The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments (original) (raw)
Abstract
RNA editing alters plant mitochondrial and chloroplast transcripts by converting specific cytidines to uridines, which usually results in a change in the amino acid sequence of the translated protein. Systematic studies have experimentally identified sites of RNA editing in organellar transcriptomes from several species, but these analyses have not kept pace with rate of genome sequencing. The PREP (predictive RNA editors for plants) suite was developed to computationally predict sites of RNA editing based on the well-known principle that editing in plant organelles increases the conservation of proteins across species. The PREP suite provides predictive RNA editors for plant mitochondrial genes (PREP-Mt), for chloroplast genes (PREP-Cp), and for alignments submitted by the user (PREP-Aln). These servers require minimal input, are very fast, and are highly accurate on all seed plants examined to date. PREP-Mt has proved useful in several research studies and the newly developed PREP-Cp and PREP-Aln servers should be of further assistance for analyses that require knowledge of the location of sites of RNA editing. The PREP suite is freely available at http://prep.unl.edu/.
OVERVIEW
RNA editing is a generic term comprising a variety of processes that alter the DNA-encoded sequence of a transcribed RNA by inserting, deleting or modifying nucleotides in the transcript. These various processes have been observed sporadically throughout eukaryotes and in some viruses, although the mechanisms and outcomes of editing are generally lineage specific (1). In plants, RNA editing affects mitochondrial and plastid transcripts of all major lineages of land plants (i.e. angiosperms, gymnosperms, ferns, lycophytes, hornworts, mosses and liverworts) and operates by the site-specific modification of cytidines to uridines and, in some groups, uridines to cytidines (2–4). These C-to-U and U-to-C changes are generally found at codon positions that effect a change in the encoded amino acid (5). Therefore, it is important to know where sites of RNA editing exist in the transcriptome in order to understand the proper structure and function of the translated proteins.
To discover the location of RNA edit sites in plant organellar transcriptomes, comprehensive experimental analyses have been carried out for several species. For chloroplasts, this list now includes over 10 angiosperms (e.g. 6–8), as well as a gymnosperm, a hornwort and a fern (9–11). The chloroplasts of ferns and hornworts contain hundreds of C-to-U and U-to-C edit sites (10,11), whereas angiosperm and gymnosperm chloroplasts harbor only a few dozen C-to-U sites and no U-to-C sites at all (6–9). For plant mitochondria, four angiosperms have been examined and all of them have several hundred C-to-U sites but no U-to-C sites (5,12–14).
Unfortunately, these systematic analyses of plant organellar transcriptomes have not kept pace with the rate of genome sequencing. There are now over 100 plastid and 20 mitochondrial genomes from land plants available in the sequence databases, and edit sites clearly abound in almost all of them. Of course, it is neither the aim of many of these genome sequencing projects to experimentally identify sites of RNA editing, nor is it always practical to do so for every newly sequenced genome. In recent years, several studies have taken various computational approaches to predict edit sites, with varying degrees of success (15–18). Some methods attempt to predict sites using information in the immediate sequence context (15,17), but these generally suffer from low specificity resulting in a large number of false positives. This is due to the low frequency of editing in angiosperm mitochondrial genes, where <10% of the cytidines in protein-coding genes are actually edited (5,12–14). Other approaches utilize evolutionary information and have achieved better results. The predictive RNA editor for plant mitochondrial genes (PREP-Mt) identifies sites based on the principle that editing increases protein conservation among species (16). The most successful program to date, CURE, relies on the shared ancestry of edit sites, only considering cytidine positions that are known to be edited in other species (18).
All of the current methods have focused on the abundance of data from angiosperm mitochondria, so it is unclear whether they will be generally applicable for more divergent plant groups or for chloroplast editing. To address the need for a chloroplast predictor, the predictive RNA editor for plant chloroplast genes (PREP-Cp) was developed by adapting the PREP-Mt methodology. PREP-Cp behaves almost identically to PREP-Mt; the only difference is that PREP-Cp translates and aligns an input sequence to a pre-defined alignment of chloroplast homologs, whereas PREP-Mt aligns to a homologous mitochondrial alignment. And for times when the pre-defined alignments from PREP-Mt and PREP-Cp are not adequate, the predictive RNA editor for user-defined alignments (PREP-Aln) provides an alternative. PREP-Aln applies the PREP-Mt methodology to a custom alignment submitted by the user containing a mix of RNA sequences (with known edit sites) and DNA sequences (in which sites will be predicted). This flexibility allows the user to potentially increase prediction accuracy by taking advantage of newly published editing data or by increasing sampling from a targeted lineage of interest. This suite of web servers should greatly expand our ability to identify potential sites of RNA editing in plant organellar transcripts.
WEBSITE USAGE
The PREP suite of servers was designed for ease of use. Inputs and outputs are intended to be intuitive and straightforward, and predictions are returned nearly instantaneously. A help file is available that describes the input parameters and the output in more detail. Sample data are also provided.
To use PREP-Mt or PREP-Cp, the user is required to submit a protein-coding sequence (with introns spliced out) and to select its gene identity and the codon position of the first nucleotide. The user is also given the option to provide a name for the input sequence, such as the species name from which the sequence originated, and to define a cutoff value (C). The cutoff value sets the minimum score that a predicted edit site must receive before it is reported as a prediction. Because edit sites receive a score between 0 and 1, C must also be a number in this range. Lower C will increase the number of predicted edit sites. The upside is that this increases the number of true edit sites that will be found, but at the same time, the number of incorrect predictions will also be higher. Higher C, in contrast, will make fewer incorrect predictions but will also find fewer true sites. If no cutoff value is selected, the server defaults to optimal values described below. PREP-Mt and PREP-Cp are also able to process multiple sequences at once. To use batch mode, the user must upload a tab-delimited text file containing five parameters on a single line for each sequence to be tested. The five parameters correspond to the inputs described above for single submissions and must be placed in the following order: (i) a sequence name; (ii) the gene name; (iii) the codon position of the first nucleotide; (iv) a cutoff value; and (v) the protein-coding sequence.
The input for PREP-Aln is slightly different because it allows users to define their own custom alignments to be used for prediction, in contrast to PREP-Mt and PREP-Cp that rely on pre-defined alignments. To use PREP-Aln, the user must provide a codon-based nucleotide alignment in FASTA format. In other words, the alignment should consist of protein-coding sequence only with gaps placed between codons and gap lengths in multiples of three. An alignment of this nature can be generated by using an alignment of the translated protein sequences to guide the positioning of gaps in the nucleotide alignment. Several online resources, such as PAL2NAL (http://coot.embl.de/pal2nal/) and RevTrans (http://www.cbs.dtu.dk/services/RevTrans/), can produce codon alignments automatically. The alignment submitted to PREP-Aln must also contain at least one RNA sequence (preferably more) and any number of DNA sequences. All RNA sequences in the alignment must be flagged by adding ‘_RNA’ to the end of their definition lines.
The output for all programs is simple (Figure 1). For each predicted edit site, information is provided about its location, effect and score. The ‘Nt Pos’, ‘AA Pos’ and ‘Align Col’ columns list the location of the predicted site in the nucleotide sequence, predicted protein sequence and resulting alignment, respectively. The ‘Effect’ column shows how the edit site changes the codon and the encoded amino acid. The ‘Score’ of the site is a rough indicator of the confidence of prediction and is equal to the proportion of sites in the alignment that have the same amino acid at that position as the edited version of the input sequence. In addition to the on-screen output, downloadable files are provided. These files include a tab-delimited version of the on-screen results and FASTA files of the edited RNA and protein sequence. Most importantly, the alignment used to guide the prediction of edit sites is made available. Users of PREP-Mt and PREP-Cp should check this alignment carefully to ensure that the input sequence aligned properly to the pre-defined alignment of homologs. When using PREP-Aln or batch mode of PREP-Mt and PREP-Cp, the downloadable data files for all examined sequences are combined and stored as compressed archives in.tgz and.zip formats.
Figure 1.

Sample output from a PREP analysis. Prediction results show the location, effect and score of each edit site. Downloadable files are also provided, including a tab-delimited version of prediction results, FASTA files of the edited RNA and protein sequence and the alignment used for prediction.
PREDICTION METHOD
As mentioned, the PREP suite of programs identifies potential sites of RNA editing based on the evolutionary principle that editing increases protein conservation among species. This is a fundamental quality of RNA editing in plants that was noticed upon its discovery in 1989 (19–21) and has been repeatedly observed in nearly all subsequent studies. Full details of the PREP-Mt methodology have been published previously (16). Essentially, all three programs perform the same series of steps: (i) an input sequence is translated using the standard genetic code; (ii) the translated sequence is aligned to a set of homologous proteins; (iii) the alignment is examined column-by-column to determine if an editing event could increase the similarity of the input sequence to the sequences in the pre-defined alignment. An edit site is predicted if a C-to-U change in a codon causes it to produce an amino acid that is found in more of the homologous proteins than the amino acid coded for by the unedited codon. If a cutoff value is specified by the user, the score of the edited version of the codon must also be >C.
The major difference between each server is in the set of homologous proteins used for comparison to the input sequence. For PREP-Aln, the protein homologs derive from the RNA-tagged sequences in the input file provided by the user. PREP-Aln pulls out all of the DNA sequences from the input alignment, and then builds the homologous protein alignment by translating the RNA sequences remaining in the input alignment. PREP-Aln then compares each of the pulled DNA sequences to the translated RNA alignment. For PREP-Mt and PREP-Cp, the set of homologous proteins is determined by the user when the gene name parameter is specified. These alignments of known mitochondrial or chloroplast proteins have been pre-generated from data available in GenBank and literature sources. The mitochondrial alignments were described previously and consist predominantly of six species with widespread transcriptomic sequence data (Figure 2A), and three species (Marchantia polymorpha, Chara vulgaris, Chaetosphaeridium globosum) that lack RNA editing (16). To create the chloroplast alignments, chloroplast genomes from seed plants whose transcriptomes have been extensively examined for editing (Figure 2B) were downloaded from GenBank. The known positions of edit sites were used to reconstruct mature, edited RNA sequences and these sequences were translated using the standard genetic code. Homologous proteins were aligned with ClustalW and manually adjusted when necessary to produce a collection of 35 alignments representing all chloroplast genes with evidence for editing in at least one of the seed plants in this study (Figure 2B).
Figure 2.

Seed plants with extensive editing data for (A) mitochondrial genes and (B) chloroplast genes. For each species is listed the number of genes with editing information, along with the number of edited (Pos) and unedited (Neg) cytidines found in those genes. The chronogram shows evolutionary relationships and approximate divergence times for species. Divergence times are listed in millions of years (MYA) and were taken from published analyses (22,23). Species in black were used to generate the sets of homologous protein alignments and to optimize the cutoff value. Species in red were used for the unseen tests only.
PERFORMANCE EVALUATION
To evaluate predictive performance, PREP-Mt and PREP-Cp were subjected to several tests. Prediction results for all cytidines were compared with their known editing status and then classified as true positives (TP) or true negatives (TN) when correct, and false positives (FP) or false negatives (FN) when incorrect. Performance was evaluated using several standard measures including sensitivity (Sens), specificity (Spec), positive predictive value (PPV), and Matthews Correlation Coefficient (MCC):
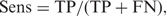

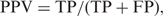
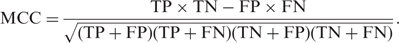
It was not necessary to independently test the performance of PREP-Aln. This is because PREP-Aln produces results that are identical to PREP-Mt and PREP-Cp if given the same set of inputs and homologous sequences.
Optimization of the cutoff value
As already mentioned, the cutoff value, C, affects the number of edit sites that are predicted. Lower values produce more TP but also more FP, whereas higher values produce fewer of both. To examine the effect of the cutoff value on PREP-Mt and PREP-Cp predictive performance, genes from species listed in black in Figure 2 were subjected to leave-one-out cross-validation over a range of C (Figure 3). As observed previously (16), PREP-Mt performed well over a broad range of cutoff values from 0 to 0.6 (Figure 3A). PPV increased slightly over this range indicating a mild increase in the efficiency of prediction, whereas sensitivity dropped a little due to a small reduction in the number of true edit sites identified. MCC remained steady at 82–83% suggesting that the trade-off between sensitivity and PPV was quite balanced over this range, with a slight peak of performance at C = 0.2. PREP-Mt performance diminished noticeably at cutoff values >0.6. The performance results of PREP-Cp show that it behaves quite differently (Figure 3B). PREP-Cp performed increasingly better with higher cutoff values, achieving maximal overall performance at C = 0.8 with a MCC of 76%. The need for a high cutoff for accurate prediction in seed plant chloroplasts may result from the fact that there are so few true edit sites present, making it critical to keep FP as low as possible.
Figure 3.

Effect of the cutoff value. Performance of (A) PREP-Mt Score and (B) PREP-Cp Score are shown for a series of cutoff values ranging from 0 to 1. Performance was visualized by plotting sensitivity (Sens), positive predictive value (PPV) and Matthews Correlation Coefficient (MCC) for each cutoff value. Maximal performance measures are shown with large open circles. Maximal MCC values were used to determine optimal cutoff values in the tests of unseen data.
Predictive performance
There are now four published methods to predict sites of RNA editing in plant mitochondrial genes (15–18). Two of these approaches (15,17) were not tested here for two reasons: (i) they are not available online, and (ii) their published specificity is low, which results in a large number of false positive predictions due to the fact that the vast majority of cytidines in the mitochondrial transcriptome are not edited. Performance of the two online resources, PREP-Mt and CURE, has been compared previously (18). In that publication, the CURE method was shown to perform slightly better than PREP-Mt on angiosperms with extensive editing data. However, because the CURE method relies on the shared ancestry of edit sites, it is unclear whether it will perform as well on unseen data from species that are more distantly related to the angiosperms used for training.
To test the performance of PREP-Mt and CURE on unseen data, unpublished editing information was obtained from four species (Nicotiana tabacum, Liriodendron tulipifera, Amborella trichopoda and Ginkgo biloba) that are progressively more distantly related to the angiosperms used for training (Figure 2A). These new data were evaluated with PREP-Mt using a cutoff value of 0.2 (based on optimizations shown in Figure 3) and with CURE using default settings (Table 1). For Nicotiana, which is most closely related to the training species, CURE performs slightly better overall with a MCC of 87% versus 85% for PREP-Mt, consistent with previous results (18). However, CURE performance gets progressively worse as evolutionary distance increases: MCC drops to 80%, 71% and 47% for Liriodendron, Amborella and Ginkgo, respectively. In contrast, MCC for PREP-Mt stays >80% for all species and achieves the highest score for Ginkgo, the most evolutionarily distant species tested. Both methods consistently return very high specificity values (98–99%), which is a critical requirement for any predictor of editing to keep FP predictions low. The major difference between the two methods is in their ability to detect edit sites. The sensitivity for CURE falls from a high of 84% for Nicotiana to only 30% for Ginkgo, whereas PREP-Mt sensitivity ranges from 78% to 86% for all four species. The wide variation in results for CURE suggests that it may be overoptimized for the data used for training. PREP-Mt does not seem to suffer the same problem, and therefore may be more reliable in general.
Table 1.
Performance of PREP-Mt and CURE on unseen data
| Species | Software | TP | TN | FP | FN | Sens | Spec | PPV | MCC |
|---|---|---|---|---|---|---|---|---|---|
| Nicotiana | CURE | 430 | 6189 | 35 | 84 | 0.837 | 0.994 | 0.925 | 0.870 |
| PREP-Mt | 428 | 6173 | 51 | 86 | 0.833 | 0.992 | 0.894 | 0.852 | |
| Liriodendron | CURE | 583 | 6006 | 37 | 233 | 0.714 | 0.994 | 0.940 | 0.800 |
| PREP-Mt | 676 | 5986 | 57 | 140 | 0.828 | 0.991 | 0.922 | 0.858 | |
| Amborella | CURE | 500 | 5762 | 47 | 335 | 0.599 | 0.992 | 0.914 | 0.712 |
| PREP-Mt | 655 | 5729 | 80 | 180 | 0.784 | 0.986 | 0.891 | 0.814 | |
| Ginkgo | CURE | 162 | 1996 | 14 | 379 | 0.299 | 0.993 | 0.920 | 0.472 |
| PREP-Mt | 462 | 1976 | 34 | 79 | 0.854 | 0.983 | 0.931 | 0.865 | |
| Overall | CURE | 1675 | 19 953 | 133 | 1031 | 0.619 | 0.993 | 0.926 | 0.733 |
| PREP-Mt | 2221 | 19 864 | 222 | 485 | 0.821 | 0.989 | 0.909 | 0.847 |
PREP-Cp was also subjected to a series of performance tests. PREP-Cp performance was first examined on species present in the pre-defined sets of protein alignments using leave-one-out cross-validation (Table 2). These results derive from the cutoff value optimization tests performed previously (Figure 2B). Results using the optimal C = 0.8 are shown. Overall results from PREP-Cp are quite good, although there is some variation in individual species performance. MCC is highest for Atropa (87%) and lowest for Pinus (62%). This variability is not due to an inability of PREP-Cp to find edit sites because sensitivity is high (81–96%) for all seven species. Rather, the variability of performance among species can be largely attributed to the number of FP predictions. Nicotiana and Atropa have the highest MCC and the fewest FP, whereas Pinus and Phalaenopsis have the lowest MCC and the most FP. PREP-Cp was evaluated on unseen data as well (Table 3). Results for N. sylvestris and Saccharum are very good, which is not surprising because their editing profiles are very similar to N. tabacum and Zea, respectively. Pisum performs more poorly, again the result of a high level of FP. The variability in performance of chloroplast editing may, in part, be due to the incomplete nature of the experimental analyses performed for some species. Several of the FP predictions made by PREP-Cp may in fact be real edit sites that have yet to be verified experimentally. PREP-Cp thus provides a useful platform to search for novel edit sites in chloroplast transcriptomes.
Table 2.
Performance of PREP-Cp on training data
| Species | TP | TN | FP | FN | Sens | Spec | PPV | MCC |
|---|---|---|---|---|---|---|---|---|
| Arabidopsis | 26 | 2711 | 15 | 3 | 0.897 | 0.994 | 0.634 | 0.751 |
| N. tabacum | 31 | 3747 | 4 | 7 | 0.816 | 0.999 | 0.886 | 0.849 |
| Atropa | 29 | 3674 | 7 | 2 | 0.935 | 0.998 | 0.806 | 0.867 |
| Zea | 25 | 4577 | 12 | 1 | 0.962 | 0.997 | 0.676 | 0.805 |
| Oryza | 22 | 3624 | 10 | 3 | 0.880 | 0.997 | 0.688 | 0.776 |
| Phalaenopsis | 34 | 4739 | 21 | 8 | 0.810 | 0.996 | 0.618 | 0.705 |
| Pinus | 24 | 1894 | 31 | 3 | 0.889 | 0.984 | 0.436 | 0.616 |
| Overall | 191 | 24 966 | 100 | 27 | 0.876 | 0.996 | 0.656 | 0.756 |
Table 3.
Performance of PREP-Cp on unseen data
| Species | TP | TN | FP | FN | Sens | Spec | PPV | MCC |
|---|---|---|---|---|---|---|---|---|
| Pisum | 22 | 4405 | 24 | 5 | 0.815 | 0.995 | 0.478 | 0.621 |
| Nicotiana sylvestris | 32 | 3672 | 2 | 1 | 0.970 | 0.999 | 0.941 | 0.955 |
| Saccharum | 23 | 3285 | 3 | 0 | 1.000 | 0.999 | 0.885 | 0.940 |
| Overall | 77 | 11 362 | 29 | 6 | 0.928 | 0.997 | 0.726 | 0.819 |
CONCLUSIONS
The PREP suite is a family of web servers dedicated to predicting sites of RNA editing in plant organellar genes. The PREP-Mt server was developed several years ago (16) to find mitochondrial edit sites, and it has been used in a number of genomic (24), evolutionary (25–28) and phylogenetic (29–31) studies that required knowledge of the location of sites of RNA editing. PREP-Mt performs well on all seed plants tested to date, whereas CURE performed poorly on unseen data from several angiosperms and a gymnosperm. Given the strong performance of PREP-Mt, the same methodology has now been applied by the PREP-Cp server to the problem of chloroplast RNA editing. PREP-Cp performs well and is the first and only online tool to predict edit sites in chloroplast genes. The third server, PREP-Aln, is also unique and allows the user to supply custom alignments for prediction, allowing greatest flexibility for advanced users.
AVAILABILITY
The PREP suite is available at http://prep.unl.edu/. It is free for all to use and there is no login requirement.
FUNDING
University of Nebraska Lincoln (by start-up funds). Funding for open access charge: University of Nebraska Lincoln (by start-up funds).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The author thanks G. J. Young for providing mitochondrial editing data for Amborella, Liriodendron and Ginkgo; Y. Sugiyama for providing mitochondrial editing data for Nicotiana; M. Sugiura for providing chloroplast editing data for Pisum; and J. C. Gray for providing the Pisum chloroplast genome sequence.
REFERENCES
- 1.Gott JM, Emeson RB. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000;34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- 2.Hiesel R, Combettes B, Brennicke A. Evidence for RNA editing in mitochondria of all major groups of land plants except the Bryophyta. Proc. Natl Acad. Sci. USA. 1994;91:629–633. doi: 10.1073/pnas.91.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malek O, Lättig K, Hiesel R, Brennicke A, Knoop V. RNA editing in bryophytes and a molecular phylogeny of land plants. EMBO J. 1996;15:1403–1411. [PMC free article] [PubMed] [Google Scholar]
- 4.Freyer R, Kiefer-Meyer MC, Kössel H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc. Natl Acad. Sci. USA. 1997;94:6285–6290. doi: 10.1073/pnas.94.12.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giegé P, Brennicke A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl Acad. Sci. USA. 1999;96:15324–15329. doi: 10.1073/pnas.96.26.15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maier RM, Neckermann K, Igloi GL, Kössel H. Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995;251:614–628. doi: 10.1006/jmbi.1995.0460. [DOI] [PubMed] [Google Scholar]
- 7.Hirose T, Kusumegi T, Tsudzuki T, Sugiura M. RNA editing sites in tobacco chloroplast transcripts: editing as a possible regulator of chloroplast RNA polymerase activity. Mol. Gen. Genet. 1999;262:462–467. doi: 10.1007/s004380051106. [DOI] [PubMed] [Google Scholar]
- 8.Corneille S, Lutz K, Maliga P. Conservation of RNA editing between rice and maize plastids: are most editing events dispensable? Mol. Gen. Genet. 2000;264:419–424. doi: 10.1007/s004380000295. [DOI] [PubMed] [Google Scholar]
- 9.Wakasugi T, Hirose T, Horihata M, Tsudzuki T, Kössel H, Sugiura M. Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: the pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc. Natl Acad. Sci. USA. 1996;93:8766–8770. doi: 10.1073/pnas.93.16.8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K. RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res. 2003;31:2417–2423. doi: 10.1093/nar/gkg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf PG, Rowe CA, Hasebe M. High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene. 2004;339:89–97. doi: 10.1016/j.gene.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 12.Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics. 2002;268:434–445. doi: 10.1007/s00438-002-0767-1. [DOI] [PubMed] [Google Scholar]
- 13.Handa H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003;31:5907–5916. doi: 10.1093/nar/gkg795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mower JP, Palmer JD. Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol. Genet. Genomics. 2006;276:285–293. doi: 10.1007/s00438-006-0139-3. [DOI] [PubMed] [Google Scholar]
- 15.Cummings MP, Myers DS. Simple statistical models predict C-to-U edited sites in plant mitochondrial RNA. BMC Bioinformatics. 2004;5:132. doi: 10.1186/1471-2105-5-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mower JP. PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinformatics. 2005;6:96. doi: 10.1186/1471-2105-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson J, Gopal S. Correction: genetic algorithm learning as a robust approach to RNA editing site site prediction. BMC Bioinformatics. 2006;7:406. doi: 10.1186/1471-2105-7-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du P, Li Y. Prediction of C-to-U RNA editing sites in plant mitochondria using both biochemical and evolutionary information. J. Theor. Biol. 2008;253:579–586. doi: 10.1016/j.jtbi.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Covello PS, Gray MW. RNA editing in plant mitochondria. Nature. 1989;341:662–666. doi: 10.1038/341662a0. [DOI] [PubMed] [Google Scholar]
- 20.Gualberto JM, Lamattina L, Bonnard G, Weil JH, Grienenberger JM. RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature. 1989;341:660–662. doi: 10.1038/341660a0. [DOI] [PubMed] [Google Scholar]
- 21.Hiesel R, Wissinger B, Schuster W, Brennicke A. RNA editing in plant mitochondria. Science. 1989;246:1632–1634. doi: 10.1126/science.2480644. [DOI] [PubMed] [Google Scholar]
- 22.Wikström N, Savolainen V, Chase MW. Evolution of the angiosperms: calibrating the family tree. Proc. R. Soc. London B. 2001;268:2211–2220. doi: 10.1098/rspb.2001.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magallon SA, Sanderson MJ. Angiosperm divergence times: the effect of genes, codon positions, and time constraints. Evolution. 2005;59:1653–1670. doi: 10.1554/04-565.1. [DOI] [PubMed] [Google Scholar]
- 24.Chaw SM, Shih AC, Wang D, Wu YW, Liu SM, Chou TY. The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, Bpu sequences, and abundant RNA editing sites. Mol. Biol. Evol. 2008;25:603–615. doi: 10.1093/molbev/msn009. [DOI] [PubMed] [Google Scholar]
- 25.Parkinson CL, Mower JP, Qiu YL, Shirk AJ, Song K, Young ND, dePamphilis CW, Palmer JD. Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol. Biol. 2005;5:73. doi: 10.1186/1471-2148-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez L, Picardi E, Quagliariello C. RNA editing has been lost in the mitochondrial cox3 and rps13 mRNAs in Asparagales. Biochimie. 2007;89:159–167. doi: 10.1016/j.biochi.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol. Biol. 2007;7:135. doi: 10.1186/1471-2148-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Touzet P, Delph LF. The effect of breeding system on polymorphism in mitochondrial genes of Silene. Genetics. 2008 doi: 10.1534/genetics.108.092411. doi: 10.1534/genetics.108.092411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen G, Seberg O, Davis JI, Stevenson DW. RNA editing and phylogenetic reconstruction in two monocot mitochondrial genes. Taxon. 2006;55:871–886. [Google Scholar]
- 30.Zhu XY, Chase MW, Qiu YL, Kong HZ, Dilcher DL, Li JH, Chen ZD. Mitochondrial matR sequences help to resolve deep phylogenetic relationships in rosids. BMC Evol. Biol. 2007;7:217. doi: 10.1186/1471-2148-7-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duvall MR, Robinson JW, Mattson JG, Moore A. Phylogenetic analyses of two mitochondrial metabolic genes sampled in parallel from angiosperms find fundamental interlocus incongruence. Am. J. Bot. 2008;95:871–884. doi: 10.3732/ajb.2007310. [DOI] [PubMed] [Google Scholar]