Fibrillar Amyloid-β Peptides Activate Microglia via TLR2: Implications for Alzheimer’s Disease (original) (raw)
. Author manuscript; available in PMC: 2009 Nov 15.
Abstract
Microglial activation is an important pathological component in brains of patients with Alzheimer’s disease (AD), and fibrillar amyloid-β (Aβ) peptides play an important role in microglial activation in AD. However, mechanisms by which Aβ peptides induce the activation of microglia are poorly understood. The present study underlines the importance of TLR2 in mediating Aβ peptide-induced activation of microglia. Fibrillar Aβ1–42 peptides induced the expression of inducible NO synthase, proinflammatory cytokines (TNF-α, IL-1β, and IL-6), and integrin markers (CD11b, CD11c, and CD68) in mouse primary microglia and BV-2 microglial cells. However, either antisense knockdown of TLR2 or functional blocking Abs against TLR2 suppressed Aβ1–42-induced expression of proinflammatory molecules and integrin markers in microglia. Aβ1–42 peptides were also unable to induce the expression of proinflammatory molecules and increase the expression of CD11b in microglia isolated from TLR2−/− mice. Finally, the inability of Aβ1–42 peptides to induce the expression of inducible NO synthase and to stimulate the expression of CD11b in vivo in the cortex of TLR2−/− mice highlights the importance of TLR2 in Aβ-induced microglial activation. In addition, ligation of TLR2 alone was also sufficient to induce microglial activation. Consistent to the importance of MyD88 in mediating the function of various TLRs, antisense knockdown of MyD88 also inhibited Aβ1–42 peptide-induced expression of proinflammatory molecules. Taken together, these studies delineate a novel role of TLR2 signaling pathway in mediating fibrillar Aβ peptide-induced activation of microglia. The Journal of Immunology, 2008, 181: 7254–7262.
Alzheimer’s disease (AD)3 is a neurodegenerative disorder resulting in progressive neuronal death and memory loss. Neuropathologically, the disease is characterized by the presence of both neurofibrillary tangles and neuritic plaques composed of aggregates of amyloid-β (Aβ) protein, a 40–43 aa proteolytic fragment derived from the amyloid precursor protein (1, 2). The importance of Aβ in AD has been shown by means of several transgenic animal studies. The overexpression of mutant amyloid precursor protein in mice results in senile plaques formation and synapse loss and correlative memory deficits, as well as behavioral and pathological abnormalities similar to those found in AD patients (1, 2). Although deposition of Aβ peptides is one of the primary causes of neuronal loss in AD (2, 3), the mechanism by which Aβ causes neuronal loss has been poorly characterized.
Microglia are considered as CNS-resident professional macrophages and sensor cells that respond to many pathological events. Localized activation of microglia has been implicated in the pathogenesis of a variety of neurodegenerative diseases, including AD, Parkinson’s disease, Creutzfeld-Jacob disease, HIV-associated dementia, stroke, and multiple sclerosis. During activation, microglia are capable of releasing various potentially cytotoxic molecules such as NO, oxygen radicals, proteases, adhesion molecules, and proinflammatory cytokines such as TNF-α, IL-1β, LT-α, and IL-6 (4–8). It is believed that excessive production of these neurotoxic proinflammatory molecules plays an important role in enhancing the degenerative process in the inflamed CNS of AD patients. Therefore, understanding mechanisms that regulate microglial activation is an important area of investigation that may enhance the possibility of finding a primary or an adjunct therapeutic approach against incurable neurodegenerative disorders.
TLR, the mammalian homologs of the Drosophila Toll protein, serves as an important link between innate and adaptive immunity (9, 10). TLRs respond mainly to bacteria, bacterial products, virus and flagellin by transmitting a ligand-induced transmembrane signal that induces the expression of various cytokines such as TNF-α, IL-1, IL-6, and IL-12 for host responses. At least 11 different TLRs have been described to date, which display distinct ligand specificities (11–16). Although all the major CNS cell types express TLRs, microglia are the only cells in the CNS that express almost all the TLRs known to date. However, theoretically CNS microglia should not come in contact to bacterial products barring couple of pathological conditions such as meningitis and brain abscess, suggesting that TLRs might be involved in pathogen-in-dependent brain pathologies as well.
We wondered whether TLRs were playing any role in Aβ-induced microglial activation. In this study, we report that fibrillar Aβ1–42 increased the expression of both proinflammatory molecules and TLR2 in microglia. However, antisense knockdown of TLR2 suppressed fibrillar Aβ1–42-induced expression of proin-flammatory molecules and integrin markers in microglia. Furthermore, microglia derived from TLR2 knockout mice were not activated by fibrillar Aβ1–42. These in vitro data were substantiated by in vivo findings in which intracortical injection of fibrillar Aβ1–42 in TLR2 knockout mice did not exhibit any microglial activation.
Materials and Methods
Reagents
FBS and DMEM/F-12 were obtained from Mediatech. LPS (Escherichia coli) and polyinosinic-polycytidylic acid (poly(I-C)) were purchased from Sigma-Aldrich. Human Aβ1–42 and Aβ42-1 reverse peptides were obtained from Bachem Bioscience. Lipoteichoic acid (LTA) from Bacillus subtilis (TLR2 ligand) and FSL1 (follistatin-like 1 ligand for TLR2/6) have been obtained from Invivogen. Anti-mouse TLR2 Abs were obtained from eBioscience. Abs against mouse CD11b and inducible NO synthase (iNOS) were purchased from Calbiochem. TLR2−/− mice and littermate controls were purchased from Jackson ImmunoResearch Laboratories. Phosphorothioate-labeled antisense and scrambled oligodeoxynucleotides were synthesized in the DNA-synthesizing facility of Invitrogen.
The following antisense oligonucleotide (ASO) and scrambled oligonu-cleotide (ScO) were used to target MyD88 and different TLRs genes: MyD88 (ASO) 5′-GGC CGC CAC GGG CGT CCG AG-3′, (ScO) 5′-GGA CCC CGA GGG CCG CGC TG-3′; TLR1 (ASO) 5′-GGT AGG TCC TTG GGC ACT CTG-3′, (ScO) 5′-GGC TCC TTT AGG GCC ATG GTG-3′; TLR2 (ASO) 5′-CTG GAG CGG CCA TCA CAC ACC-3′, (ScO) 5′-CAT CGC ACG CAG CCGAGC CAT-3′; TLR3 (ASO) 5′-GGC TGC AGT CAG CTA CGT TG-3′, (ScO) 5′-TGC AGC TAG TGC TAG GCG TC-3′; TLR4 (ASO) 5′-GCC AGG AGC CAG GGA GGC A-3′, (ScO) 5′-ACG GCG ACG GCA AGC GGA G-3′; TLR6 (ASO) 5′-GCA GAG GCT ATC CCA GAG GG-3′, (ScO) 5′-GCG GAA TGC CCG GAA CGT AG-3′; TLR7 (ASO) 5′-GCA GTC CAC GAT CAC ATG G-3′, (ScO) 5′-ATC GGA TCC GAC TGA CCA G-3′; and TLR9 (ASO) 5′-GGA GGG ACA AGG GGT GCA G-3′, (ScO) 5′-AGG GCG GGA TGG AGC GAG A-3′.
Preparation of fibrillar Aβ
Fibrillar Aβ1–42 (Bachem Bioscience) was prepared by incubating freshly solubilized peptides at 50 µM in sterile distilled water at 37°C for 5 days (17).
Isolation of mouse microglia
Primary microglia were isolated from mixed glial cultures according to the procedure of Giulian and Baker (18). Briefly, on day 7–9, the mixed glial cultures were washed three times with DMEM/F-12 and subjected to shaking at 240 rpm for 2 h at 37°C on a rotary shaker. The floating cells were washed and seeded on to plastic tissue culture flasks and incubated at 37°C for 2 h. The attached cells were removed by trypsinization and seeded onto new plates for further studies. The 90–95% of this preparation was found to be positive for Mac-1 surface Ag (19, 20). Mouse BV-2 microglial cells, a gift from V. Bocchini (University of Perugia, Perugia, Italy) were also maintained and induced as indicated.
Semiquantitative RT-PCR analysis
Total RNA was isolated from mouse BV2 microglia and mouse primary microglial cells using RNA-Easy Qiagen kit following the manufacturer’s protocol. To remove any contaminating genomic DNA, total RNA was digested with DNase. Semiquantitative RT-PCR was conducted as described earlier (16) using oligo(dT)12–18 as primer and MMLV reverse transcriptase (Clontech Laboratories) in a 20-µl reaction mixture. The resulting cDNA was appropriately diluted, and diluted cDNA was amplified using Titanium Taq polymerase and the following primers: iNOS (sense) 5′-CAG CTC CTC ACT GGG ACA GCA CAG A-3′, (antisense) 5′-CTT CCA GCC TGG CCA GAT GTT CCT C-3′; IL-1′ (sense) 5′-ATG GCA ACT GTT CCT GAA CTC AAC T-3′, (antisense) 5′-CAG GAC AGG TAT AGA TTC TTT CCT TT-3′; TNF-α (sense) 5′-TTC TGT CTA CTG AAC TTC GGG GTG ATC GGT CC-3′, (antisense) 5′-GTATGA GAT AGC AAA TCG GCT GAC GGT GTG GG-3′; IL-6 (sense) 5′-CGT CCC CTG GCA TTC CTA GTG GTG-3′, (antisense) 5′TGA TCC AGG CGT GAC ATC-3′; IL-15 (sense) 5′-GGG CTG TGT CAG TGT AGG TCT CCC T-3′, (antisense) 5′-CCA GCT CCT CAC ATT CCT TGC AGC C-3′; CD11b (sense) 5′-CAG ATC AAC AAT GTG ACC GTA TGG G-3′, (antisense) 5′-CAT CAT GTC CTT GTA CTG CCG CTT G-3′; MyD88 (sense) 5′-GCT GCT GGC CTT GTT AGA CCG TGA G-3′, (antisense) 5′-GAC GTC ACG GTC GGA CAC ACA CAA C-3′; TLR1 (sense) 5′-GGA CCT ACC CTTT GCA AAC AA-3′, (antisense) 5′-GGT GGC ACA AGA TCA CCT TT-3′; TLR2 (sense) 5′-TGC TTT CCT GCT GGA GAT TT-3′, (antisense) 5′-TGT AAC GCA ACA GCT TCA GG-3′; TLR3 (sense) 5′-TTG TCT TCT GCA CGA ACC TG-3′, (antisense) 5′-GGC AAC GCA AGG ATT TTA TT-3′; TLR4 (sense) 5′-ACC TGG CTG GTT TAC ACG TC-3′, (antisense) 5′-CTG CCA GAG ACA TTG CAG AA-3′; TLR5 (sense) 5′-GAG CTC AAT GGG GGA CCA GAA CAC-3′, (antisense) 5′-CGG CAG TAC TGA CAC TTG TTG CGG-3′; TLR6 (sense) 5′-CCA AGA ACA AAA GCCC CTG AG-3′, (antisense) 5′-TGT TTT GCA ACC GAT TGT GT-3′; TLR7 (sense) 5′-GGA AAT TGC CCT CGA TGT TA-3′, (antisense) 5′-CAA AAA TTT GGC CTC CTC AA-3′; TLR8 (sense) 5′-GAA GCA TTT CGA GCA TCT CC-3′, (antisense) 5′-GAA GAC GAT TTC GCC AAG AG-3′; TLR9 (sense) 5′-ACT GAG CAC CCC TGC TTC TA-3′, (antisense) 5′-AGA TTA GTC AGC GGC AGG AA-3′; and GAPDH (sense) 5′-GGT GAA GGT CGG AGT CAA CG-3′, (antisense) 5′-GTG AAG ACG CCA GTG GAC TC-3′.
Amplified products were electrophoresed on a 1.8% agarose gels and visualized by ethidium bromide staining. Message for the GAPDH gene was used to ascertain that an equivalent amount of cDNA was synthesized from different samples.
Real-time PCR analysis
It was performed using the ABI Prism 7700 sequence detection system (Applied Biosystems) as described earlier (21). Briefly, reactions were performed in 96-well optical reaction plates on cDNA equivalent to 50 ng of DNase-digested RNA in a volume of 25 µl, containing 12.5 µl of TaqMan Universal Master mixture and optimized concentrations of FAM-labeled probe, forward and reverse primers, following the manufacturer’s protocol. All primers and FAM-labeled probes for mouse TNF-α, iNOS, IL-6, IL-1β, and GAPDH were obtained from Applied Biosystems. The mRNA expression of different genes was normalized to the label of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software and analyzed by ANOVA.
Immunofluorescence analysis
Immunofluorescence analysis was performed as previously described (21). Briefly, wells containing 4–5 × 105 cells/well were fixed with 4% para-formaldehyde for 20 min followed by treatment with cold ethanol (−20°C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS-T (PBS-Tween 20) for 30 min and incubated in PBS-T containing 1% BSA and goat anti-CD11b (1/100), and mouse anti-iNOS (1/75). After three washes in PBS-T (15 min each), wells were further incubated with Cy5 and Cy2 (Jackson ImmunoResearch Laboratories). For negative controls, a set of culture wells was incubated under similar conditions without primary Abs. The samples were mounted and observed under an Olympus IX81 fluorescence microscope.
Microinjection of Aβ into the cortex of wild-type and TLR2−/− mice
Wild-type and TLR2−/− B6.129 mice (8- to 10-wk-old) were anesthetized with i.p. injection of ketamine and xylazine and underwent cortical operations in a Kopf small animal stereotaxic instrument (Kopf Instruments). Briefly, the animal was mounted in a stereotaxic frame on a heating blanket. Body temperature was maintained at 37 ± 0.5°C during the time of surgery. A midsagittal incision was made to expose the cranium, and a hole <0.5 mm in diameter was drilled with a dental drill over the frontal cortex according to the following coordinates: 1.5 mm anterior to bregma, 1.5 mm lateral to bregma, and 1 mm ventral (see Fig. 8_A_). Either Aβ1–42 (1 µg) or Aβ42–1 (1 µg) in 2 µl of saline solution was injected using a 5-µl syringe (Hamilton) over a period of 3 min, and the needle was held in place for another minute before withdrawing it from the skull to prevent reflux up the needle tract. Similarly, control mice received 2 µl of saline solution. The incision was closed with surgical staples and covered with a mixture of Bacitracin and Hurricane (20% benzocaine).
FIGURE 8.
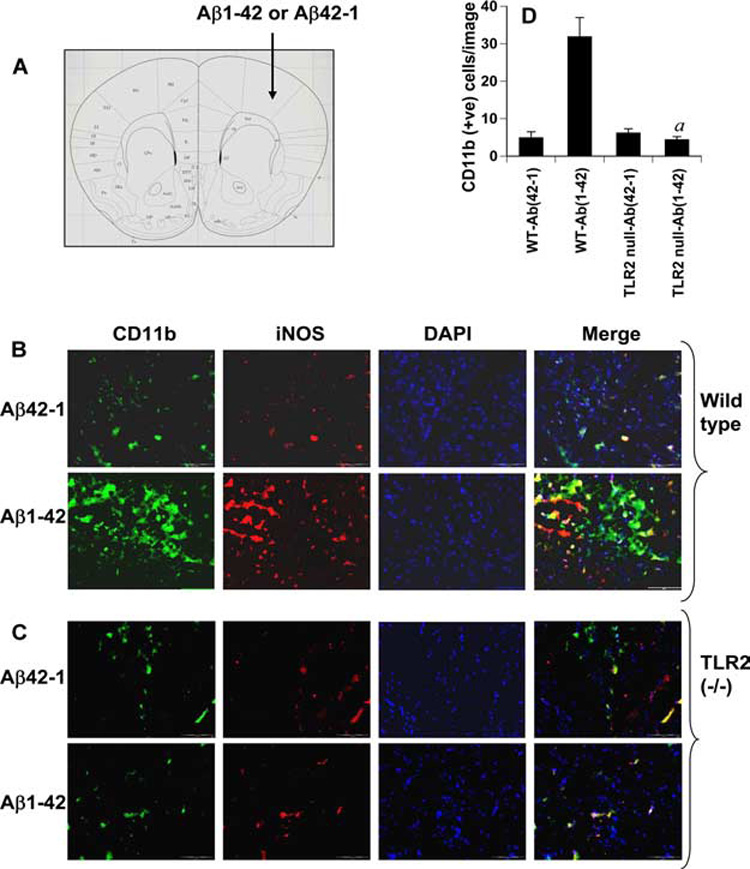
Fibrillar Aβ-mediated activation of microglia in vivo in the cortex depends on TLR2. One microgram of either Aβ1–42 or Aβ42–1 dissolved in 2 µl of saline solution was stereotaxically injected into the cortex (A) of wild-type (B) and TLR2−/− (C) mice. After 24 h of microinjection, cortical sections of both wild-type and knockout mice were double-immunolabeled with Abs against CD11b and iNOS. DAPI (4′,6-diamidino-2-phenylindole) was used to visualize nucleus. Setting of the fluorescent microscope was strictly kept unaltered during the experiment. D, CD11b-positive cells were counted in four cortical sections (two images per slide) of each of four different mice by an Olympus IX81 fluorescence microscope using the MicroSuite imaging software. a, p < 0.001 vs wild-type Aβ1–42.
Histological analysis
After 24 h of microinjection, six mice from each of the following groups (saline and Aβ) were anesthetized with a mixture of ketamine (66.6 mg/kg) and xylazine (6.66 mg/kg) by i.p. injection. The cortex was dissected from each mice model after perfusion with PBS (pH 7.4) and then with 4% (w/v) paraformaldehyde solution in PBS (22). The tissues were further fixed for at least 2 h in the same fixative at room temperature followed by three PBS washes (15 min each). Routine histology was performed to obtain morphological details of cortex tissues. Paraformaldehyde-fixed tissues were embedded in paraffin, and serial sections (5 µm) were cut. Sections were stained with Abs against CD11b and iNOS.
Statistics
Data are expressed as mean ± SD. Statistical comparisons were made using Student’s t test (SAS system). Differences between mean were considered significant at p < 0.05.
Results
Effect of Aβ on the mRNAs expression of different TLRs in mouse BV-2 microglial cells and primary microglia
Microglial activation plays an important role in the pathogenesis of AD. It has been reported that microglia are the only cells in the CNS that express most of the TLRs (TLR1-TLR9) known to date (23). Therefore, we decided to explore the role of TLRs in Aβ-mediated microglial activation. Because the fibrillar form of Aβ is commonly found in the senile plaques in the brain of AD patients, at first we examined whether fibrillar Aβ1–42 was capable of modulating the expression of TLRs in microglia. As demonstrated in Fig. 1_A_, fibrillar Aβ markedly stimulated the expression of TLR2, TLR4, TLR6, TLR7, and TLR9, but not TLR1, TLR3, TLR5, and TLR8, in BV-2 microglial cells. We compared the expression pattern of different TLRs by Aβ with that of LPS and poly(I-C), which are known stimuli of various TLRs. Similar to Aβ, bacterial LPS also stimulated the expression of TLR2, TLR4, TLR6, TLR7, and TLR9 in microglial cells (Fig. 1_A_). However, among three stimuli tested, only poly(I-C) was capable of increasing the expression of TLR3 and TLR8 (Fig. 1_A_). Poly(I-C) also stimulated the expression of TLR2, TLR6, TLR7, and TLR9 in microglial cells (Fig. 1_A_). Next we investigated whether Aβ was also capable of increasing the expression of different TLRs in primary microglia. Similar to that observed in BV-2 microglial cells, Aβ increased the expression of TLR2, TLR4, TLR6, TLR7, and TLR9 in primary mouse microglia (Fig. 1_B_). However, in contrast to BV-2 microglial cells, Aβ stimulated the expression of TLR1 in primary microglia (Fig. 1_B_). These results suggest that fibrillar Aβ increases the expression of various TLRs in microglia and that TLRs may be involved in Aβ-induced microglial pathology.
FIGURE 1.
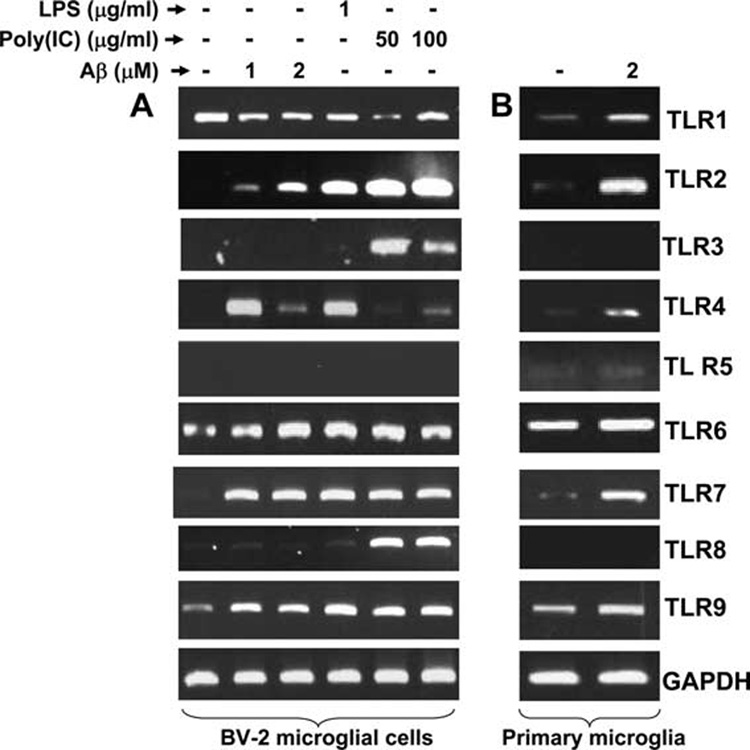
Effect of fibrillar Aβ1–42 peptides on the expression of different TLRs in mouse BV-2 microglial cells and primary microglia. Mouse BV-2 microglial cells (A) and primary microglia (B) were stimulated with fibrillar Aβ1–42, LPS, and poly(I-C) under serum-free conditions. After 6 h of treatment, total RNA was analyzed for the expression of different TLRs by semiquantitative RT-PCR. Results represent three independent experiments.
Effect of Aβ on the mRNA expression of different proinflammatory molecules and integrins in microglial cells
Aβ peptides are known to activate microglia. A number of studies have shown that Aβ peptides induce the expression of proinflammatory molecules and increase the level of cell surface integrins in microglia (8, 24–27). To investigate whether the interaction of fibrillar Aβ1–42 with microglial cells could also induce the expression of proinflammatory molecules and cell surface integrins in our culture condition, we examined the mRNA expression of these molecules after treatment with fibrillar Aβ1–42. Cells were also stimulated with LPS and poly(I-C) as positive controls. It is evident from Fig. 2_A_ that fibrillar Aβ1–42 peptides at different doses tested induced the mRNA expression of proinflammatory molecules (iNOS, TNF-α, IL-1β, IL-6, and IL-15) and microglial integrin markers (CD11a, CD11b, CD11c, and CD68) in microglial cells. We tested the specificity of the effect by using fibrillar reverse (Aβ42-1) peptides. As expected, reverse Aβ42-1 peptides were unable to increase the expression of TNF-α in microglial cells (Fig. 2_B_). These results suggest that fibrillar Aβ1–42 is capable of increasing the expression of proinflammatory molecules and integrin markers in microglia.
FIGURE 2.
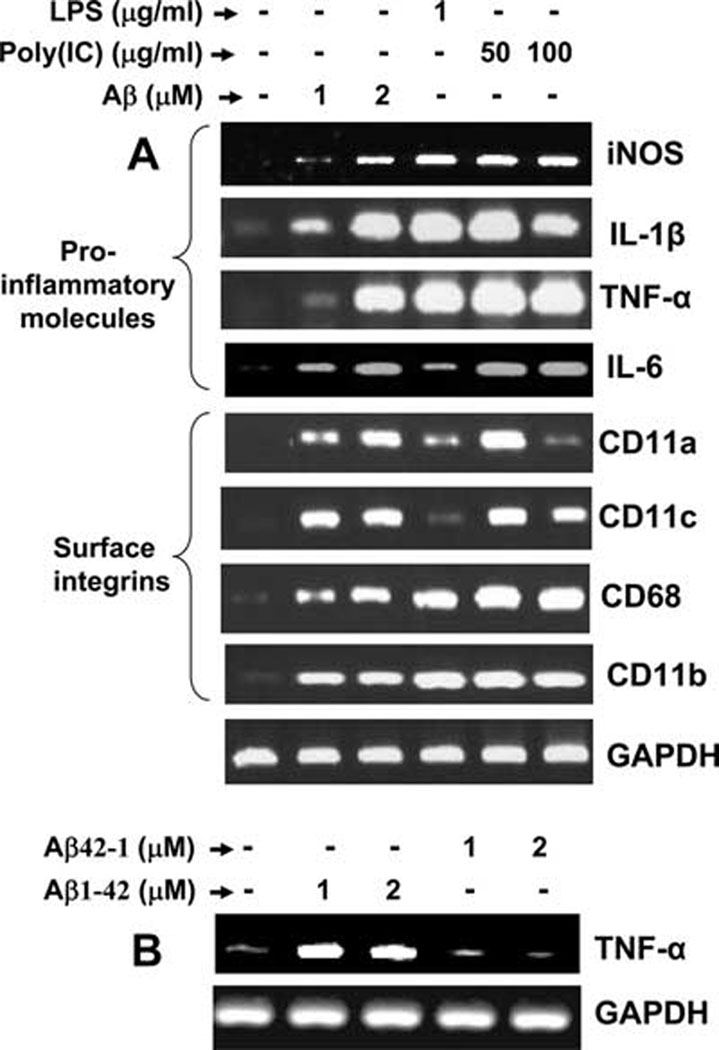
Microglial cells express different proinflammatory cytokines, iNOS, and microglial cell surface markers after stimulation with Aβ1–42. A, Mouse BV-2 microglial cells were stimulated with fibrillar Aβ1–42, LPS, and poly(I-C) under serum-free conditions for 6 h followed by analysis of various proinflammatory molecules (iNOS, IL-1β, TNF-α, and IL-6) and surface integrins (CD11a, CD11b, CD11c, and CD68) by semiquantitative RT-PCR. B, Cells were stimulated with different concentrations of fibrillar Aβ1–42 and Aβ42-1 (reverse) peptides. After 6 h of stimulation, the mRNA expression of TNF-α was analyzed by RT-PCR. Results represent three independent experiments.
Antisense knockdown of various TLRs on fibrillar Aβ1–42-induced expression of proinflammatory molecule in mouse BV-2 microglial cells
Next we sought to determine which, if any, TLRs were functional and able to play a role in initiating an innate immune response in microglial cells after Aβ1–42 challenge. ASOs provide the most effective tool with which to investigate the in vivo functions of different proteins in primary nondividing cells. Therefore, we adopted the antisense knockdown technique to investigate the role of TLRs in Aβ1–42-mediated expression of TNF-α in microglia. At first, we selected and screened-specific ASO and ScO sequences against different mouse TLRs. Cells receiving either ASO or ScO were analyzed for the expression of different TLRs by RT-PCR. As shown in Fig. 3_A_, ASO, but not ScO, against TLR1 knocked down the expression of TLR1 in microglial cells. However, antisense knockdown of TLR1 had no effect on Aβ-induced expression of TNF-α (Fig. 3_A_). As expected, ASO, but not ScO, against TLR2 suppressed the expression of TLR2 (Fig. 3_B_). Interestingly, in contrast to antisense knockdown of TLR1, ASO against TLR2 abrogated Aβ-induced expression of TNF-α in microglial cells (Fig. 3_B_). Similarly, ASOs, but not ScOs, derived against TLR4 (Fig. 3_C_), TLR6 (Fig. 3_D_), TLR7 (Fig. 3_E_), and TLR9 (Fig. 3_F_) knocked down the expression of respective TLRs. However, antisense knockdown of TLR4, TLR6, TLR7, and TLR9 had no effect on Aβ-induced expression of TNF-α in microglial cells (Fig. 3, C–F). These results suggest the possible involvement of TLR2 in Aβ-induced expression of TNF-α in microglial cells.
FIGURE 3.
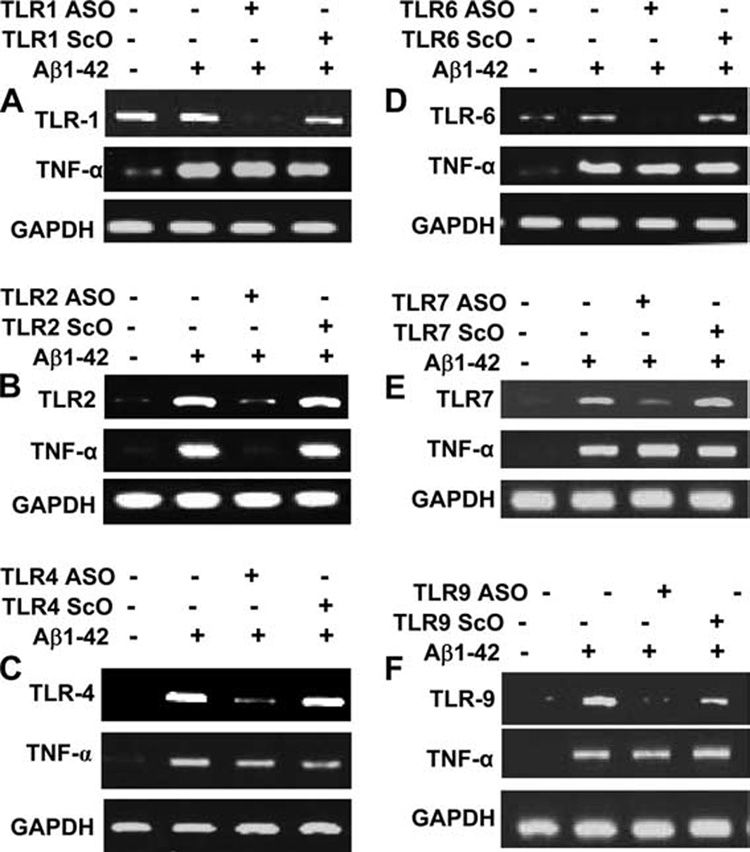
Antisense knockdown of TLR2, but not other TLRs, leads to impaired expression of TNF-α in response to fibrillar Aβ1–42 in mouse BV-2 microglial cells. Cells received 0.5 µM of either ASO or ScO against TLR1 (A), TLR2 (B), TLR4 (C), TLR6 (D), TLR7 (E), and TLR9 (F). After 48 h of incubation, cells were stimulated with 2 µM fibrillar Aβ1–42 under serum-free conditions. Total RNA was analyzed for the expression of TNF-α and respective TLR by semiquantitative RT-PCR. Results represent three independent experiments.
To address whether knockdown of TLR2 affects specifically Aβ-induced microglial expression of TNF-α or other inducers of microglial activation also require TLR2 for the expression of microglial TNF-α, we examined the effect of antisense knockdown of TLR2 on LPS- and poly(I-C)-induced expression of TNF-α. As expected, LPS alone induced the expression of TNF-α (Fig. 4_A_). Although consistent to an earlier report (13), LPS markedly induced the mRNA expression of TLR2 in microglial cells (Fig. 1_A_), yet in contrast to suppressing Aβ-induced expression of TNF-α (Fig. 3_B_), ASO against TLR2 did not affect LPS-induced microglial expression of TNF-α (Fig. 4_A_). Because LPS is known to function via TLR4, we examined whether antisense knockdown of TLR4 abrogates LPS-induced expression of TNF-α. It is clearly evident from Fig. 4_B_ that ASO, but not ScO, against TLR4 markedly suppressed LPS-induced expression of TNF-α. Similarly, antisense knockdown of TLR2 also had no effect on poly(I-C)-induced expression of TNF-α (Fig. 4_C_). However, as expected, antisense knockdown of TLR3, the receptor for poly(I-C), inhibited poly(I-C)-mediated expression of TNF-α in microglial cells (Fig. 4_D_). It appears that although LPS and poly(I-C) induced microglial TNF-α via TLR4 and TLR3, respectively, fibrillar Aβ1–42 peptides involved TLR2 to induce TNF-α in microglia.
FIGURE 4.
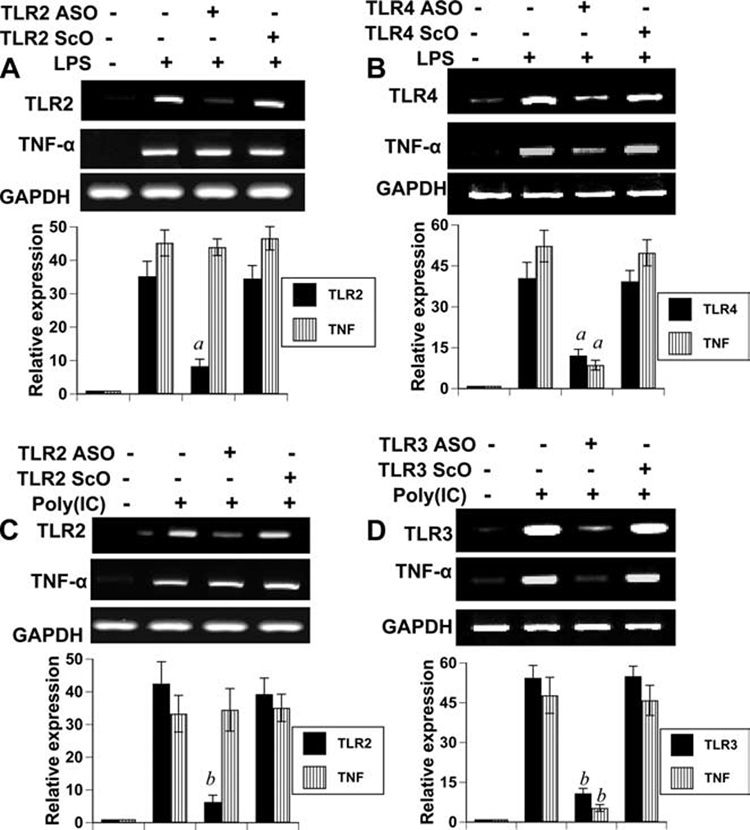
Effect of antisense knockdown of TLR2, TLR3, and TLR4 on LPS- and poly(I-C)-induced expression of TNF-α in mouse BV-2 microglial cells. Cells received 0.5 µM of either ASO or ScO against TLR2 (A) and TLR4 (B). After 48 h of incubation, cells were stimulated with 1 µg/ml LPS for 6 h followed by analysis of TNF-α by RT-PCR. Cells received 0.5 µM of either ASO or ScO against TLR2 (C) and TLR3 (D). After 48 h of incubation, cells were stimulated with 100 µg/ml poly(I-C) for 6 h followed by analysis of TNF-α by RT-PCR. The relative expression (bottom panels) of TLRs or TNF-α (TLR/GAPDH or TNF-α/GAPDH) was measured after scanning the bands with a Fluor Chem 8800 Imaging System (Alpha Innotech Corporation). Results represent mean ± SD of three independent experiments. a, p < 0.001 vs LPS and b, p < 0.001 vs poly(I-C).
Antisense knockdown of TLR2 suppresses Aβ-induced expression of proinflammatory molecules in mouse primary microglia
In addition to TNF-α, Aβ peptides are also capable of inducing the expression of other proinflammatory molecules and integrin markers in microglia (Fig. 2). Therefore, next we investigated whether fibrillar Aβ1–42 peptides need TLR2 to induce the expression of proinflammatory molecules and integrin markers in mouse primary microglia. As expected, fibrillar Aβ peptides induced the mRNA expression of proinflammatory molecules (iNOS, IL-1β, and IL-6) (Fig. 5) and integrin markers (CD11a, CD11b, and CD68) (Fig. 5_A_) in primary microglia. However, consistent to that observed in BV-2 microglial cells, ASO, but not ScO, against TLR2 attenuated the Aβ1–42-induced expression of TLR2, iNOS, IL-1β, IL-6, CD11a, CD11b, and CD68 in mouse primary microglia (Fig. 5).
FIGURE 5.
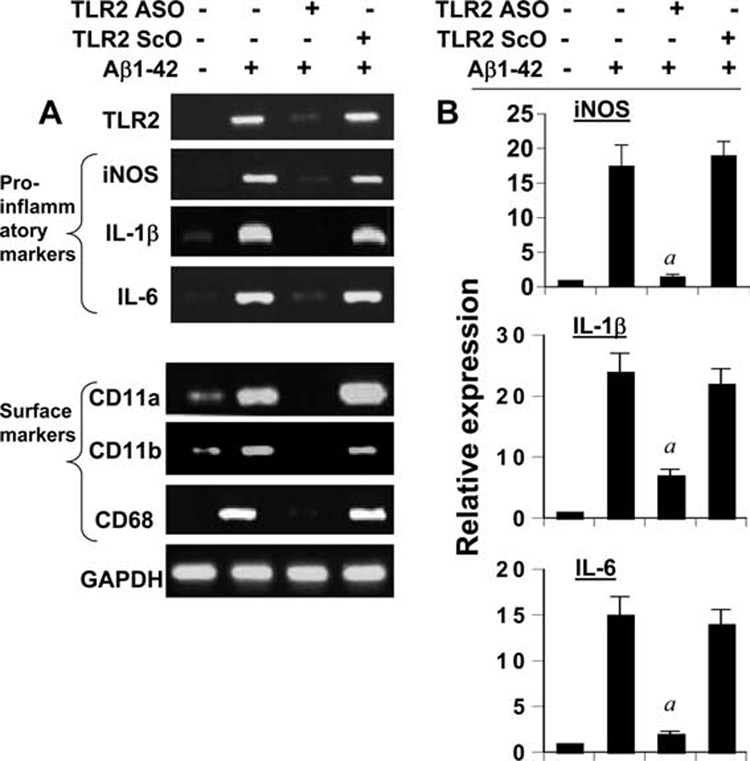
Antisense knockdown of TLR2 suppresses Aβ-induced expression of proinflammatory molecules and surface integrins in primary mouse microglia. Cells received 0.5 µM of either ASO or ScO against TLR2. A, After 48 h of incubation, cells were stimulated with 2 µM fibrillar Aβ1–42 for 6 h under serum-free conditions followed by analysis of proinflammatory molecules (iNOS, IL-1β, and IL-6) and microglial integrin markers (CD11a, CD11b, and CD68) by semiquantitative RT-PCR. B, The expression of proinflammatory molecules (iNOS, IL-1β, and IL-6) was also analyzed by quantitative real-time PCR. Results are mean ± SD of three different experiments. a, p < 0.001 vs only Aβ1–42.
Neutralization of TLR2 blocks Aβ-induced expression of proinflammatory molecules in mouse primary microglia
To further confirm these results, we used anti-TLR2-neutralizing Ab to examine the possible involvement of TLR2 in Aβ-induced expression of TNF-α, iNOS, and CD11b in mouse primary microglia. As shown in Fig. 6_A_, anti-TLR2 functional blocking Ab significantly inhibited the mRNA expression of TNF-α and iNOS in Aβ-stimulated microglia. In contrast, isotype control IgG had no such inhibitory effect on the mRNA expression of TNF-α and iNOS (Fig. 6_A_). Immunohistochemistry results also indicate that fibrillar Aβ1–42 peptides markedly increased the surface expression of CD11b in microglia and that neutralization of TLR2 resulted in a significant reduction of CD11b protein expression in Aβ-stimulated microglia (Fig. 6_B_). As expected, isotype control IgG was unable to suppress Aβ-induced expression of CD11b protein in microglia (Fig. 6_B_). Consistently, Aβ1–42 peptides also induced the expression of iNOS protein in CD11b-positive microglia that was negated by functional blocking Abs against TLR2, but not control IgG (Fig. 6_B_). These results suggest that TLR2 is functional in microglia and that Aβ peptides transduce proinflammatory signal in microglia via TLR2.
FIGURE 6.
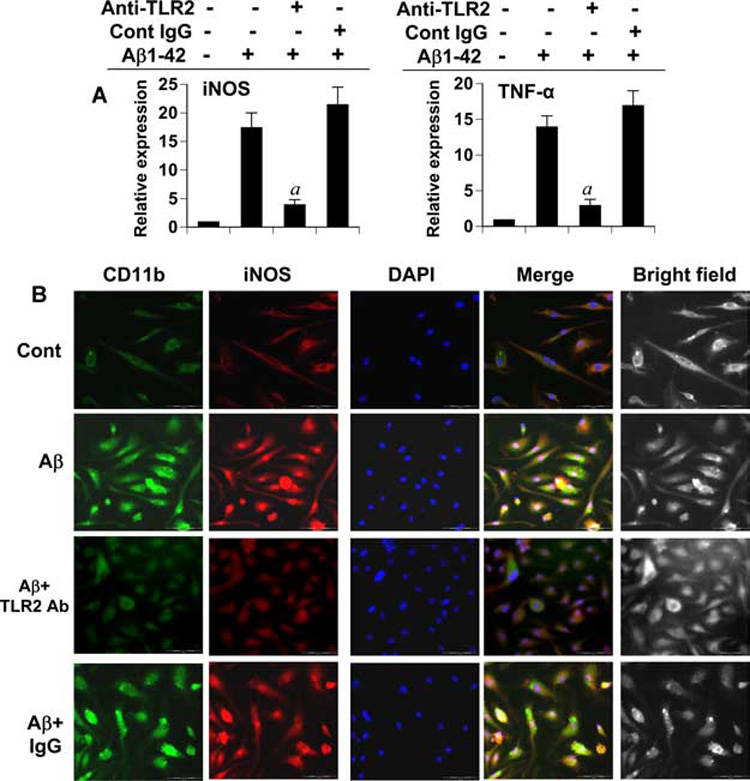
Effect of functional blocking Abs against TLR2 on the expression of proinflammatory molecules in primary mouse microglia. A, Cells preincubated with either functional blocking Abs against TLR2 or control IgG for 30 min were stimulated by 2 µM fibrillar Aβ1–42 for 6 h followed by analysis of iNOS and TNF-α mRNAs by quantitative real-time PCR. Results are mean ± SD of three different experiments. a, p < 0.001 vs only Aβ1–42. B, Cells preincubated with either TLR2 blocking Ab or control IgG for 30 min were stimulated by fibrillar Aβ. After 24 h, cells were double-immunolabeled with Abs against CD11b and iNOS. DAPI (4′,6-diamidino-2-phenylindole) was used to visualize nucleus. Results represent three independent experiments.
Effect of Aβ peptides on the expression of proinflammatory molecules in microglia isolated from TLR2−/− mice
To further confirm the involvement of TLR2 in Aβ-induced microglial activation, we isolated primary microglia from wild-type and TLR2 knockout mouse pups. As observed in Fig. 7_A_, Aβ1–42 peptides induced the mRNA expression of iNOS and TNF-α in microglia isolated from wild-type, but not TLR2−/− mice. Double-labeled immunofluorescence results in Fig. 7_B_ also demonstrate that Aβ peptides were able to induce the expression of iNOS protein in CD11b-positive primary microglia isolated from wild-type, but not TLR2−/− mice. Recently Lotz et al. (28) have shown that soluble Aβ1–40 peptides increase the release of proinflammatory molecules (NO and TNF-α) in microglia stimulated by Pam3Cys, an agonist of TLR2. However, Aβ1–40 peptides induce the production of proinflammatory molecules independent of TLR2 (28). Because in our experiment Aβ1–42 requires TLR2 for the induction of proinflammatory molecules in microglia, we investigated whether Aβ1–40 also requires TLR2. It is clearly evident as shown in Fig. 7_C_ that Aβ1–40 peptides induced the expression of TNF-α in microglia isolated from both wild-type and TLR2−/− mice. These results suggest that fibrillar Aβ1–42, but not Aβ1–40, induces proinflammatory molecules in microglia via TLR2.
FIGURE 7.
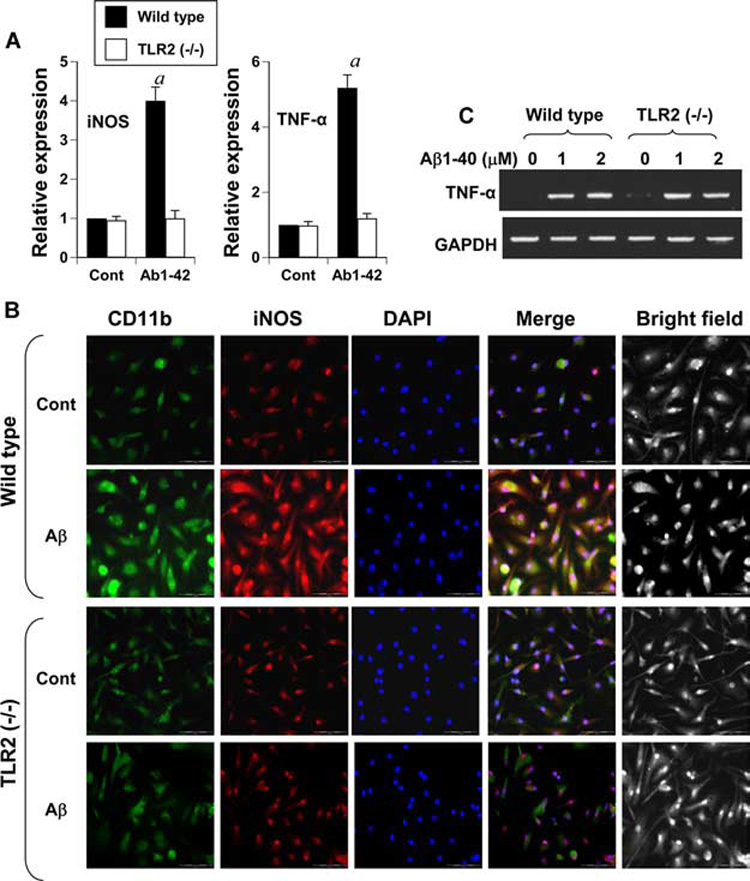
Immunofluorescence and gene expression analysis of microglial activation markers in Aβ-treated wild-type and TLR2−/− microglia. Microglia isolated from wild-type and TLR2−/− mice were stimulated with 2 µM fibrillar Aβ1–42 under serum-free conditions. A, After 6 h of treatment, the mRNA expression of iNOS and TNF-α was analyzed by quantitative real-time PCR. Results are mean ± SD of three different experiments. a, p < 0.001 vs control. B, After 24 h of treatment with 2 µM fibrillar Aβ1–42, cells were double-immunolabeled with Abs against CD11b and iNOS. DAPI (4′,6-diamidino-2-phenylindole) was used to visualize nucleus. Results represent three independent experiments. C, Microglia isolated from wild-type and TLR2−/− mice were stimulated with different concentrations of Aβ1–40 under serum-free conditions. After 6 h of treatment, the mRNA expression of TNF-α was analyzed by semiquantitative PCR.
Is activation of microglia by Aβ in vivo in the CNS dependent on TLR2
Using various approaches, the experiments presented have shown that fibrillar Aβ1–42 peptides increase the expression of proinflammatory molecules in cultured microglia through TLR2 signaling pathway. Next to address whether fibrillar Aβ1–42 peptides require TLR2 to induce iNOS in vivo in the CNS, fibrillar Aβ1–42 peptides were microinjected into the cortex of wild-type and TLR2−/− mice. We also used Aβ42–1 (reverse) peptides as control. As expected, microinjection of fibrillar Aβ1–42 (Fig. 8_B_, bottom), but not Aβ42–1 peptides (Fig. 8_B_, top), into the cortex (Fig. 8_A_) of 8- to 10-wk-old male B6.129 (wild-type) mice induced the expression of iNOS and CD11b proteins. In fact, most of the CD11b-positive cells in (Aβ1–42)-microinjected cortex of wild-type mice colocalized with iNOS (Fig. 8_B_, bottom). However, both Aβ42–1 and Aβ1–42 peptides failed to increase the expression of iNOS and CD11b proteins in the cortex of TLR2−/− mice (Fig. 8_C_). Quantitation of CD11b-positive cells in cortical sections also shows that Aβ1–42 but not Aβ42–1 peptides induced microgliosis and that Aβ1–42 peptides induced microglial activation via TLR2 (Fig. 8_D_). These in vivo results clearly demonstrate that TLR2 is critical for (Aβ1–42)-induced microglial activation.
Determining whether ligation of TLR2 alone is sufficient to induce the expression of proinflammatory molecules in microglia
In addition to engaging TLR2, fibrillar Aβ peptides may also involve multiple signaling cascades to activate microglia (32). Therefore, we investigated whether ligation of TLR2 alone was sufficient for the activation of microglia. FSL1 and LTA are known ligands of TLR2 (29–31). Therefore, microglial cells were stimulated with different concentration of LTA-1 and FSL1 for 6 h followed by analysis of mRNA expression by RT-PCR. As illustrated in Fig. 9_A_, LTA-1 alone dose-dependently enhanced the expression of iNOS, IL-1β, and IL-6 as compared with untreated control. Similarly, FSL1 also dose-dependently increased the expression of iNOS, IL-1β, and IL-6 in BV-2 microglial cells (Fig. 9_B_). Taken together, these results clearly demonstrate that TLR2 is functional in microglial cells and that engagement of TLR2 alone is sufficient for microglial activation.
FIGURE 9.
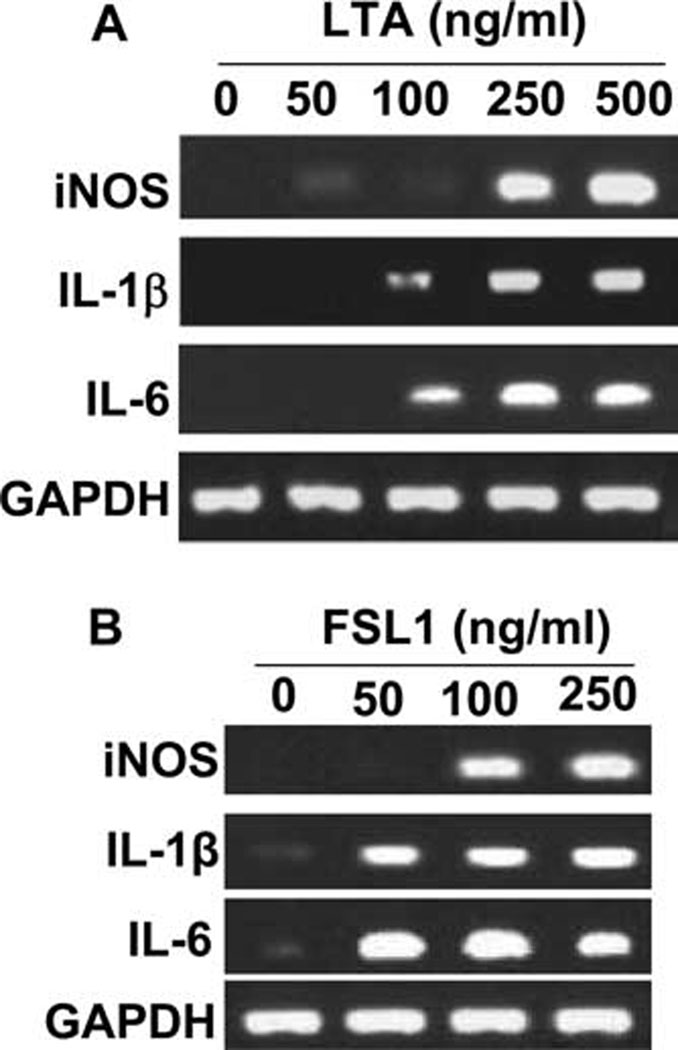
LTA and FSL1, ligands of TLR2, induce the expression of proinflammatory molecules in mouse BV-2 microglial cells. Cells were stimulated with different doses of LTA (A) and FSL1 (B) under serum-free condition for 6 h followed by analysis of proinflammatory molecules (iNOS, IL-1β, and IL-6) by semiquantitative RT-PCR. Results represent three independent experiments.
Does Aβ-induced expression of proinflammatory molecules in microglia depend on MyD88
TLR signal transduction is mediated via different adaptor proteins (33–37). MyD88, the most important adaptor molecule for most TLRs including TLR2, serves to bridge TLRs to the downstream signaling elements (MAPKs or NF-κB) and leads to the expression of proinflammatory molecules (33, 34, 38). Therefore, to investigate whether fibrillar Aβ1–42 up-regulate the expression of proinflammatory molecules in primary mouse microglia through the adaptor molecule MyD88, we examined the effect of antisense knockdown of MyD88 on Aβ-induced expression of proinflammatory molecules by RT-PCR and quantitative real-time PCR. Primary microglia constitutively expressed MyD88 and we did not observe any increase in its expression after Aβ challenge (Fig. 10_A_). As expected, ASO, but not ScO, against MyD88 markedly suppressed the expression of MyD88 (Fig. 10_A_). Consistent to the involvement of TLR2 in Aβ-induced expression of proinflammatory molecules in microglia, antisense knockdown of MyD88 attenuated the mRNA expression of iNOS and TNF-α in Aβ-stimulated primary mouse microglia (Fig. 10). These results suggest that fibrillar Aβ1–42 peptides use the TLR signal transduction pathway for the induction of proinflammatory molecules in microglia.
FIGURE 10.

Antisense knockdown of MyD88 suppresses Aβ-induced expression of proinflammatory molecules in mouse BV-2 microglial cells. Cells received 0.5 µM of either ASO or ScO against MyD88. After 48 h of incubation, cells were stimulated with 2 µM fibrillar Aβ1–42 for 6 h followed by the analysis of proinflammatory molecules (iNOS and TNF-α) by semiquantitative RT-PCR (A) and quantitative real-time PCR (B). Results are mean ± SD of three different experiments. a, p < 0.001 vs only Aβ1–42.
Discussion
Although activated microglia scavenge dead cells from the CNS and secrete different neurotrophic factors for neuronal survival, once microglia are activated in neurodegenerating microenvironment, it always goes beyond control and eventually detrimental effects override beneficial effects. Therefore, activation of microglia has become a hallmark of different neurodegenerative diseases including AD. In the brain of AD patients, activated microglia exhibiting augmented level of cell surface markers are found to be associated with the plaque region (39–41). In cell culture experiments, Aβ peptides have been shown to activate microglia and induce different proinflammatory molecules. However, mechanisms by which Aβ peptides cause microglial activation are poorly understood.
Generally, TLRs are known to be ligated by different pathogen-associated molecules. In this study, we provide the first demonstration that nonpathogen neurotoxin fibrillar Aβ1–42 peptides associated with AD pathology have the capability to engage and use specific TLR to transduce downstream intracellular signaling cascades for microglial activation. Several lines of evidence support the conclusion that Aβ peptides activate microglia via TLR2. Our conclusion is based on the several observations. First, fibrillar Aβ1–42 peptides increased the expression of different TLRs (TLR2, TLR4, TLR6, TLR7, and TLR9), proinflammatory molecules (iNOS, IL-1β, TNF-α, and IL-6), and microglial integrin markers (CD11a, CD11b, CD11c, and CD68) in mouse primary microglia and BV-2 microglial cells. Second, antisense knockdown of only TLR2, but not other TLRs, suppressed Aβ-induced expression of proinflammatory molecules and integrin markers in microglia. Third, neutralizing Abs against TLR2, but not control IgG, negated Aβ-mediated increase in proinflammatory molecules and CD11b in microglia. Fourth, Aβ peptides remained unable to increase iNOS, TNF-α, and CD11b in microglia derived from TLR2 knockout mice in contrast to marked increase in expression of these proinflammatory molecules in microglia isolated from wild-type mice. Fifth, microinjection of fibrillar Aβ1–42 peptides increased the expression of iNOS and CD11b in vivo in the cortex of wild-type, but not TLR2−/− mice. Sixth, ligation of TLR2 alone was sufficient to induce the expression of proinflammatory molecules in microglia. Seventh, the adaptor molecule MyD88 is a critical for signaling by most TLRs including TLR2 (42, 43). Our MyD88 antisense knockdown studies also show that Aβ-mediated induction of proinflammatory molecules in microglia requires MyD88. Taken together, TLR2 signaling is necessary for fibrillar Aβ-mediated activation of microglia.
Our finding of reduced Aβ1–42-mediated expression of proin-flammatory molecules in TLR2−/− microglia contrasts with a recent report in which soluble Aβ1–40 peptides induce iNOS in TLR2−/− microglia, suggesting that Aβ1–40-mediated microglial activation is independent of TLR2 (28). We have also observed that Aβ1–40 peptides induce the expression of TNF-α in microglia isolated from both wild-type and TLR2−/− mice. This discrepancy may be due to the high hydrophobicity and aggregatibility properties of the fibrillar Aβ1–42 peptides in comparison to Aβ1–40 peptide, which is a soluble form of Aβ. It is also known that increased adsorption of insoluble aggregates of Aβ1–42, but not Aβ1–40, to Aβ deposition in senile plaques occurs in the brain of AD patients and that deposition of Aβ1–42, but not Aβ1–40, plays a central role in the pathogenesis of AD (44). Recent hypothesis also suggests that compound with high hydrophobicity may contribute to immunostimulatory effect through the interaction with TLRs (45). A more recent study also supports this hypothesis. It has been found that endogenous, nonpathogenic compound hemozoin, derived from heme by malarial pathogen, interacts with TLR9 and leads to downstream signaling cascades in dendritic cells due to its high hydrophobic properties similar to other ligands such as lipoproteins or DNA (46). Therefore, although soluble Aβ1–40 peptides do not require any TLR to induce iNOS in microglia, hydrophobic fibrillar Aβ1–42 peptides use TLR2 for microglial activation.
However, at present, we do not know mechanisms by which fibrillar Aβ1–42 peptides engage TLR2 on microglia. According to the stated hydrophobicity theory, highly hydrophobic fibrillar Aβ1–42 may directly bind to TLR2 on microglial surface and initiate intracellular signaling cascades. Conversely, several reports indicate that fibrillar form of Aβ peptides are able to bind to several cell surface receptors such as the RAGE (receptor for advanced glycation end-products), the NMDA-R (_N_-methyl-D-aspartate receptor), the P75 neurotrophin receptor (P75NTR), the CLAC-P/collagen type XXV (collagen-like Alzheimer amyloid plaque component precursor/collagen XXV), and the scavenger type A receptor (SR-A) (28, 47–52). Therefore, Aβ may bind to RAGE or SR-A, which may associate or interact with TLR2. Recently, Koenigsknecht and Landreth (53) have shown that fibrillar Aβ1–42 peptides associate or interact with the multicomponent Aβ receptor complex consisting of CD36, α6β1-integrin, and CD47, but not RAGE, and participate in both cellular adhesion and initiation of intracellular signaling events. Importantly, it was also observed that disrupting the interaction of Aβ fibrils with any component of the receptor complex inhibits Aβ stimulation (48). Therefore, it is also possible that TLR2 is an integral component of this receptor complex that plays a critical role in fibrillar Aβ-induced microglial activation. At present, works are underway in our laboratory to delineate such mechanisms.
In summary, we have demonstrated that fibrillar Aβ1–42 peptides activate microglia via TLR2 signaling pathway. Although the local concentration of fibrillar Aβ peptides present in the brain microenvironment of AD patients may differ from the concentration we used in microglia and the in vitro situation of mouse microglia in culture may not truly resemble the in vivo situation of microglia in the brain of AD patients, our results suggest that specific targeting of TLR2 may be an important step for the attenuation of microglial activation in the CNS during AD pathology.
Footnotes
1
This study was supported by Grant IIRG-07-58684 from Alzheimer’s Association and Grants NS39940 and NS48923 from the National Institutes of Health.
3
Abbreviations used in this paper: AD, Alzheimer’s disease; iNOS, inducible NO synthase; Aβ, amyloid-β; LTA, lipoteichoic acid; poly(I-C), polyinosinic-polycytidylic acid; ASO, antisense oligonucleotide; ScO, scrambled oligonucleotide.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu. Rev. Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 2.Sudoh S, Kawamura Y, Sato S, Wang R, Saido TC, Oyama F, Sakaki Y, Komano H, Yanagisawa K. Presenilin 1 mutations linked to familial Alzheimer’s disease increase the intracellular levels of amyloid β-protein 1–42 and its N-terminally truncated variant(s) which are generated at distinct sites. J. Neurochem. 1998;71:1535–1543. doi: 10.1046/j.1471-4159.1998.71041535.x. [DOI] [PubMed] [Google Scholar]
- 3.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid β peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J. Leukocyte Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 5.Murphy GM, Jr, Yang L, Cordell B. Macrophage colony-stimulating factor augments β-amyloid-induced interleukin-1, interleukin-6, and nitric oxide production by microglial cells. J. Biol. Chem. 1998;273:20967–20971. doi: 10.1074/jbc.273.33.20967. [DOI] [PubMed] [Google Scholar]
- 6.Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate A β1–-42-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J. Neuroimmunol. 2001;113:49–62. doi: 10.1016/s0165-5728(00)00404-5. [DOI] [PubMed] [Google Scholar]
- 7.Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 8.Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid β and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J. Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]
- 9.Lemaitre B. The road to Toll. Nat. Rev. 2004;4:521–527. doi: 10.1038/nri1390. [DOI] [PubMed] [Google Scholar]
- 10.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 11.Akira S, Takeda K. Toll-like receptor signalling. Nat. Rev. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 12.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 13.Ishii KJ, Coban C, Akira S. Manifold mechanisms of toll-like receptor-ligand recognition. J. Clin. Immunol. 2005;25:511–521. doi: 10.1007/s10875-005-7829-1. [DOI] [PubMed] [Google Scholar]
- 14.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 15.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 16.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 17.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giulian D, Baker TJ. Peptides released by ameboid microglia regulate astroglial proliferation. J. Cell Biol. 1985;101:2411–2415. doi: 10.1083/jcb.101.6.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J. Biol. Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J. Biol. Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jana M, Pahan K. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radical Biol. Med. 2005;39:823–831. doi: 10.1016/j.freeradbiomed.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. Antineuroinflammatory effect of NF-κB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J. Immunol. 2004;173:1344–1354. doi: 10.4049/jimmunol.173.2.1344. [DOI] [PubMed] [Google Scholar]
- 23.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 24.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. β-Amyloid proteindependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 25.Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Floden AM, Combs CK. β-Amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J. Neurosci. 2006;26:4644–4648. doi: 10.1523/JNEUROSCI.4822-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindberg C, Selenica ML, Westlind-Danielsson A, Schultzberg M. β-Amyloid protein structure determines the nature of cytokine release from rat microglia. J. Mol. Neurosci. 2005;27:1–12. doi: 10.1385/JMN:27:1:001. [DOI] [PubMed] [Google Scholar]
- 28.Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R. Amyloid β peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J. Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- 29.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Tolllike receptor 2. J. Biol. Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 30.Kirschning CJ, Schumann RR. TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr. Top. Microbiol. Immunol. 2002;270:121–144. doi: 10.1007/978-3-642-59430-4_8. [DOI] [PubMed] [Google Scholar]
- 31.Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, Gobel UB, Weber JR, Schumann RR. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- 32.Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, et al. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer’s disease. J. Exp. Med. 1996;184:1425–1433. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Hise AG, Kalsow CM, Pearlman E. Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect. Immun. 2006;74:5325–5332. doi: 10.1128/IAI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J. Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 35.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 36.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 37.Raubenheimer EJ, van Nickerk JP, Hauman CH. Salivary myoepithelium: distribution, structure, functions and pathologic proliferations. J. Dent. Assoc. S. Afr. 1987;42:631–637. [PubMed] [Google Scholar]
- 38.Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J. Immunol. 2006;176:6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 40.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 41.McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirology. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- 42.Akira S, Hoshino K. Myeloid differentiation factor 88-dependent and-independent pathways in Toll-like receptor signaling. J. Infect. Dis. 2003;187 Suppl. 2:S356–S363. doi: 10.1086/374749. [DOI] [PubMed] [Google Scholar]
- 43.Bergstrand G, Larsson S, Bergström M, Eriksson L, Edner G. Cerebrospinal fluid circulation: evaluation by single-photon and positron emission tomography. AJNR Am. J. Neuroradiol. 1983;4:557–559. [PMC free article] [PubMed] [Google Scholar]
- 44.Tamaoka A, Sawamura N, Fukushima T, Shoji S, Matsubara E, Shoji M, Hirai S, Furiya Y, Endoh R, Mori H. Amyloid β protein 42(43) in cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurologic. Sci. 1997;148:41–45. doi: 10.1016/s0022-510x(96)00314-0. [DOI] [PubMed] [Google Scholar]
- 45.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 46.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 2005;201:19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004;18:203–205. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- 48.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, Turnbull JE. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer’s β-secretase. J. Cell Biol. 2003;163:97–107. doi: 10.1083/jcb.200303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yates SL, Kocsis-Angle J, Embury P, Brunden KR. Inflammatory responses to amyloid fibrils. Methods Enzymology. 1999;309:723–733. doi: 10.1016/s0076-6879(99)09048-5. [DOI] [PubMed] [Google Scholar]
- 51.Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp. Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 52.Lue LF, Yan SD, Stern DM, Walker DG. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr. Drug Targets. 2005;4:249–266. doi: 10.2174/1568007054038210. [DOI] [PubMed] [Google Scholar]
- 53.Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J. Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]