Differential Roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice (original) (raw)
. Author manuscript; available in PMC: 2010 Mar 25.
Abstract
Recent studies have shown that Toll-like receptors (TLRs) are involved in cerebral ischemia/reperfusion (I/R) injury. This study was to investigate the role of TLR2 and TLR4 in acute focal cerebral I/R injury. Cerebral infarct size, neurological function and mortality were evaluated. NFκB binding activity, phosphorylation of IκBα, Akt and ERK1/2 were examined in ischemic cerebral tissue by EMSA and Western blots. Compared to wild type (WT) mice, in TLR4 knockout (TLR4KO) mice, brain infarct size was decreased (2.6 ± 1.18% vs 11.6 ± 1.97% of whole cerebral volume, p<0.05) and neurological function was maintained (7.3 ± 0.79 vs 4.7 ± 0.68, p<0.05). However, compared to TLR4KO mice, TLR2 knockout (TLR2KO) mice showed higher mortality (38.2% vs 13.0%, p<0.05), decreased neurological function (2.9 ± 0.53 vs 7.3 ± 0.79, p<0.05) and increased brain infarct size (19.1 ± 1.33% vs 2.6 ± 1.18%, p<0.05). NFκB activation and IκBα phosphorylation were attenuated in TLR4KO mice (1.09 ± 0.02 and 1.2 ± 0.04) compared to TLR2KO mice (1.31 ± 0.02 and 2.2 ± 0.32) after cerebral ischemia. Compared to TLR4KO mice, in TLR2KO mice, the phosphorylation of Akt (0.2 ± 0.03 vs 0.9 ± 0.16, p<0.05) and ERK1/2 (0.8 ± 0.06 vs 1.3 ± 0.17) evoked by cerebral I/R was attenuated. The present study demonstrates that TLR2 and TLR4 play differential roles in acute cerebral I/R injury. Specifically, TLR4 contributes to cerebral I/R injury, while TLR2 appears to be neuroprotective by enhancing the activation of protective signaling in response to cerebral I/R.
Keywords: TLR2, TLR4, cerebral, ischemia/reperfusion, mouse
Introduction
A growing body of evidence suggests that cerebral ischemia/reperfusion (I/R) leads to a robust in situ inflammatory response, which contributes to cerebral I/R injury (De Simoni et al., 2002; Stoll, 2002), but, the cellular and molecular mechanisms associated with cerebral I/R injury remain to be elucidated. Toll-like receptors (TLRs), a conserved receptor family, have been demonstrated to play a critical role in the induction of innate and inflammatory responses (Aderem and Ulevitch, 2000; Akira, et al., 2001; Anderson, 2000). Activation of TLRs initiates transcription of genes associated with immune responses and inflammation (Aderem and Ulevitch, 2000; Akira, et al., 2001; Anderson, 2000). Recently, TLR2 and TLR4 have been implicated in cerebral I/R injury (Lehnardt et al., 2007; Hua et al., 2007; Caso et al., 2007). Expression of TLR2 and TLR4 was increased in mouse brain after cerebral ischemia (Lehnardt S, 2007; Hua F., 2007). In addition, TLR2 and TLR4 gene deficiency attenuated brain damage induced by cerebral ischemia in mice (Lehnardt et al., 2007; Hua et al., 2007; Caso et al., 2007). However, the molecular mechanisms underlying the involvement of TLR2 and TLR4 in cerebral I/R injury are unknown. In the present study, we observed that brain infarct size was significantly less in TLR4 knockout mice (TLR4KO), but increased in TLR2 knockout mice (TLR2KO) 24 hours after cerebral I/R. In addition, deficiency of TLR4, but not TLR2 attenuated the activation of nuclear factor kappa B (NFκB) signaling induced by cerebral I/R. The phosphatidylinositide 3-kinase / protein kinase B (PI3K/Akt) pathway, a well known protective pathway, was activated in TLR4KO mice, but inhibited in TLR2KO mice after cerebral I/R. Our data indicates that cerebral I/R activated NFκB signaling through TLR4-mediated signaling. TLR4 deficiency protects against acute cerebral ischemic damage. In contrast to TLR4, TLR2 deficiency does not attenuate the NFκB activation, instead, it inhibits the activation of PI3K/Akt signaling, thereby exacerbating the brain damage induced by cerebral I/R. The present study demonstrates that TLR2 and TLR4 play differential roles in acute cerebral I/R injury. Specifically, TLR4 contributes to cerebral I/R injury, while TLR2 appears to be neuroprotective.
Results
Mortality and neurological deficits associated with focal cerebral I/R
Mortality was monitored in WT, TLR4KO and TLR2KO mice within 24 hrs after cerebral I/R. As shown in Figure 1A, six of twenty-eight WT mice (21.43%) died and three of twenty-three TLR4KO mice died (13.04%). While mortality was reduced in the TLR4KO I/R mice the difference did not achieve statistical significance. In contrast, thirteen of thirty-four TLR2KO mice (38.24%) died within the same time period, which is significantly greater than that in TLR4KO mice (p< 0.05). As expected, there were no deaths in sham control mice. Neurological score evaluation is an index for the degree of neurological deficits associated with stroke. Figure 1B shows that the neurological score was significantly greater in TLR4KO mice (7.3 ± 0.79), compared to WT I/R mice (4.7 ± 0.68), but decreased in TLR2KO mice (2.9 ± 0.53) compared to TLR4KO 24 hours after cerebral I/R. There was no neurological deficit in the sham control mice (data not shown).
Figure 1. Differential mortality (A) and neurological function (B) in TLR4KO and TLR2KO mice following cerebral I/R.

(A) Mortality was monitored for 24 hrs after cerebral I/R. Mortality was increased in TLR2KO mice compared with TLR4KO mice and WT mice within 24 hours after cerebral I/R. # compared with WT and TLR4KO, p<0.05; (B) Neurological function was evaluated using a scoring system ranging from 0 to 15, with 15 being a perfect score and 0 being death due to cerebral I/R. Neurological function was maintained in TLR4KO mice, but it was decreased in TLR2KO mice compared with WT mice after cerebral I/R. a: p<0.05 compared with WT; b: p<0.01 compared with TLR4KO.
Differential effect of TLR2 and TLR4 deficiency on cerebral infarct size after I/R
Infarct size was evaluated in the mice 24 hours after cerebral I/R by the TTC staining method. As shown in Figure 2, brain infarct size was less in TLR4KO mice (2.6 ± 1.18% vs 11.6 ± 1.97%, p<0.05), compared to WT mice. But infarct size was increased in TLR2KO mice (19.1 ± 1.33% vs 2.6 ± 1.18%, p<0.05).
Figure 2. Differential effects of TLR2 and TLR4 on cerebral infarct size.

Infarct size was determined by TTC staining and expressed as the percentage of actual infarct volume in the total cerebral volume. The infarct size was significantly less in TLR4KO mice, but greater in TLR2KO mice compared with WT mice. Representative brain sections stained with TTC are shown above the graph. a: p<0.01 compared with WT; b: p<0.01 compared with TLR4KO.
Differential effect of TLR2 and TLR4 on brain histopathology following acute I/R
H&E-stained sections of mouse brains from all experimental mice subjected to sham surgery showed normal histology (Fig. 3A). Pan-necrosis when observed in any cerebral ischemic animals whether WT and gene knockout consisted of shrinkage of all cellular elements with pyknosis of the nuclei. All WT mice subjected to I/R showed acute pan-necrosis in the distribution of the middle cerebral artery. Some nuclei were in the early stages of fragmentation (Fig. 3B). Rare neutrophils were present in parenchyma. Microscopic hemorrhage at the border of the infarct was also observed. There was a sharp border between the necrotic brain and viable brain. The areas of brain consistently infarcted in WT animals were thalamus and caudate-putamen with more variable involvement of the overlying neocortex and the hippocampus. In some cases there was selective acute loss of neurons in cortex or hippocampus rather than pan-necrosis. TLR4KO mice showed variable effects after being subjected to I/R and had smaller areas of acute infarction (Fig. 3C). All TLR2KO mice subjected to I/R showed larger areas of pan-necrosis in the distribution of the middle cerebral artery (Fig. 3D). These areas of infarction involve thalamus, caudate-putamen and large areas of the hemispheric neocortex. Hippocampus showed either pan-necrosis or selective neuronal loss.
Figure 3. Differential effect of TLR2 and TLR4 deficiency on cerebral histopathology after I/R injury.

Brain sections were obtained 1.5 mm behind the Bregma in the coronal plane and stained with hematoxylin–Eosin (H–E) staining using standard methods. The brain regions illustrated are all from parietal cortex. (A). WT shame (WT-S): Slides show the normal morphology in the brain. (B). WT mice subjected to cerebral I/R: (WT-I/R): Illustrated is an area of shrinkage of all cellular elements with pyknosis of the nuclei. Some nuclei are in the early stages of fragmentation. (C). TLR4KO mice subjected to cerebral I/R (T4KO-I/R): Scattered acute neuronal damage is illustrated; in general infracted areas were smaller. (D). TLR2KO mice subjected to cerebral I/R (T2KO-I/R): Slides show pan-necrosis similar to what is described for (B) in the distribution of the middle cerebral artery. Areas of acute infarction such as this were much more extensive than that in TLR4KO mice. All photographs were taken at the same magnification. Magnification bar shows 25 µm.
Activation of NFkB following acute cerebral I/R injury
NFκB is an important transcription factor downstream in the TLR2 and TLR4-mediated signaling pathways which responds to a variety of stimuli (Hoshino et al., 2002; Porter and Janicke, 1999; Toshchakov et al., 2002). EMSA results showed that, in WT mice, NFκB DNA binding activity was increased after cerebral I/R, compared with sham control (1.26 ± 0.03 vs 1.05 ± 0.01, Fig. 4A, p<0.05). The NFκB DNA binding activity in TLR4KO mice did not change, and was significantly lower than that in WT mice after cerebral I/R (1.09 ± 0.02 vs 1.26 ± 0.03, Fig. 4A, p<0.05). However, in TLR2KO mice, the NFκB DNA binding activity increased after cerebral I/R, and was significantly higher than that in TLR4KO mice (1.31 ± 0.02 vs 1.09 ± 0.02, Fig. 4A, p<0.05). Phosphorylation of the inhibitor of kappa B (IκBα) induces nuclear translocation and activation of NFκB. We assessed the levels of phosphorylated-IκBα (p-IκB) in brain tissue. Compared to sham control, p-IκB in WT mice was significantly increased after cerebral I/R (1.7 ± 0.10 vs 0.8 ± 0.07, Fig. 4B, p<0.05), and was significantly greater than that in TLR4KO (Fig. 4B). The levels of p-IκBα in TLR2KO mice were significantly increased and greater than that observed in TLR4KO mice after cerebral I/R (2.2 ± 0.32 vs 1.2 ± 0.04, Fig. 4B, p<0.05).
Figure 4. NFκB activation in the brain of TLR2KO and TLR4KO mice subjected to cerebral I/R.
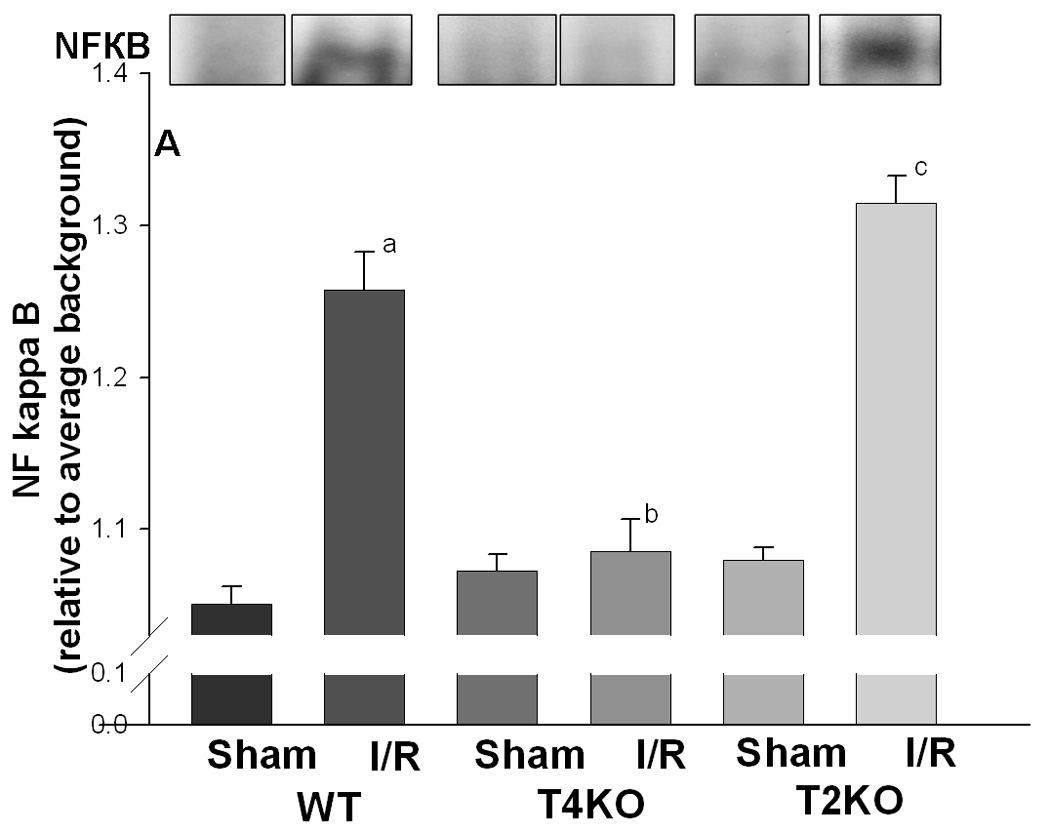

Cellular proteins were isolated from brain tissues and evaluated by EMSA (for NFκB DNA binding activity, Fig. 4A) and Western blot (for IκB with specific antibody against p-IκB and IκB, Fig. 4B). (A): NFκB DNA binding activity was significantly increased after cerebral I/R in WT mice compared with sham control. The NFκB DNA binding activity in TLR4KO mice was significantly less than that in WT mice as well as in TLR2KO mice 24 hour after cerebral I/R. The NFκB DNA binding activity In TLR2KO mice was not significantly different from that in WT mice. (B) The levels of p-IκBα were significantly increased in WT mice and TLR2KO after cerebral I/R. p-IκBα levels were significantly lower in TLR4KO mice after cerebral I/R. Note: WT: wild type mice; T4KO: TLR4 knockout mice; T2KO: TLR2 knockout mice. I/R: cerebral ischemia/reperfusion. WT-sham: n=3; WT-I/R: n=5; T4KO-sham: n=3; T4KO-I/R: n=5; T2KO-sham: n=3; T2KO-I/R: n=5. a. p<0.05 compared with WT-S; b. p<0.05 compared with WT-I/R; c p<0.05 compared with T4KO-I/R.
Differential activation of the PI3K/Akt pathway in TLR2KO and TLR4KO mice following acute cerebral I/R
Activation of the PI3K/Akt pathway plays a role in neuroprotection and may cross talk with TLRs pathways (Ojaniemi et al., 2003; Romaskova et al., 1999). We examined the levels of phosphorylated Akt (p-Akt) in brain tissue. As shown in figure 5, the levels of p-Akt (0.9 ± 0.16 vs 0.5 ± 0.06, p<0.05) in TLR4KO mice were significantly higher than that in WT mice subjected to cerebral I/R. In contrast, the levels of p-Akt in TLR2KO mice did not increase after cerebral I/R and was significantly lower than that in TLR4KO mice subjected to cerebral I/R (0.2 ± 0.03 vs 0.9 ± 0.16, p<0.05).
Figure 5. Differential activation of the PI3K/Akt pathway in TLR2KO and TLR4KO mice after cerebral I/R.

Cellular proteins were isolated from brain tissues and subjected to Western blot with specific antibody against p-Akt. The levels of p-Akt were significantly greater in TLR4KO mice compared with WT mice after cerebral I/R, while the levels of p-Akt were significantly lower in TLR2KO mice after cerebral I/R compared with TLR4KO mice and WT mice. Note: WT: wild type mice; T4KO: TLR4 knockout mice; T2KO: TLR2 knockout mice. I/R: cerebral ischemia/reperfusion. WT-sham: n=3; WT-I/R: n=5; T4KO-sham: n=3; T4KO-I/R: n=5; T2KO-sham: n=3; T2KO-I/R: n=5. a: p<0.05 compared with WT-S,; b: p<0.05compared with WT-I/R,; c: p<0.05 compared with T4KO-I/R.
ERK1/2 activation following acute cerebral I/R
The extracellular signal regulated protein kinases (ERK) are members of the mitogen activated protein kinase (MAPK) family. ERK1 and ERK2, which are activated by the MAPK/ERK kinase-1/2 (MEK1/2), are emerging as important regulators of neuronal responses to both functional and pathologic stimuli, which play a beneficial, neuroprotective role in many systems (Hetman et al., 2004; Cavanaugh, 2004). We examined the levels of phosphorylated ERK1/2 (p-ERK1/2). As shown in figure 6, cerebral I/R increased the level of p-ERK1/2 in all brain tissues. However, the level of p-ERK1/2 in TLR2KO mice (0.8 ± 0.06) was significantly less than that in TR4KO (1.3 ± 0.17) and WT mice (1.2 ± 0.14) after cerebral I/R (Fig. 6).
Figure 6. TLR2 deficiency attenuated the increased activation of ERK1/2 signaling associated with cerebral I/R.

Cellular proteins were isolated from brain tissues and subjected to Western blot with specific antibody against p-ERK1/2. Cerebral I/R increased the phosphorylation of ERK1/2. However, the levels of p-ERK1/2 in TLR2KO mice were significantly lower than that in TLR4KO and WT mice. Note: WT: wild type mice; T4KO: TLR4 knockout mice; T2KO: TLR2 knockout mice. I/R: cerebral ischemia/reperfusion. WT-sham: n=3; WT-I/R: n=5; T4KO-sham: n=3; T4KO-I/R: n=5; T2KO-sham: n=3; T2KO-I/R: n=5. a: p<0.05 compared with WT-Sham,; b: p<0.05 compared with T4KO-Sham,; c: p<0.05 compared with T4KO-I/R.
Discussion
Toll-like receptors (TLRs) are a family of signal transduction molecules which are critical for the induction of immunity and in host defense (Medzhitov et al., 1997). TLR4, the first mammalian TLR identified, is involved in the activation of many signaling proteins, which result in transcription of genes encoding inflammation-associated molecules and cytokines (Lu et al., 2008). TLR2 is another member of the TLR family that has been studied in substantial detail in recent years (Kirschning and Schumann, 2002). Activation of TLR2 and TLR4 recruits an adaptor protein called myeloid differentiation factor-88 (MyD88). MyD88 recruits IL-1 receptor associated receptor kinases (IRAKs) and results in nuclear translocation of nuclear transcription factor kappa B (NFκB), which then initiates transcription of genes associated with innate immune responses and inflammation. This pathway is called the TLR4-mediated MyD88-dependent pathway, which is common to most TLRs (except TLR3) and shared by both TLR2 and TLR4. Since TLRs-mediated signaling triggers and regulates immune/inflammatory responses, which play a critical role in cell death after I/R, TLRs have been reported to play a role in the pathological process of I/R injury in many organs, including the liver (Zhang et al., 2005), kidney (Leemans et al., 2005), heart (Hua et al., 2007a), intestine (Aprahamian et al., 2008) and brain (Hua et al., 2007b; Hua et al., 2008). While many findings indicate that TLR4 contributes to ischemic injury and TLR4 deficiency has a protective effect on ischemic organs (Jin et al., 2007; Shen et al., 2005; Hua et al., 2007a; Hua et al., 2007b), reports about the role of TLR2 in ischemic injury are inconsistent. For instance, some indicate that TLR2 contributes to ischemic injury in liver (Zhang et al., 2005), kidney (Leemans et al., 2005), heart (Sakata et al., 2007) and brain (Ziegler et al., 2007; Lehnardt et al., 2007), however, other reports indicate that TLR2 does not contribute to liver I/R Injury (Shen et al., 2005), rather it protects the small-bowel from I/R injury (Aprahamian, et al., 2008). The molecular mechanisms underlying these effects are not clear and remain to be investigated. In present study, we elucidated the role and mechanisms of TLR2 and TLR4 in the pathophysiological process of cerebral I/R injury by inducing focal cerebral I/R in TLR2KO and TLR4KO mice.
Our data show that infarct size was smaller and neurological functional deficits were attenuated in TLR4KO mice compared to WT mice after acute cerebral I/R, which is consistent with the previous studies (Caso et al., 2008; Cao et al., 2007). In TLR2KO mice, we observed increased brain damage, decreased neurological function and higher mortality related to cerebral I/R compared to WT mice and TLR4KO mice (Fig. 1, Fig. 2 and Fig. 3). Our finding that TLR2 deficiency aggravates brain damage after acute cerebral I/R is consistent with previous reports indicating that TLR2 does not contribute to I/R Injury (Shen XD., et al., 2005), instead, protects tissue from I/R injury (Aprahamian, CJ., 2008). However, this is in contrast to previous reports regarding the contribution of TLR2 in I/R injury (Ziegler et al., 2007; Lehnardt et al., 2007).
NFκB is an important nuclear transcription factor regulated by TLRs, which initiates transcription of genes associated with immune responses and inflammation (Hoshino et al., 2002; Porter and Janicke, 1999; Toshchakov et al., 2002). Inhibition of NFκB activity has been shown to have a therapeutic effect on experimental cerebral ischemia (Schneider et al., 1999; Tounai et al., 2007). We have reported that cerebral ischemia increases TLR4 expression, phosphorylation of IκBα and activation of NFκB in the hippocampus after transient global cerebral ischemia (Hua et al., 2007b). In the present study, we found that cerebral I/R increased levels of phosphorylation of IκBα and NFκB activation in WT mice. TLR4 deficiency attenuated NFκB activation signaling (Fig. 4), which is consistent with our previous report (Hua et al., 2007b). In contrast, in TLR2KO mice, the levels of NFκB DNA binding activity and p-IκBα still increased after cerebral I/R, and were significantly higher than that in TLR4KO mice. These results demonstrated that the activation of NFκB signaling induced by cerebral I/R can be attenuated by the deficiency of TLR4, but not TLR2. There is a possibility that the as yet unidentified endogenous ligands produced in ischemic brain are specific for TLR4, not TLR2. However, other possible roles of TLR2 in the process of cerebral I/R injury cannot be ruled out.
Recent evidence suggested that there is cross-talk between the TLR and the PI3K/Akt signaling pathways (Ojaniemi et al., 2003; Romaskova and Makarov, 1999). PI3K/Akt signaling has been shown to prevent brain tissue from ischemic injury (Endo et al., 2006; Shioda et al., 2008). Phosphorylation of Akt (p-Akt), an important physiologic mediator of the PI3K pathway (Martin et al., 2005), activates several downstream targets of the PI3K pathway and acts as a crucial regulator of many cellular functions, including cell survival and apoptosis. Whether the PI3K/Akt signaling pathway is responsible for the involvement of TLR2 and TLR4 in cerebral I/R injury is not clear. In the present study, we have observed higher levels of p-Akt in TLR4KO mice compared with WT mice after cerebral I/R (Fig. 5). However, the levels of p-Akt in TLR2KO mice were significantly lower than that in TLR4KO as well as WT mice (Fig. 5). Our findings suggest that there are interactions between TLR2, TLR4 and PI3K/Akt signaling networks in cerebral I/R injury. TLR4 deficiency enhances the activation of PI3K/Akt signaling in ischemic brain tissue. TLR2 may positively regulate PI3K/Akt signaling and TLR2 deficiency may inhibit the activation of PI3K/Akt signaling.
It has also been reported that the mitogen activated protein kinases (MAPK) play a prominent role in regulating cell proliferation, differentiation and adaptation (Harper and Wilkie, 2003). The MAPKs comprise a ubiquitous group of signaling proteins including: the extracellular signal regulated protein kinases (ERK), c-Jun N-terminal kinases and p38 MAPK, which have been implicated in neuronal injury and disease (Harper and Wilkie, 2003). ERK1 and ERK2, which are activated by the MAPK/ERK kinase-1/2 (MEK1/2), play a beneficial, neuroprotective role in many systems (Hetman and Gozdz, 2004; Cavanaugh., 2004). Our data showed that cerebral I/R increased the activation of ERK1/2 in WT, TLR2KO and TLR4KO mice. However, the level of p-ERK1/2 in TLR2KO mice is significantly less than that observed in WT mice and TLR4KO mice (Fig. 6), which indicates that the activation of ERK1/2 was attenuated to some extent, in TLR2 KO mice, but not in TLR4KO mice.
In summary, the most significant finding that emerged from the present study was that TLR2 and TLR4 play differential roles and differentially modulate cellular signaling pathways in acute cerebral I/R injury. TLR4-mediated signaling appears to play a role in mediating the pathophysiology of cerebral I/R injury, while TLR2-mediated signaling appears to be neuroprotective by enhancing the activation of protective signaling, such as PI3K/Akt signaling and ERK1/2 signaling, in response to cerebral I/R. These data suggest that differential modulation of TLR2/4 may represent a new and novel approach to attenuating cerebral I/R injury.
Experimental Procedures
Animals
TLR2 gene knockout mice (TLR2KO, B6.129-TLR2tm1kir/J, n=40, male), TLR4 gene knockout mice (TLR4KO, C57BL/10ScCr, n=29, male) and wild type mice (WT, C57BL/10ScSn, n=36, male) with body weights between 25~30g were obtained from the Jackson Laboratory and maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU). The mice were normal in size, and did not display any gross physical or behavioral abnormalities. We have compared the infarct size 24 hours after cerebral I/R between C57BL/10J (WT control for TLR4KO) and C57BL/6J (WT control for TLR2KO) mice. There is no significantly difference between the strains (data not shown). The C57BL/10ScSn mice were used as WT control for the rest experiments in present study. The experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, revised 1996). Animal care and experimental protocols were approved by the ETSU Committee on Animal Care (protocol number P060605-AS).
Experimental design
Mice were assigned to the following six groups: wild type sham control (WS), wild type mice subjected to focal cerebral ischemia/reperfusion (W-I/R), TLR2 knockout sham control (T2KO-S), TLR2 knockout mice subjected to focal cerebral ischemia/reperfusion (T2KO-I/R), TLR4 knockout sham control (T4KO-S) and TLR4 knockout mice subjected to focal cerebral ischemia/reperfusion (T4KO-I/R). Mortality and neurological scores were evaluated in the mice which were subjected to one hour of ischemia followed by reperfusion for 24 hrs before being killed for tissue assay.
Focal cerebral ischemia/reperfusion
Focal cerebral ischemia/reperfusion (I/R) was induced by occlusion of the middle cerebral artery (MCAO) on the left side according to previously published methods (Hua F., et al. 2008) with modifications. Briefly, mice were subjected to anesthesia by 5.0% Isoflurane and maintained by inhalation of 1.5% to 2% Isoflurane driven by 100% oxygen flow. Mice were ventilated (110 breaths/min with volume 0.5ml) and body temperature was regulated at 37.0°C by surface water heating. Following the skin incision, the left common carotid artery (CCA), the external carotid artery (ECA) and the internal carotid artery (ICA) were carefully exposed. Microvascular aneurysm clips were applied to the left CCA and the ICA. A silicon rubber-coated 6-O monofilament (Doccol Corp. CA, USA) was introduced into an arteriotomy hole, fed distally into the ICA and advanced 11 mm from the carotid bifurcation. The ICA clamp was removed and focal cerebral ischemia started. After ischemia for 60 minutes, the filament and the CCA clamp were gently removed (reperfusion starts). The ischemic and reperfusion conditions were confirmed by regional cerebral blood flow (rCBF) detected by the PeriFlux system 5000 (PE5001, Jarfalla, Sweden). The collar suture at the base of the ECA stump was tightened. The skin was closed, anesthesia discontinued, and the animal allowed to recover in pre-warmed cages. Control mice underwent a neck dissection and coagulation of the external carotid artery, but no occlusion of the middle cerebral artery.
Evaluation of neurological score
All mice were scored by a blinded investigator using a neurological evaluation instrument described previously (Garcia et al., 1995) with modification. Briefly, the scoring system included five principle tasks: spontaneous activity over a 3-minute period (0–3), symmetry of movement (0–3), open-field path linearity (0–3), beam walking on a 3cm × 1 cm beam (0–3), and response to vibrissae touch (1–3) (Shimamura et al., 2006; Hua et al., 2008). The scoring system ranged from 0 to 15, in which 15 is a perfect score and 0 is death due to cerebral I/R injury.
Assessment of cerebral infarct volume size
The infarct size was determined as described previously (Reglodi et al., 2002). Twenty-four hours after I/R, mice were anesthetized by 5.0% Isoflurane and killed by perfusing with ice cold phosphate buffered saline (PBS) via the ascending aorta. Brains were removed and sectioned coronally into 2-mm-thick slices. The slices were stained with 2% triphenyltetrazolium chloride (TTC) solution at 37°C for 15 min followed by fixation with 10% formalin neutral buffer solution (pH 7.4). The infarct areas were traced and quantified with an image-analysis system. Unstained areas (pale color) were defined as ischemic lesions. The areas of infarction and the areas of both hemispheres were calculated for each brain slice. The actual infarct volume adjusted for edema was calculated by dividing the infarct volume by the edema index, which was calculated by dividing the total volume of the left hemisphere by the total volume of the right hemisphere. Infarct volumes are expressed as percentage of the total brain volume ± S.E.M.
Histological observation
Twenty-four hours after I/R, mice were sacrificed and perfused with ice cold phosphate buffered saline (PBS) via the ascending aorta followed by 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight. The brains were embedded in paraffin and cut into sections (7µm thickness). Brain sections obtained 1.5 mm behind the Bregma in the coronal plane were stained with hematoxylin-Eosin (H–E) using standard methods. The normal morphology and the presence and nature of ischemic damage were verified by a neuropathologist who was unaware of the experimental design or results of the TTC assay.
Western Blots
Twenty-four hours after I/R, mice were sacrificed and brains were removed. Cellular proteins were prepared from ischemic cerebral hemispheres (left hemispheres), electrophoresed with SDS-polyacrylamide gel and transferred onto Hybond ECL membranes (Amersham Pharmacia, Piscataway, NJ) (Hua et al. 2008). The ECL membranes were incubated with the appropriate primary antibody followed by incubation with peroxidase-conjugated secondary antibodies. The signals were detected with the ECL system (Amersham Pharmacia). The same membranes were probed with anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase, Biodesign, Saco, Maine) after being washed with stripping buffer. The signals were quantified by scanning densitometry and computer-assisted image analysis. The primary antibodies used in the present study were anti-phospho-Akt, anti-Akt1, anti-phospho-IκBα, anti-IκBα, anti-phospho-ERK1/2 and anti-ERK2 (Cell Signaling Technology, Danvers, MA 01923). The secondary antibodies used in the present study were anti-rabbit (Sigma-Aldrich, St. Louis, MO 63178) and anti-mouse (Cell signaling Technology, Danvers, MA 01923).
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were isolated from ischemic cerebral hemispheres as previously described (Hua F., et al. 2007). NFκB binding activity was examined by EMSA in a 15 µl binding reaction mixture containing 15 µg of nuclear proteins and 35 fmol [γ-32P] labeled double-stranded NFκB consensus oligonucleotide. The signals were quantified by scanning densitometry and computer-assisted image analysis. The results were expressed as the ratio of the integrated density volume (IDV) of NFκB to the average IDV of background.
Statistical analysis
Comparison for mortality was accomplished with the Chi-square test. Continuous scale measurements were expressed by the mean and standard error of the mean (S.E.M) for each group. Group comparisons for neurological function (neurological score) were accomplished with the t-test (applied to score values) and by the chi-square test (Stat-Exact software). One way ANOVA and the least significant difference procedure were used to assess the effectiveness of intervention and group differences for infarct size, levels of NFκB, IκBα, p-Akt and p-ERK1/2. Probability levels of 0.05 or smaller were considered significant.
Acknowledgements
This work was supported by AHA postdoctoral fellowship 0625348B and AHA Scientist Development Grant 0830481N to FH; ETSU RDC Grant to RLK; NIH RO1GM53552 to DLW and NIH RO1HL071837 to CL. The authors wish to thank Dr. Donald G. Stein for his critical reading of the manuscript.
Abbreviation
TLRs
Toll-like receptors
I/R
Ischemia/reperfusion
NFκB
Nuclear factor kappa B
IκBα
Inhibitor of kappa B alpha
PI3K/Akt
phosphatidylinositide 3-kinase / protein kinase B
ERK
Extracellular signal regulated protein kinases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 3.Anderson KV. Toll signaling pathways in the innate immune response. Current Opinion in Immunology. 2000;12:13–19. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 4.Aprahamian CJ, Lorenz RG, Harmon CM, Dimmit RA. Toll-like receptor 2 is protective of ischemia-reperfusion-mediated small-bowel injury in a murine model. Pediatr Crit Care Med. 2008;9:105–109. doi: 10.1097/01.PCC.0000288717.44702.C0. [DOI] [PubMed] [Google Scholar]
- 5.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 6.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 7.Cavanaugh JE. Role of extracellular signal regulated kinase 5 in neuronal survival. Eur J Biochem. 2004;271:2056–2059. doi: 10.1111/j.1432-1033.2004.04131.x. [DOI] [PubMed] [Google Scholar]
- 8.De Simoni MG, Milia P, Barba M, De Luigi A, Parnetti L, Gallai V. The inflammatory response in cerebral ischemia: focus on cytokines in stroke patients. Clin Exp Hypertens. 2002;24:535–542. doi: 10.1081/ceh-120015330. [DOI] [PubMed] [Google Scholar]
- 9.Endo H, Nito C, Kamada H, Nishi T, Chan PH. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1479–1489. doi: 10.1038/sj.jcbfm.9600303. [DOI] [PubMed] [Google Scholar]
- 10.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 11.Harper SJ, Wilkie N. MAPKs: new targets for neurodegeneration. Expert Opin Ther Targets. 2003;7:187–200. doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- 12.Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- 13.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 14.Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol. 2007a;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- 15.Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, Kalbfleisch JH, Browder IW, Li C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007b;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin X, Qin Q, Tu L, Zhou X, Lin Y, Qu J. Toll-like receptors (TLRs) expression and function in response to inactivate hyphae of Fusarium solani in immortalized human corneal epithelial cells. Mol Vis. 2007;17:1953–1961. [PMC free article] [PubMed] [Google Scholar]
- 18.Kirschning CJ, Schumann RR. TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr Top Microbiol Immunol. 2002;270:121–144. doi: 10.1007/978-3-642-59430-4_8. [DOI] [PubMed] [Google Scholar]
- 19.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 21.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Martin S, Dicou E, Vincent JP, Mazella J. Neurotensin and the neurotensin receptor-3 in microglial cells. J Neurosci Res. 2005;81:322–326. doi: 10.1002/jnr.20477. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 24.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 25.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 26.Reglodi D, Tamas A, Somogyvari-Vigh A, Szanto Z, Kertes E, Lenard L, Arimura A, Lengvari I. Effects of pretreatment with PACAP on the infarct size and functional outcome of rat permanent focal cerebral ischemia. Peptides. 2002;23:2227–2234. doi: 10.1016/s0196-9781(02)00262-0. [DOI] [PubMed] [Google Scholar]
- 27.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 28.Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, Tacheuchi O, Akira S, Mann DL. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292:H503–H509. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 29.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 30.Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–1800. doi: 10.1111/j.1600-6143.2005.00932.x. [DOI] [PubMed] [Google Scholar]
- 31.Shimamura N, Matchett G, Yatsushige H, Calvert JW, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 ameliorates focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Stroke. 2006;37:1902–1909. doi: 10.1161/01.STR.0000226991.27540.f2. [DOI] [PubMed] [Google Scholar]
- 32.Shioda N, Han F, Morioka M, Fukunaga K. Bis (1-oxy-2-pyridinethiolato) oxovanadium (IV) enhances neurogenesis via phosphatidylinositol 3-kinase/Akt and extracellular signal regulated kinase activation in the hippocampal subgranular zone after mouse focal cerebral ischemia. Neuroscience. 2008;155:876–887. doi: 10.1016/j.neuroscience.2008.05.056. [DOI] [PubMed] [Google Scholar]
- 33.Stoll G. Inflammatory cytokines in the nervous system: multifunctional mediators in autoimmunity and cerebral ischemia. Rev Neurol. 2002;158:887–891. [PubMed] [Google Scholar]
- 34.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 35.Tounai H, Hayakawa N, Kato H, Araki T. Immunohistochemical study on distribution of NF-kappaB and p53 in gerbil hippocampus after transient cerebral ischemia: effect of pitavastatin. Metab Brain Dis. 2007;22:89–104. doi: 10.1007/s11011-006-9040-3. [DOI] [PubMed] [Google Scholar]
- 36.Zhang JX, Wu HS, Wang H, Zhang JH, Wang Y, Zheng QC. Protection against hepatic ischemia/reperfusion injury via downregulation of toll-like receptor 2 expression by inhibition of Kupffer cell function. World J Gastroenterol. 2005;11:4423–4426. doi: 10.3748/wjg.v11.i28.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ziegler G, Harhausen D, Schepers C, Hoffmann O, Röhr C, Prinz V, König J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]