tremor, gait abnormalities, reduced spon taneous movements, and weight and breathing abnormalities, and they die by 10−12 weeks of age50. Transgenic models in which mice overexpress human MECP2 also exist, including _MECP2_Tg1 mice (which produce twice as much MeCP2 as wild-type controls) and _MECP2_Tg3 mice (which produce three times as much MeCP2 as wild-type mice). _MECP2_Tg1 males are normal until 10−12 weeks of age, when they develop a progressive neurological disorder that includes a forepaw stereotypy, seizures, aggression, hypoactivity, spasticity, kyphosis (spinal curvature), poor grooming and ataxia; they die by 12 months of age51. _MECP2_Tg3 males model the human MECP2 triplication disorder45, are smaller than their wild-type littermates at birth, develop the most severe progressive neurological disorder, and die between 3 and 6 weeks of age51. Recent data from a _Mecp2_Flox/Y mouse model, in which mice express 50% less Mecp2 mRNA, and approximately 40% less protein, than wild-type mice, suggest that even this reduction in MeCP2 dosage is sufficient to cause neurological phenotypes, and predict that mild reductions in MeCP2 protein function might cause neuropsychiatric phenotypes in humans52.
The fact that loss of MeCP2 function leads to neurological syndromes that overlap those caused by gain of MeCP2 function suggests that MeCP2 modulates crucial neuronal functions. Roles for MeCP2 in histone-deacetylase-dependent transcriptional repression, assembly of secondary chromatin structure and regulation of RNA splicing have been demonstrated, but how these functions contribute to disease is not well understood51. Recent expression data from the hypothalami of _Mecp2_-null and _MECP2_Tg3 mice show that loss of MeCP2 function has an effect on gene expression that is opposite to that of gain of function, with MeCP2 gain primarily leading to transcriptional activation53. This study provided in vivo confirmation of an earlier genome-wide analysis that correlated MeCP2-binding sites with activation of nearby genes in a neuroblastoma cell line54. Whether this activation is direct or indirect is not yet known. Hippocampal glutamate-utilizing neurons that lack MeCP2 have a 46% reduction in synaptic response, and neurons producing twice the normal amount of MeCP2 have a twofold increase in synaptic response55. These observations were explained by concomitant changes in the formation of glutamate-utilizing synapses55.
MeCP2 is phosphorylated on the serine residue at position 421 in response to neuronal activity, and this modification is necessary for MeCP2 to modulate dendritic growth and spine maturation56. Furthermore, MeCP2 regulates the expression of the gene encoding brain-derived neurotrophic factor (Bdnf), an activity-dependent gene53,56,57. _Mecp2_308/Y mice (which have an early truncation of the protein at amino acid 308) model human Rett syndrome and have heightened physiological responses to stress that are manifested by increased anxiety and increased levels of corticosterone58. The gene encoding corticotropin-releasing hormone (Crh) is a direct target of MeCP2, and altered Crh expression occurs only in those brain regions where Crh is normally expressed58.
Collectively, these findings demonstrate that MeCP2 modulates gene expression in neurons in response to changing physiological states, underscore its important role in neuronal plasticity, and indicate that disorders resulting from MeCP2 loss of function or gain of function are caused by a failure of synaptic homeostasis (Fig. 2d, e).
Cytoskeletal modulation and molecular homeostasis
Dendrites and dendritic spines are crucial for synaptic function. For example, modulation of the cytoskeleton must occur during development of the nervous system and throughout life to regulate the trafficking and anchoring of neurotransmitter receptors in the postsynaptic density. The list of genes that are crucial for the regulation of dendritic spines and synapse structure, and that are altered in human mental retardation and autism syndromes, is growing and now includes MECP2, FMR1, UBE3A, SHANK3 (which encodes a synaptic scaffolding protein), NLGN3 (neuroligin 3) and NLGN4, and NRXN1 (neurexin 1)59. Other genes that contribute to the development and modulation of synapses are those important to regulation of the cytoskeleton. The disorders discussed next are examples of how altered protein dosage leads to failure of cytoskeletal dynamics and dysfunctional neuronal systems.
Williams–Beuren syndrome (WBS) is caused by a hemizygous deletion of around 25 genes at 7q11.23 (ref. 60). Typical features include learning problems, mental retardation, visuospatial deficits, relative sparing of expressive language, inappropriate hypersociability, attention problems, dysmorphic facies, and cardiovascular and connective-tissue abnormalities60. Reciprocal duplication of this region causes language delay, decreased social interaction and repetitive behaviours60,61. Epilepsy and abnormal cortical migration have also been observed62.
The cardiovascular phenotype that is commonly seen in WBS, supravalvular aortic stenosis, is caused by haploinsufficiency of the elastin gene63. Deletion of CYLN2 (which encodes cytoplasmic linker protein 115, CLIP115), LIMK1 (a LIM kinase) and FZD9 (the receptor frizzled 9) may contribute to the cognitive phenotypes observed in WBS64. Frizzled 9 is a WNT receptor that is selectively expressed in the hippocampus65. _Fzd9_-hemizygous mice show increased apoptosis in the developing hippocampal dentate gyrus, a decreased seizure threshold, and impaired visuospatial learning65. Individuals with WBS have overall smaller brain volumes with decreased subcortical white matter, increased cortical folding and abnormal cell-packing densities in some regions, raising the possibility that FZD9 contributes to such phenotypes64.
In mice, Cyln2 is expressed by many neurons and encodes the highly conserved protein CLIP115, which specifically associates with the ends of growing microtubules to regulate microtubule dynamics64. Cyln2 hemizygosity leads to growth deficiency, altered brain morphology, altered hippocampal synaptic plasticity and specific motor coordination deficits in mice66. Microtubule dynamics are not altered, but increased accumulation of CLIP170 (another cytoplasmic linker protein) and dynactin occurs at microtubule ends in fibroblasts cultured from these animals66. More work is necessary to understand the contribution of the CLIP115 protein to cognitive function.
LIMK1 encodes a serine/threonine kinase that is expressed primarily in the central nervous system67. LIMK1 functions downstream of RHO signalling to regulate actin dynamics through inactivation of the actin-depolymerization factor cofilin by phosphorylation67. Brain tissue from _Limk1_-knockout mice showed decreased cofilin phosphorylation, decreased accumulation of actin filaments in dendritic spines, abnormal clusters of actin filaments in dendrites, altered spine morphology, and altered presynaptic and postsynaptic function (including enhanced hippocampal long-term potentiation)67. In regard to behaviour, the gene-knockout mice showed abnormal conditioned and spatial learning67. The importance of LIMK1 to spine morphogenesis and synaptic function underscores the role of synaptic homeostasis in neurological disorders67–69.
These results suggest that loss or gain of CLIP115 or LIMK1 results in neurons with decreased ability to dynamically regulate cytoskeletal structure in response to the requirements of synaptic strengthening, remodelling and pruning processes.
Homeostatic control mechanisms control network function
The overlap of neurodevelopmental and psychiatric phenotypes (such as mental retardation, epilepsy, autism and other abnormal behaviours) that results from either loss or gain of the same proteins or RNA molecules supports an emerging theme that normal cognition and behaviour depend on tight neuronal homeostatic control mechanisms. Neurons must constantly respond to changes in their excitability that depend on the stage of development and environmental factors. This plasticity can be affected by a variety of molecular processes that influence synaptic output, including chromatin state, RNA biogenesis, transcription, translation, protein turnover and cytoskeletal dynamics.
Why is it that neurons in vivo are less able than neurons in vitro to compensate for what seem to be minor alterations in gene dosage? One possibility is that neuronal functions that are crucial for cognition, language and social development are particularly vulnerable. The extent of phenotypic overlap in these areas, irrespective of the molecular or cellular deficits, is curious. This is not to say that the disorders or phenotypes discussed here are homogeneous; rather, overlapping cognitive, behavioural and social domains are commonly affected. Such overlap is not observed with other neurological signs, such as neuromuscular weakness or dystonia. Another possibility is that compensatory changes within individual neurons are insufficient to restore normal activity to a network. We suggest a model in which altered proteins that are themselves integral to neuronal homeostasis (for example, FMRP or MeCP2), or loss or gain of function of proteins that trigger homeostatic responses, lead to dysfunctional neuronal networks with weakened synaptic flexibility (Fig. 3). We further propose that this lack of synaptic flexibility is what eventually causes the overlapping phenotypes.
Figure 3. Homeostatic responses could result in a compensated neuronal network with decreased flexibility.
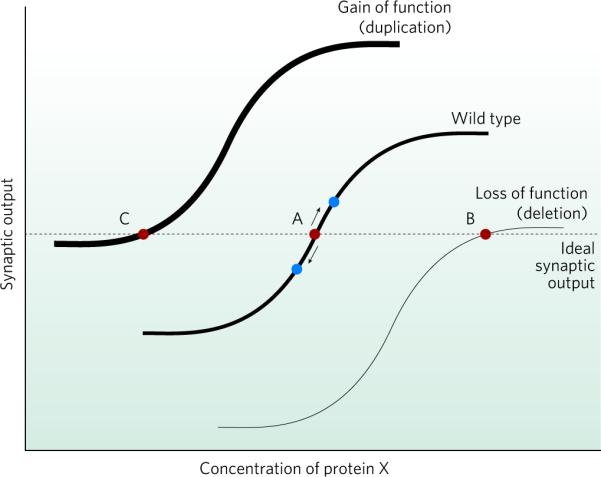
The curves show how synaptic output varies with the concentration of a given protein (X). Synaptic output is the product of synapse number and the synaptic strength per individual synapse. Protein X represents any protein important to synaptic strength and learning, for example, the AMPA receptor. The horizontal dashed line represents the ideal level of synaptic output (red circles). The middle curve (wild type) represents the normal situation. Point A is the concentration of X necessary to produce an ideal synaptic output. The neuron is flexible; in response to any learning event or stimulus, it can rapidly increase or decrease synaptic output (blue circles) with minimal change in the concentration of protein X (because the neuron exists in an ideal state represented by point A being on the steep portion of the curve). In cases in which there is a permanent loss or gain of the activity of a specific molecule (thin and thick curves, respectively), homeostatic changes in protein X occur to restore appropriate synaptic output (points B and C, respectively). At these new steady states, however, the neuron no longer retains the flexibility necessary to adequately respond to a learning or extinction stimulus (because points B and C are on flatter regions of the curve). Therefore, the dynamic ability of the neuronal system to respond is weakened, and phenotypes such as mental retardation and autism can result.
The ability of a neuronal network to be flexible is limited by its inhibitory and excitatory connectivities, the intrinsic variability of cells, and its developmental stage and epigenetic history (acquired changes in patterns of gene expression that are not due to alterations in DNA sequence). A plausible explanation for periods of normal development before disease onset in neurodevelopmental or psychiatric disorders is that cells are able to compensate within a network for periods of time before an accumulation of variable and stochastic events causes system dysfunction. For example, it is interesting that girls with Rett syndrome have a period of normal development but that boys with the _MECP2_-duplication syn drome do not. Loss of MeCP2 function ultimately leads to clinical syn dromes that overlap with those caused by gain of MeCP2 function, despite these changes in MeCP2 resulting in opposite molecular signatures in the neurons. This is not surprising given the multitude of processes that contribute to homeostatic regulation of neuronal function and the large number of genes whose expression is altered on either loss or gain of the protein53. Within the context of our model we predict that changes induced by either loss or gain of protein function dynamically weaken the responsiveness of neuronal networks to stimuli (Fig. 3). This model could also explain the fluctuating nature of many psychiatric diseases.
Future challenges
To gain better insight into how neuron-specific defects compromise network function, improved techniques for studying neuronal networks in vivo are essential. It will also be crucial to evaluate whether and how the homeostatic capacity of a network is compromised following an initial compensatory process or in response to a primary defect. To determine whether multiple neuronal networks impinge on social behaviour, cognition and communication to cause a disorder such as autism, or whether subtle defects or changes within single networks are sufficient, will require specific investigations aimed at defining spatial and cell-specific threshold effects. Determining how to increase the synaptic flexibility of a network irrespective of the primary molecular defect might prove to be a fruitful way to manage the shared symptoms in this class of cognitive and behavioural disorders. Another therapeutic approach might involve dampening the homeostatic changes that occur in response to a primary defect.
The observation that functional loss or gain of proteins or RNAs involved in diverse processes leads to mental retardation, autism and other neuropsychiatric symptoms predicts that there are likely to be hundreds to thousands of genes whose alteration results in these common neurological phenotypes. A recent search by us of the Online Mendelian Inheritance in Man (OMIM) database yielded 305 genes known to be associated with mental retardation, 51 with autism and 75 with a behavioural disorder.
Another important prediction based on this theory of failed neuronal homeostasis in neuropsychiatric disease is that disorders such as schizophrenia, bipolar disorder, anxiety disorders and obsessive–compulsive disorder will have two main aetiologies. One will be genetic, with such disorders being caused by less severe alterations either in some of the genes or pathways that cause severe neurodevelopmental disorders such as autism (and whose effects often converge on the synapse) or in genes that have a crucial role in modulating synaptic plasticity in adolescence and into adulthood. The identification of individuals with significant psychiatric morbidity (for example, psychosis) among those with a mutation in MECP2, maternal uniparental disomy in PWS, or the 22q11.2 deletion syndrome supports this hypothesis. The second aetiology is likely to involve acquired epigenetic changes that result in altered gene expression or RNA processing, often in the same pathways that cause autism or similar neuropsychiatric disease in children. The fact that there are probably hundreds, if not thousands, of such genes may explain the poor yield of specific gene associations in psychiatric diseases, the small number of cases resulting from a defect in any one gene, and the variable penetrance and expressivity of these alleles in kindreds. It is probable that alteration of the same gene can cause anxiety in one individual, obsessive–compulsive disorder in another, psychosis in another, and autism in yet another. Undoubtedly, these disorders are modified by genetic background and life experience, through which synaptic plasticity and epigenetic changes may be either protective or predisposing to disease.
The identification of molecular pathways common to mental retardation and neuropsychiatric disorders will lead to better understanding of neuronal networks and the underpinnings of cognition, language, behaviour, social interaction and emotion. For therapeutic trials, it will be important to group patients by pathophysiological mechanism of disease rather than simply by which gene is affected. Finally, the identification of drugs that perturb or restore neuronal and synaptic homeostasis may significantly improve the quality of life for patients and their families.
Acknowledgements
In memory of our mentor, Ralph D. Feigin. We are grateful to C. Rosenmund for careful reading of the manuscript, discussions and helping us to articulate our hypothesis. We are indebted to the Howard Hughes Medical Institute, the National Institute of Neurological Disorders and Stroke (grant number 1R01 NS057819-01 to H.Y.Z., and grant numbers T32 NS43124 and 1K08 NS062711-01 to M.B.R.) and the Simons Foundation for supporting our research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Verkerk AJ, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 2.Garber K, Smith KT, Reines D, Warren ST. Transcription, translation and fragile X syndrome. Curr. Opin. Genet. Dev. 2006;16:270–275. doi: 10.1016/j.gde.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Comery TA, et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl Acad. Sci. USA. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peier AM, et al. (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum. Mol. Genet. 2000;9:1145–1159. doi: 10.1093/hmg/9.8.1145. [DOI] [PubMed] [Google Scholar]
- 5.Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 6.Darnell JC, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 7.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Dolen G, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [This paper provides genetic evidence that supports the mGluR theory of fragile X pathogenesis and proposes a potential therapeutic strategy.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamoto M, et al. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl Acad. Sci. USA. 2007;104:15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [This paper provides a mechanism underlying abnormal AMPA-receptor surface expression in excitatory synapses in fragile X syndrome and suggests that the key principle responsible for fragile X syndrome is that synaptic activation cannot stimulate the additional local protein synthesis necessary for synaptic plasticity to occur.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 12.McBride SM, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 14.Gothelf D, et al. Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. Am. J. Psychiatry. 2007;164:663–669. doi: 10.1176/ajp.2007.164.4.663. [DOI] [PubMed] [Google Scholar]
- 15.Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 16.Ensenauer RE, et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am. J. Hum. Genet. 2003;73:1027–1040. doi: 10.1086/378818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yobb TM, et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am. J. Hum. Genet. 2005;76:865–876. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paylor R, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl Acad. Sci. USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long JM, et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 20.Yagi H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 21.Hiroi N, et al. A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proc. Natl Acad. Sci. USA. 2005;102:19132–19137. doi: 10.1073/pnas.0509635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nature Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- 23.Stark KL, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nature Genet. 2008;40:751–760. doi: 10.1038/ng.138. [This paper suggests a novel pathophysiological mechanism for the cognitive and psychiatric phenotypes observed in the human 22q11.2 deletion syndrome: abnormal miRNA biogenesis.] [DOI] [PubMed] [Google Scholar]
- 24.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol. Life Sci. 2007;64:947–960. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battaglia A. The inv dup(15) or idic(15) syndrome: a clinically recognisable neurogenetic disorder. Brain Dev. 2005;27:365–369. doi: 10.1016/j.braindev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Sahoo T, et al. Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nature Genet. 2008;40:719–721. doi: 10.1038/ng.158. [This paper confirms the cause of PWS — deficiency of non-coding RNA molecules important for normal RNA processing — and suggests a novel role for snoRNAs in cognitive and psychiatric disease.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miura K, et al. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis. 2002;9:149–159. doi: 10.1006/nbdi.2001.0463. [DOI] [PubMed] [Google Scholar]
- 28.Gallagher RC, Pils B, Albalwi M, Francke U. Evidence for the role of PWCR1/HBII-85 C/D box small nucleolar RNAs in Prader–Willi syndrome. Am. J. Hum. Genet. 2002;71:669–678. doi: 10.1086/342408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding F, et al. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE. 2008;3:e1709. doi: 10.1371/journal.pone.0001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elsea SH, Girirajan S. Smith–Magenis syndrome. Eur. J. Hum. Genet. 2008;16:412–421. doi: 10.1038/sj.ejhg.5202009. [DOI] [PubMed] [Google Scholar]
- 31.Girirajan S, et al. How much is too much? Phenotypic consequences of Rai1 overexpression in mice. Eur. J. Hum. Genet. 2008;16:941–954. doi: 10.1038/ejhg.2008.21. [DOI] [PubMed] [Google Scholar]
- 32.Smith AC, et al. Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am. J. Med. Genet. 1986;24:393–414. doi: 10.1002/ajmg.1320240303. [DOI] [PubMed] [Google Scholar]
- 33.Chen KS, et al. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nature Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 34.Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith–Magenis syndrome. Nature Genet. 2003;33:466–468. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- 35.Imai Y, et al. Cloning of a retinoic acid-induced gene, GT1, in the embryonal carcinoma cell line P19: neuron-specific expression in the mouse brain. Brain Res. Mol. Brain Res. 1995;31:1–9. doi: 10.1016/0169-328x(95)00020-s. [DOI] [PubMed] [Google Scholar]
- 36.Bi W, et al. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith–Magenis syndrome. Hum. Genet. 2004;115:515–524. doi: 10.1007/s00439-004-1187-6. [DOI] [PubMed] [Google Scholar]
- 37.Bi W, et al. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith–Magenis syndrome. Hum. Mol. Genet. 2005;14:983–995. doi: 10.1093/hmg/ddi085. [DOI] [PubMed] [Google Scholar]
- 38.Bi W, et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum. Mol. Genet. 2007;16:1802–1813. doi: 10.1093/hmg/ddm128. [DOI] [PubMed] [Google Scholar]
- 39.Potocki L, et al. Molecular mechanism for duplication 17p11.2 — the homologous recombination reciprocal of the Smith–Magenis microdeletion. Nature Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- 40.Potocki L, et al. Characterization of Potocki–Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17)(p11.2p11.2). J. Clin. Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 43.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 45.del Gaudio D, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- 46.Van Esch H, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meins M, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J. Med. Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friez MJ, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–e1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- 49.Smyk M, et al. Different-sized duplications of Xq28, including MECP2, in three males with mental retardation, absent or delayed speech, and recurrent infections. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:799–806. doi: 10.1002/ajmg.b.30683. [DOI] [PubMed] [Google Scholar]
- 50.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 51.Collins AL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 52.Samaco RC, et al. A partial loss of function allele of methyl-CpG-binding protein predicts a human neurodevelopmental syndrome. Hum. Mol. Genet. 2008;17:1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yasui DH, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl Acad. Sci. USA. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chao H-T, Zoghbi H, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [This paper provides evidence that either loss or gain of MeCP2 alters excitatory synaptic function, leading to overlapping abnormal neurological phenotypes.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinowich K, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 58.McGill BE, et al. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl Acad. Sci. USA. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bourgeron T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb. Symp. Quant. Biol. 2007;72:645–654. doi: 10.1101/sqb.2007.72.020. [DOI] [PubMed] [Google Scholar]
- 60.Berg JS, et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams–Beuren syndrome region. Genet. Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- 61.Somerville MJ, et al. Severe expressive-language delay related to duplication of the Williams–Beuren locus. N. Engl. J. Med. 2005;353:1694–1701. doi: 10.1056/NEJMoa051962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Torniero C, et al. Cortical dysplasia of the left temporal lobe might explain severe expressive-language delay in patients with duplication of the Williams–Beuren locus. Eur. J. Hum. Genet. 2007;15:62–67. doi: 10.1038/sj.ejhg.5201730. [DOI] [PubMed] [Google Scholar]
- 63.Ewart AK, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nature Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- 64.Tassabehji M. Williams–Beuren syndrome: a challenge for genotype–phenotype correlations. Hum. Mol. Genet. 2003;12(special no 2):R229–R237. doi: 10.1093/hmg/ddg299. [DOI] [PubMed] [Google Scholar]
- 65.Zhao C, et al. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development. 2005;132:2917–2927. doi: 10.1242/dev.01871. [DOI] [PubMed] [Google Scholar]
- 66.Hoogenraad CC, et al. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nature Genet. 2002;32:116–127. doi: 10.1038/ng954. [DOI] [PubMed] [Google Scholar]
- 67.Meng Y, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 68.Heredia L, et al. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid β-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer's disease. J. Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lim MK, et al. Parkin interacts with LIM kinase 1 and reduces its cofilin-phosphorylation activity via ubiquitination. Exp. Cell Res. 2007;313:2858–2874. doi: 10.1016/j.yexcr.2007.04.016. [DOI] [PubMed] [Google Scholar]