Ligand activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) inhibits chemically induced skin tumorigenesis (original) (raw)
Abstract
Peroxisome proliferator-activated receptor (PPAR)β/δ-null mice exhibit enhanced tumorigenesis in a two-stage chemical carcinogenesis model as compared with wild-type mice. Previous work showed that ligand activation of PPARβ/δ induces terminal differentiation and inhibits proliferation of primary keratinocytes, and this effect does not occur in the absence of PPARβ/δ expression. In the present studies, the effect of ligand activation of PPARβ/δ on skin tumorigenesis was examined using both in vivo and ex vivo skin carcinogenesis models. Inhibition of chemically induced skin tumorigenesis was observed in wild-type mice administered GW0742, and this effect was likely the result of ligand-induced terminal differentiation and inhibition of replicative DNA synthesis. These effects were not found in similarly treated PPARβ/δ-null mice. Ligand activation of PPARβ/δ also inhibited cell proliferation and induced terminal differentiation in initiated/neoplastic keratinocyte cell lines representing different stages of skin carcinogenesis. These studies suggest that topical administration of PPARβ/δ ligands may be useful as both a chemopreventive and/or a chemotherapeutic approach to inhibit skin cancer.
Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors and part of the nuclear hormone receptor superfamily. PPARs regulate expression of genes involved in a myriad of biological functions, including cell proliferation, immune responses, differentiation, fatty acid metabolism and energy homeostasis (1–6). PPARs modulate gene expression by undergoing conformational change after ligand binding allowing release of corepressors, recruitment of coactivators and subsequent heterodimerization with the retinoid X receptor-α. The ligand-bound PPAR–retinoid X receptor-α heterodimer complex binds specific peroxisome proliferator response elements in target genes and increases their expression (1–6). The PPAR subfamily consists of three distinct members, PPARα, PPARβ/δ and PPARγ. The biological roles of PPARα and PPARγ have been extensively characterized, whereas the physiological function of PPARβ/δ remains less clear.
PPARβ/δ is the most ubiquitously expressed PPAR isotype (7,8) with particularly high expression in the intestine, liver and keratinocytes (9). PPARβ/δ is activated by fatty acids (10), tetradecylthioacetic acid (TTA) (11), GW501516 (12), GW0742 (12) and L-165041 (13). PPARβ/δ agonists are being evaluated as therapeutic agents as there is strong evidence that ligand activation of this receptor can modulate lipid and glucose homeostasis, making this PPAR an excellent target for treating diseases including diabetes, dyslipidemias, metabolic syndrome and obesity. For example, the PPARβ/δ agonist GW501516 markedly increases serum high-density lipoproteins levels, whereas lowering serum triglyceride levels in obese animals (14,15). Ligand activation of PPARβ/δ also increases fatty acid oxidation in skeletal muscle by upregulation of fatty acid metabolizing enzymes (16–19). In addition to modulating lipid homeostasis, ligand activation of PPARβ/δ can improve insulin sensitivity through a mechanism that requires PPARβ/δ (20–22). Recent evidence also suggests that PPARβ/δ may be constitutively active since it can be coimmunoprecipitated with its obligatory heterodimerization partner in nuclear extracts (9). It is also worth noting that there is evidence that PPARβ/δ has anti-inflammatory activities in a number of different models systems (reviewed in ref. 23; supplementary Table I is available at Carcinogenesis Online). Thus, there is good reason for targeting PPARβ/δ to prevent/treat metabolic diseases.
Despite the high value of targeting PPARβ/δ for treatment of various metabolic disorders, there remains a concern about the role of PPARβ/δ in carcinogenesis due to considerable controversy (reviewed in refs 2,24). Despite some reports indicating that ligand activation of PPARβ/δ potentiates tumorigenesis (reviewed in refs 2,24), there is a large body of evidence that do not support this view including the observations made by a number of independent laboratories that PPARβ/δ mediates terminal differentiation and/or inhibition of cell proliferation (reviewed in refs 2,24; 23; supplementary Table II is available at Carcinogenesis Online) and that PPARβ/δ has anti-inflammatory activities (reviewed in ref. 23; 23; supplementary Table I is available at Carcinogenesis Online). Definitive evidence that PPARβ/δ ligands fail to increase cell growth or inhibit apoptotic signaling was also recently reported in seven different human cancer cell lines (25,26). Collectively, these and many other related observations (reviewed in refs 2,24) suggest that ligand activation of PPARβ/δ attenuates tumorigenesis, although this hypothesis must be examined critically.
The first evidence suggesting that PPARβ/δ inhibits skin tumorigenesis was the observation that mice lacking PPARβ/δ expression exhibited enhanced epidermal hyperplasia in response to the tumor promoter 12-_O_-tetradecanoylphorbol-13-acetate (TPA) (27,28). It is worth noting that these observations were made in two distinct null mouse models generated by different laboratories and gene targeting strategies. Consistent with these observations, mice lacking PPARβ/δ expression exhibit enhanced skin tumorigenesis in the two-stage [7,12-dimethylbenz[_a_]anthracene (DMBA)/TPA] chemical carcinogenesis bioassay (29). An earlier onset of tumor formation, increased tumor incidence and increased tumor size is found in PPARβ/δ-null mice as compared with wild-type mice (29). PPARβ/δ-null mice also exhibit enhanced epidermal hyperplasia and cell proliferation and reduced caspase 3 activity and apoptosis in response to topical treatment with TPA (29). These findings demonstrate that PPARβ/δ protects against chemically induced skin tumorigenesis, through mechanisms that include attenuation of cell proliferation and/or modulation of apoptosis. These findings also suggest that ligand activation of PPARβ/δ could inhibit chemically induced skin tumorigenesis. Indeed, reports from four independent laboratories show that ligand activation of PPARβ/δ results in the induction of terminal differentiation of keratinocytes and/or inhibition of cell proliferation (11,30–32) and that this effect is mediated by PPARβ/δ (30). Thus, the present studies examined the hypothesis that ligand activation of PPARβ/δ inhibits skin tumorigenesis through the induction of terminal differentiation using both in vivo and ex vivo model systems.
Materials and methods
Two-stage chemical carcinogenesis bioassay
Female, wild-type and PPARβ/δ-null mice in the resting phase of the hair cycle (6–8 weeks of age) were initiated with 50 μg of DMBA dissolved in 200 μl acetone. One week after initiation, wild-type and PPARβ/δ-null mice were treated 3 days/week for 48 weeks with topical application of 5 μg of TPA followed an hour later with either acetone or GW0742 (200 μl of either a 1 or 5 μM stock solution). These concentrations of ligand were chosen based on previous work showing PPARβ/δ-dependent induction of terminal differentiation in mouse skin within this concentration range (30). The onset of tumor formation, tumor size and tumor number was assessed weekly. After 48 weeks of TPA and vehicle/ligand treatment, mice were euthanized by overexposure to carbon dioxide. Skin and tumor samples from each mouse were fixed in 10% neutral-buffered formalin or 70% ethanol and then paraffin embedded, sectioned and stained with hematoxylin and eosin. A pathologist examined hematoxylin and eosin-stained sections of suspected carcinomas.
Short-term analysis of epidermal cell proliferation and terminal differentiation
Wild-type or PPARβ/δ-null mice were treated topically with vehicle control (acetone) or TPA (5 μg per mouse) followed an hour later with either vehicle control (acetone) or GW0742 (200 μl of 5 μM solution). Skin samples were collected either 6 or 24 h after this last treatment.
Replicative DNA synthesis was examined in mice 25 h post-TPA treatment as described above. Mice were injected intraperitoneally with bromodeoxyuridine (BrdU) (100 mg/kg) at the time TPA was applied and 8 h post-TPA application. Skin samples were obtained and fixed in 10% neutral-buffered formalin. Representative skin samples were embedded in paraffin and sectioned (4–6 μm) and prepared for immunohistochemical analysis of BrdU incorporation. Detection of BrdU-labeled keratinocytes was performed using an immunohistochemical kit (Exalpha Biologicals, Watertown, MA) following the manufacturer's recommended procedures. BrdU-labeled keratinocytes were quantified using light microscopy by counting the number of BrdU-labeled and unlabeled keratinocytes in a given 40× frame. Representative slides from each treatment group were examined with a minimum of two different 40× frame per slide being scored for labeling analysis for each mouse skin sample.
Keratins messenger RNA (mRNA) and protein expression were examined using quantitative real-time PCR (qPCR) analysis and immunohistochemistry, respectively. Total RNA was also isolated from skin samples using TRIZOL reagent (Invitrogen, Carlsbad, CA). For qPCR analysis, complementary DNA was generated using 2.5 μg total RNA with M-MLV Reverse Transcriptase (Promega, Madison, WI). Primers were designed for qPCR using PrimerQuestsm software (Integrated DNA Technologies, Coralville, IA). qPCRs were performed using SYBR green PCR master mix (Finnzymes, Espoo, Finland) in the iCycler and detected using the MyiQ™ Real-time PCR Detection System (Bio-Rad, Hercules, CA). The following conditions were used for polymerase chain reaction: 95°C for 15 s, 94°C for 10 s, 60°C for 30 s and 72°C for 30 s and repeated for 45 cycles. The polymerase chain reaction included a no template control reaction to control for contamination and/or genomic amplification. All reactions had >90% efficiency. Five representative samples from independently treated mice were used for each group and time point. Relative expression levels of mRNA were normalized to glyceraldehyde 3-phosphate dehydrogenase and analyzed for statistical significance using one-way analysis of variance (Prism 4.0).
For immunohistochemistry, antigens were retrieved using the citrate buffer epitope retrieval method and keratin proteins were detected using mouse keratin polyclonal antibodies (Covance, Berkeley, CA) and a peroxidase detection system (Vector Laboratories, Burlingame, CA). Five representative samples from independently treated mice were examined for each treatment group at each time point.
Keratinocyte ex vivo cancer models
To examine the hypothesis that ligand activation of PPARβ/δ can cause initiated or neoplastic keratinocytes to differentiate and inhibit cell proliferation, several keratinocyte cell lines were used: (i) the 308 keratinocyte cell line derived from DMBA-treated mouse skin (33,34); (ii) the SP1 keratinocyte cell line derived from a DMBA/TPA-treated mouse skin papilloma (33,34) and (iii) the Pam212 keratinocyte cell line derived from spontaneously transformed neonatal keratinocytes (35). Expression of PPARβ/δ mRNA in these cell lines was examined using qPCR analysis as described above. Analysis of neoplastic keratinocyte proliferation was performed using a colony formation assay and cell counting.
For the colony formation assay, 308 keratinocytes were seeded at low density (100 cells per well in a six-well plate) and treated with PPARβ/δ ligands under high calcium (1.4 mM) conditions in the presence or absence of 32 nM TPA for 2 weeks. This assay is based on the observations that initiated keratinocytes will form foci when exposed to tumor promoters (e.g. TPA) in the presence of differentiation-inducing conditions (e.g. high calcium) due to their relative inability to undergo terminal differentiation (36,37). Foci number and size were quantified using the binary color counting procedure from NIH image J software (version 1.37).
To determine the effect of ligand activation of PPARβ/δ on cell proliferation, 308-, SP1- and Pam212-initiated/neoplastic keratinocytes were cultured and treated with PPAR ligands. Cell number was quantified over time in triplicate, independent samples using a Z1 Coulter® particle counter (Beckman-Coulter, Fullerton, CA).
Analysis of neoplastic keratinocyte differentiation was performed by examining differentiation mRNA marker expression and cornified envelope formation. For analysis of differentiation marker mRNA, 24 h after treatment with PPAR ligands under high calcium (1.4 mM) conditions, total RNA was isolated from 308 keratinocytes using TRIZOL reagent (Invitrogen) and the manufacturer's recommended procedures. Ten micrograms of total RNA was analyzed using northern blot analysis. The following previously described complementary DNAs were used for random primed 32P-labeled probes: ADRP, SPR1A and SPR2H (30). qPCR analysis was performed using RNA from ligand-treated 308 cells as described above. To quantify cornified envelope formation, 308 or Pam212 keratinocytes were seeded at ∼90% confluency in six-well plates and treated with either GW0742 or L-165041 in low calcium (0.05 mM) or a terminal differentiation-inducing condition (1.4 mM calcium). After this treatment period, cells were collected and treated with lysis solution containing 2% sodium dodecyl sulfate and 2% β-mercaptoethanol for 5 min. Undissolved cornified envelopes observed under a hemocytometer were quantified as described previously (38).
Analysis of neoplastic keratinocyte apoptosis was performed by examining caspase 3/7 activity. Neoplastic keratinocytes at ∼50% confluency were treated with or without 1 μM GW0742 for 12 h. Following initial 12 h treatment, keratinocytes were treated with or without ultraviolet (UV) light (20 000 μJ/cm2) and maintained under prior treatment condition. Caspase 3/7 activity was measured 12 h postirradiation using the caspase 3/7 Glo reagent (Promega) following the manufacturer's recommended procedures.
Results
Ligand activation of PPARβ/δ inhibits chemically induced skin tumorigenesis
To determine if ligand activation of PPARβ/δ inhibits skin carcinogenesis, a two-stage bioassay was performed using wild-type and PPARβ/δ-null mice treated topically with the highly specific PPARβ/δ ligand GW0742 at concentrations previously shown to specifically activate PPARβ/δ (30). Consistent with past studies (29), the onset of papilloma formation was earlier after initiation in the absence of PPARβ/δ expression (Figure 1A and D). Topical administration of GW0742 did not influence the onset of papilloma formation in either genotype at a concentration of 1 μM (Figure 1A) but significantly (P ≤ 0.05) delayed papilloma formation by 5 weeks in the wild-type mice at a concentration of 5 μM (Figure 1D). This effect was not found in similarly treated PPARβ/δ-null mice (Figure 1D). The average number of wild-type mice with tumors was significantly less in response to topical application of 1 μM GW0742 from weeks 13 through 29 (P ≤ 0.05), and this was not found in similarly treated PPARβ/δ-null mice (Figure 1A). This PPARβ/δ-dependent effect was not observed in response to 5 μM GW0742 (Figure 1D). There was no significant effect observed with tumor multiplicity following topical application of 1 μM GW0742 in either genotype (Figure 1B). However, tumor multiplicity was decreased in wild-type mice by topical application of 5 μM GW0742 (Figure 1E) during early (weeks 12–23) and later stages (weeks 36–48) of the bioassay (P ≤ 0.05). This effect was not found in similarly treated PPARβ/δ-null mice up to week 42, but after week 42, tumor multiplicity was lower in PPARβ/δ-null mice treated topically with 5 μM GW0742 (Figure 1E). The average tumor size was not affected by ligand treatment in either genotype (Figure 1C and F), but the average size of skin tumors was greater in PPARβ/δ-null mice as compared with wild-type mice during the later stages of the bioassay (P ≤ 0.05). Lesions suspected of being squamous cell carcinomas were examined for histopathology. The majority of lesions suspected of being squamous cell carcinomas were later classified as keratocanthomas (Figure 2). The incidence of keratocanthomas was significantly greater in PPARβ/δ-null mice as compared with wild-type mice (Figure 2A and B). Topical administration of GW0742 decreased the incidence of keratocanthoma in wild-type mice at both 1 and 5 μM GW0742 with greater inhibition being observed with 5 μM GW0742. This effect was not found in similarly treated PPARβ/δ-null mice (Figure 2A and B). Squamous cell carcinomas were only observed in PPARβ/δ-null mice but not in wild-type mice (Figure 2A and B).
Fig. 1.

Ligand activation of PPARβ/δ inhibits chemically induced skin tumorigenesis. Two-stage chemical carcinogen testing was performed in wild-type (+/+) or PPARβ/δ-null (−/−) mice as described in Materials and Methods. Mice were treated with or without the PPARβ/δ ligand GW0742 (1 or 5 μM) during tumor promotion. The incidence and onset of lesion (lesions include papillomas, keratocanthomas and carcinomas) formation in mice treated with and without 1.0 μM GW0742 (A) or 5.0 μM GW0742 (D). Skin lesion multiplicity in mice treated with and without 1.0 μM GW0742 (B) or 5.0 μM GW0742 (E). Average skin lesion size in mice treated with and without 1.0 μM GW0742 (C) or 5.0 μM GW0742 (F).
Fig. 2.

Ligand activation of PPARβ/δ inhibits tumor progression and TPA-induced replicative DNA synthesis. Suspected carcinomas were examined microscopically and classified as either keratocanthomas or carcinomas by a pathologist. (A) Incidence of keratocanthomas and carcinomas in mice treated with and without 1 μM GW0742 (left panel) or 5 μM GW0742 (right panel). Values represent the percentage of mice with keratocanthomas or carcinomas. (B) Tumor multiplicity in mice treated with and without 1 μM GW0742 (left panel) or 5 μM GW0742 (right panel). Values represent the average number of lesions per mouse with the lesion ± SEM. (C) Wild-type (+/+) and PPARβ/δ-null (−/−) mice injected with BrdU were topically treated with acetone or TPA (5 μg per mouse) followed by topical application of 5 μM GW0742. BrdU-labeling index was determined as described in Materials and Methods. Values represent the mean ± SEM. Values with different letters are significantly different as determined by analysis of variance and post hoc testing, P ≤ 0.05.
PPARβ/δ-dependent inhibition of replicative DNA synthesis and modulation of differentiation signaling
To determine if ligand activation of PPARβ/δ can modulate epidermal cell proliferation and differentiation, mice were treated topically with the PPARβ/δ ligand, GW0742, after a single application of TPA. Topical administration of 5 μM GW0742 significantly decreased the number of BrdU-labeled keratinocytes in TPA-treated wild-type mice and this effect was not observed in similarly treated PPARβ/δ-null mice (Figure 2C). PPARβ/δ-null mice treated with TPA had a higher BrdU-labeling index as compared with similarly treated wild-type mice (Figure 2C). To examine the effect of ligand activation of PPARβ/δ on differentiation signaling, expression of early and late markers of keratinocyte terminal differentiation was examined, at both the mRNA and protein level. Expression of mRNA encoding keratin 1 was significantly reduced by topical administration of TPA and coadministration of TPA and GW0742 in both genotypes, 6 h post-application (23; supplementary Figure 1A is available at Carcinogenesis Online). No changes in the expression of keratin 1 mRNA were observed with any treatment in either genotype 24 h post-application (23; supplementary Figure 1A is available at Carcinogenesis Online). Changes in the expression of mRNA encoding keratin 10 were similar between both genotypes at both time points (23; supplementary Figure 1B is available at Carcinogenesis Online). Decreased expression of keratin 10 mRNA was observed following TPA and coadministration of TPA and GW0742, and no change was found after GW0742 treatment in both genotypes (23; supplementary Figure 1B is available at Carcinogenesis Online). While expression of mRNA encoding keratin 5 was modestly lower in response to both TPA application and coadministration of TPA and GW0742 after 6 h, this effect was not statistically significant (23; supplementary Figure 1C is available at Carcinogenesis Online). No change in the expression of mRNA encoding keratin 5 was found with any treatment in wild-type mice after 24 h, but was higher in PPARβ/δ-null mouse skin after topical application of either TPA or coadministration of GW742 with TPA (23; supplementary Figure 1C is available at Carcinogenesis Online). Expression of mRNA encoding keratin 14 was not changed by GW0742, TPA or coadministration of TPA and GW0742 in wild-type mouse skin after 6 h, but was higher in PPARβ/δ-null mouse skin after TPA or TPA and GW0742 cotreatment at this time point (23; supplementary Figure 1D is available at Carcinogenesis Online). By 24 h post-application of either TPA or TPA and GW0742, increased expression of keratin 14 was similar between genotypes (23; supplementary Figure 1D is available at Carcinogenesis Online). Expression of mRNA encoding involucrin was similar between genotypes after 6 h and was not markedly changed by any treatment (23; supplementary Figure 1E is available at Carcinogenesis Online). However, coadministration of TPA with GW0742 resulted in a PPARβ/δ-dependent increase in involucrin, as this effect was not found in similarly treated PPARβ/δ-null mice (23; supplementary Figure 1E is available at Carcinogenesis Online).
In addition to examining expression of mRNA markers of keratinocyte differentiation, immunohistochemical analysis of these proteins was also performed. Topical application of TPA caused a reduction in keratin 1 protein, in particular after 24 h in the wild-type mice (23; supplementary Figure 2A is available at Carcinogenesis Online). Expression of keratin 1 was higher in wild-type mouse skin following coadministration of GW0742 and TPA after 24 h, and this effect was not observed in PPARβ/δ-null mice (23; supplementary Figure 2A is available at Carcinogenesis Online). Expression of keratin 1 was higher in wild-type mice in all treatment groups at both the 6 and 24 h time points as compared with similarly treated PPARβ/δ-null mice (23; supplementary Figure 2A is available at Carcinogenesis Online). Expression of keratin 10 was higher in wild-type mice as compared with PPARβ/δ-null mice for all treatment groups at both the 6 and 24 h time points and topical application of GW0742 had no effect in either genotype (23; supplementary Figure 2B is available at Carcinogenesis Online). Immunohistochemical analysis of keratin 5 showed higher staining in PPARβ/δ-null mice as compared with wild-type mice for all treatment groups at the 6 h time point (23; supplementary Figure 2C is available at Carcinogenesis Online). Relative expression of keratin 5 was higher 24 h post-TPA as compared with 6 h post-TPA, although expression was similar for all treatment groups in both genotypes (23; supplementary Figure 2C is available at Carcinogenesis Online). Six hours post-TPA application, no difference in immunoreactive keratin 14 was observed between treatments or genotype (23; supplementary Figure 2C is available at Carcinogenesis Online). However, expression of keratin 14 was markedly higher in PPARβ/δ-null mice 24 h post-TPA as compared with similarly treated wild-type mice (23; supplementary Figure 2C is available at Carcinogenesis Online).
Ligand activation of PPARβ/δ inhibits mouse papilloma and carcinoma cell line growth
The observed inhibition of replicative DNA synthesis and chemopreventive effects in the chemically induced skin carcinogenesis suggests that ligand activation of PPARβ/δ could inhibit proliferation of cells in varying stages of skin carcinogenesis (e.g. initiated cells, benign papillomas and pre- and postmalignant carcinomas). Thus, the effect of ligand activation in three mouse keratinocyte tumor cell lines was examined. The 308 keratinocyte cell line has a ras mutation and was derived from mouse skin following initiation with DMBA (33,34). 308 cells can form papillomas with and without tumor promotion when grafted onto mouse skin in vivo (33,34). The SP1 keratinocyte cell line is a papilloma-like cell line derived from DMBA/TPA-treated animals with a ras mutation and produces papillomas in vivo when grafted onto mouse skin (33,34). The Pam212 keratinocyte cell line is a carcinoma-like cell line derived from spontaneous transformation of neonatal keratinocytes in culture condition and produces squamous cell carcinoma in vivo when grafted onto mouse skin (35). All three of these keratinocyte cancer lines are resistant to calcium-induced terminal differentiation (39). PPARβ/δ is expressed in the three keratinocyte cancer lines, but is noticeably lower as compared with control keratinocytes (23; supplementary Figure 3 is available at Carcinogenesis Online). Inhibition of cell proliferation is observed in all three keratinocyte cancer cell lines in response to ligand activation of PPARβ/δ (Figure 3A–C). As an indirect measure of the ability of PPARβ/δ to modulate events that promote conversion of an initiated cell into a cancerous lesion, a colony formation assay was performed using the 308 keratinocyte cell line. Unlike primary keratinocytes that undergo terminal differentiation in high (1.4 mM) calcium medium, 308 keratinocytes form proliferative foci when seeded at low density in this high calcium medium (33,34). Treatment with the PPARβ/δ ligand GW0742 or TTA inhibited foci formation and caused a significant reduction in the average size of foci in both the presence and absence of TPA (Figure 4A–C). For example, GW0742 and TTA caused a 47 and 99% decrease, respectively, in the average number of colonies as compared with control dimethyl sulfoxide cells (Figure 4B). Similarly, GW0742 and TTA caused a 16 and 76%, respectively, in average colony size as compared with control dimethyl sulfoxide cells (Figure 4B). Culturing cells in the presence of the PPARγ agonist troglitazone also inhibited colony formation and average size, whereas the PPARα agonist WY-14,643 had no effect (Figure 4A–C).
Fig. 3.
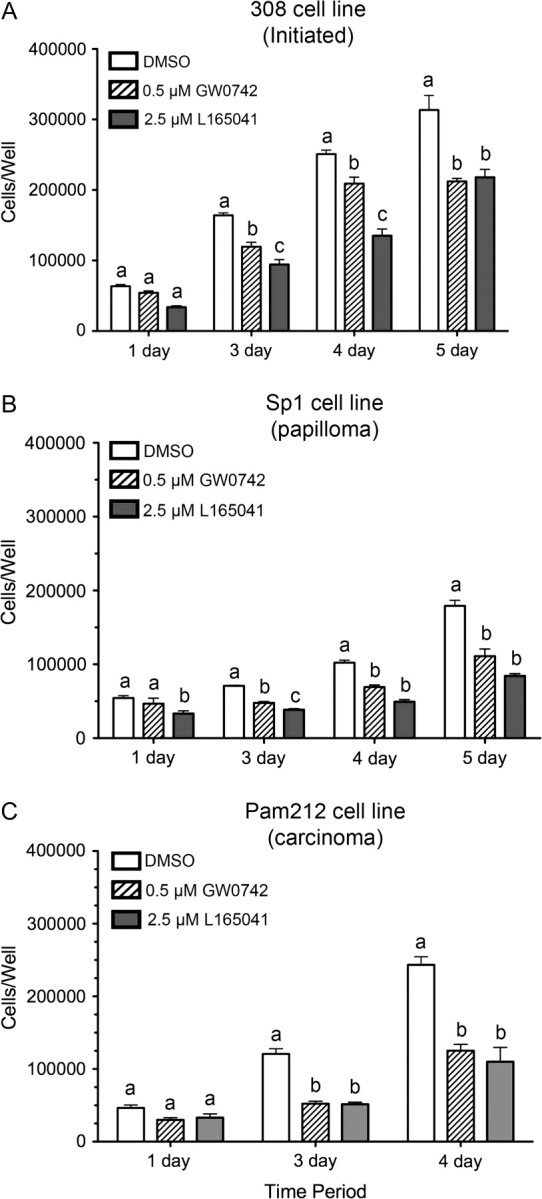
Ligand activation of PPARβ/δ inhibits cell proliferation in neoplastic keratinocyte cancer cell lines. Neoplastic keratinocytes were cultured in medium containing either GW0742 (0.5 μM) or L165041 (2.5 μM) in low (0.05 mM) calcium culture medium for 6 days prior to the start of the cell proliferation assay (n = 4 replicates per treatment group). Cell number was quantified using a Coulter counter. Cell proliferation kinetics in (A) 308 keratinocytes, (B) SP1 keratinocytes and (C) Pam212 keratinocytes. Values represent the mean ± SEM. Values with different letters are significantly different as determined by analysis of variance and post hoc testing, P ≤ 0.05.
Fig. 4.
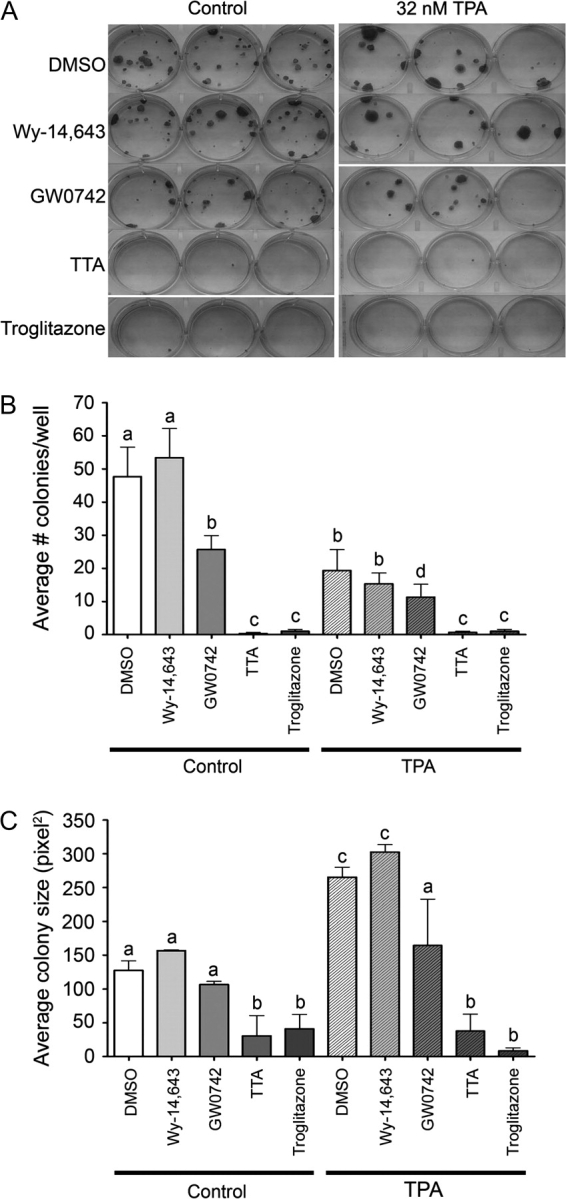
Ligand activation of PPARβ/δ inhibits colony/foci formation in an initiated keratinocyte cancer cell line. 308 keratinocytes were cultured in the presence of PPAR ligands (PPARα—50 μM Wy-14,643; PPARβ/δ—1 μM GW0742 or 10 μM TTA and PPARγ—30 μM troglitazone) under high (1.4 mM) calcium conditions in the presence or absence of 32 nM TPA. Foci were stained with rhodamine dye at the end of treatment period. (A) Representative photomicrographs of stained foci from each group. The average number of foci (B) and the average size of foci (C) in each group. Values represent the mean ± SEM. Values with different letters are significantly different as determined by analysis of variance and post hoc testing, P ≤ 0.05.
Ligand activation of PPARβ/δ induces differentiation of mouse papilloma and carcinoma cell lines
To determine if ligand activation of PPARβ/δ induces terminal differentiation in neoplastic keratinocyte cell lines, cornified envelope formation was examined in 308 and Pam212 cells. Indeed, treatment of 308 keratinocytes and Pam212 keratinocytes with PPARβ/δ ligands increased cornified envelope formation (Figure 5). To further examine the effect of ligand activation on terminal differentiation, expression of mRNA markers of differentiation was examined in the 308 keratinocyte cell line. Increased expression of mRNA encoding ADRP and SPRs was observed after treatment with PPARβ/δ ligands (Figure 6A). Additionally, expression of mRNA encoding keratin 5, keratin 6A and keratin 14 was decreased, and mRNA encoding keratin 10 was increased by GW0742 treatment in 308 cells (Figure 6B). Similarly, expression of mRNA encoding keratin 5 and keratin 14 was decreased, and mRNA encoding involucrin was increased by L165041 treatment in 308 cells (Figure 6B). Both GW0742 and L165041 increased expression of the known PPARβ/δ target gene angiopoietin-like protein 4 (Figure 6B). Since the induction of terminal differentiation can be associated with an increase in apoptotic or apoptotic-like signaling, the effect of ligand activation of PPARβ/δ on neoplastic keratinocyte apoptosis was also examined. There was no change in caspase 3/7 activity in 308, SP1 or Pam212 neoplastic keratinocytes in response to GW0742 (23; supplementary Figure 4 is available at Carcinogenesis Online). Following exposure to UV radiation, caspase 3/7 activity increased in all three neoplastic keratinocyte cell lines although Pam212 cell line keratinocyte was less responsive as compared with 308 or SP1 keratinocyte cell lines (23; supplementary Figure 4 is available at Carcinogenesis Online). Further, while UV irradiation caused a significant increase in caspase 3/7 activity, the presence of GW0742 did not significantly modulate this increase in any of the three neoplastic keratinocyte cell lines (23; supplementary Figure 4 is available at Carcinogenesis Online).
Fig. 5.

Ligand activation of PPARβ/δ induces terminal differentiation in 308 and Pam212 keratinocyte cancer cell lines. Cells were seeded at ∼90% confluency and cultured in low calcium (0.05 mM) medium or high calcium (1.4 mM) medium to induce terminal differentiation, in the presence or absence of either GW0742 (0.5 μM) or L165041 (5.0 μM). The average number of cornified envelopes was determined in (A) 308 keratinocytes and (B) Pam212 keratinocytes.
Fig. 6.

Ligand activation of PPARβ/δ increases mRNA markers of terminal differentiation in 308-initiated keratinocytes. (A) Northern blot analysis of a PPARβ/δ target gene (ADRP) and differentiation-associated genes (SPR1A and SPR2H) using RNA from 308 keratinocytes after treatment with PPAR ligands (PPARα—25 μM Wy-14,643; PPARβ/δ—0.5 μM GW0742, 10 μM TTA, 5 μM L165041 and PPARγ—10 μM troglitazone) for 24 h using culture medium containing high calcium (1.4 mM). *Significantly different than control, P ≤ 0.05. (B) qPCR analysis of a PPARβ/δ target gene (ANGPTL4) and differentiation-associated genes [keratin 5 (K5), keratin 6A (K6A), keratin 10 (K10), keratin 14 (K14) and involucrin] using RNA from 308 keratinocytes after treatment with either 0.5 μM GW0742 or 5 μM L-165041 using culture conditions as described above. Values represent the mean ± SEM. Values with different letters are significantly different as determined by analysis of variance and post hoc testing, P ≤ 0.05.
Discussion
Two-stage chemically induced skin tumorigenesis is exacerbated in PPARβ/δ-null mice (29) and ligand activation of PPARβ/δ induces terminal differentiation and inhibits cell growth in keratinocytes (11,30–32,40,41). These observations support the hypothesis that ligand activation of PPARβ/δ could be chemopreventive for chemically induced skin cancer. Results from the present study show that ligand activation of PPARβ/δ during the promotion phase of chemically induced skin cancer inhibited the onset of tumor formation, the incidence of tumors and tumor multiplicity. This clearly demonstrates that ligand activation of PPARβ/δ is chemopreventive in a chemically induced skin carcinogenesis model. Since none of the tumors examined in the wild-type mice were squamous cell carcinomas, it cannot be determined whether ligand activation of PPARβ/δ can inhibit malignant conversion. However, it is of interest to note that the average number of keratocanthomas was significantly reduced by GW0742 in the wild-type mice and this effect was not found in PPARβ/δ-null mice. While keratocanthomas are benign lesions in humans, they can progress to malignant carcinomas in mice (42). This suggests that ligand activation of PPARβ/δ could potentially inhibit malignant conversion. Consistent with this idea, squamous cell carcinomas were only found in PPARβ/δ-null mice, and administration of GW0742 had no influence on this end point. Further research is needed to determine whether malignant conversion can be inhibited by ligand activation of PPARβ/δ. Since there are a number of PPARβ/δ ligands that have been and continue to be developed, it will also be of interest to determine if all PPARβ/δ ligands will be suitable for chemoprevention and/or chemotherapy of skin cancer. Additionally, as UV radiation also contributes significantly to the incidence of human skin cancer, whether ligand activation of PPARβ/δ will be useful for preventing or treating UV-induced skin cancers should be evaluated. Interestingly, since ligand activation of PPARβ/δ does not influence UV-induced caspase 3 activity in mouse keratinocytes (40) or mouse skin cancer cell lines (present study), modulation of apoptotic signaling as a mechanism for skin cancer prevention or treatment may not be central in the PPARβ/δ/UV skin cancer model. However, it remains possible that ligand activation of PPARβ/δ may effectively induce terminal differentiation in response to UV-induced skin lesions and this idea should be examined in greater detail.
As an alternative to examining malignant conversion, the effect of ligand activation in keratinocyte cell lines representing different stages of neoplasia was also examined. Despite their known resistance to calcium-induced terminal differentiation, induction of mRNA markers of terminal differentiation was found in 308 cells and Pam212 cells in response to ligand activation of PPARβ/δ. Further, an increase in cornified envelopes was also observed in neoplastic keratinocyte cell lines after ligand activation of PPARβ/δ. This is also consistent with inhibition of cell proliferation found in 308 and Pam212 cells, the inhibition of foci number and size observed with the colony formation assay and the inhibition of chemically induced skin tumorigenesis in response to ligand activation of PPARβ/δ. This clearly demonstrates that ligand activation of PPARβ/δ can induce terminal differentiation in cell types that have DNA damage and are predisposed to develop into skin tumors and another cell type that models squamous cell carcinomas. Collectively, these findings suggest that ligand activation of PPARβ/δ can target specific cell lineages critical for the progression of chemically induced skin cancer and inhibit cell growth by inducing terminal differentiation. In particular, these findings also suggest that ligand activation of PPARβ/δ can be both chemopreventive and chemotherapeutic.
Based on results from the present studies and findings from previous work as well, the mechanism by which ligand activation of PPARβ/δ inhibits chemically induced skin tumorigenesis is likely through the induction of terminal differentiation with a concomitant inhibition of cell growth of tumor cells. For example, there is strong evidence from a number of independent laboratories supporting a role for PPARβ/δ in mediating terminal differentiation and/or inhibition of cell growth in a variety of cell types (reviewed in refs 2,24; 23; supplementary Table II is available at Carcinogenesis Online). In contrast to work by others showing increased expression of terminal differentiation markers preceding the chemopreventive influence of deleting cyclooxygenase during chemical carcinogenesis (43), earlier than normal expression of keratin 1 and keratin 10 was not found in response to ligand activation of PPARβ/δ, following acute exposure to the tumor promoter TPA in vivo. However, discordant expression of keratin 5 and keratin 14 at the mRNA level was observed in the absence of PPARβ/δ expression following TPA administration, consistent with the previously observed hyperplastic phenotype in PPARβ/δ-null mice treated with TPA (27–29,44).
There is an interesting balance between differentiation and apoptosis in keratinocytes that could be influenced by PPARβ/δ and impact skin tumorigenesis. There is evidence that when keratinocytes undergo terminal differentiation to ultimately form a cornified cell, this process is associated with increased activity of caspases including caspase 3 and caspase 14 (45,46). While caspase 3 is known to be central in the process of apoptosis and increased caspase 3 activity occurs during keratinocyte terminal differentiation (46), differentiating and differentiated keratinocytes (e.g. cornified cells) do not exhibit shrinkage, DNA fragmentation and are not phagocytosed (47,48). Additionally, caspase 14 does not participate in apoptotic signaling but rather modulates terminal differentiation (45). Thus, apoptotic signaling observed in differentiating keratinocytes is relatively unique. While there is strong evidence that ligand activation of PPARβ/δ induces terminal differentiation in keratinocytes, this process probably involves apoptotic-like signaling as well. Since it is known that increasing apoptosis by chemicals such as silymarin can inhibit skin carcinogenesis (49), it is possible that the observed inhibition of skin carcinogenesis by ligand activation of PPARβ/δ, which could be due in part to increased terminal differentiation and inhibition of cell growth, could also be enhanced by modulating apoptotic signaling as well. This idea should be examined in greater detail.
While one mechanism by which ligand activation of PPARβ/δ can inhibit tumorigenesis is through the induction of terminal differentiation and inhibition of cell growth, it is important to note that other mechanisms may also be involved. For example, there is good evidence that PPARβ/δ mediates anti-inflammatory activities in a number of cell types including colon epithelium, macrophages, cardiomyocytes, immune cells, keratinocytes, myoblasts, endothelial cells, nervous tissue and hepatocytes (reviewed in ref. 23; 23; supplementary Table I is available at Carcinogenesis Online). Additionally, recent evidence indicates a novel role for PPARβ/δ agonists in suppressing expression of tissue factor, which is central in initiating thrombosis (50). This is of interest because inhibition of thrombosis is a strategy for chemoprevention and/or chemoprevention (51–54). Whether the anti-inflammatory activities or anti-thrombotic activity of PPARβ/δ ligands contributes to the observed inhibition of chemically induced skin tumorigenesis in the present studies should be further examined. Additionally, whether combining ligand activation of PPARβ/δ with other potential therapeutics that targets other major pathways (e.g. kinases, growth factor receptors, etc.) can increase the efficacy of chemoprevention and/or chemotherapy deserves further investigation.
Supplementary material
Supplementary Tables I and II and Figures 1–4 can be found at http://carcin.oxfordjournals.org/
[Supplementary Data]
Funding
The National Institutes of Health (CA124533 to J.M.P.).
Acknowledgments
We gratefully acknowledge Drs Stuart, Henry Hennings and Ulrike Lichti for providing the keratinocyte cell lines and expert advice and Drs Andrew Billin and Timothy Willson for providing GW0742.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
BrdU
bromodeoxyuridine
DMBA
7,12-dimethylbenz[a]anthracene
mRNA
messenger RNA
PPAR
peroxisome proliferator-activated receptor
qPCR
quantitative real-time PCR
TPA
12-_O_-tetradecanoylphorbol-13-acetate
TTA
tetradecylthioacetic acid
UV
ultraviolet
References
- 1.Akiyama TE, et al. PPAR ligands: potential therapies for metabolic syndrome. Curr. Diab. Rep. 2005;5:45–52. doi: 10.1007/s11892-005-0067-3. [DOI] [PubMed] [Google Scholar]
- 2.Burdick AD, et al. The role of peroxisome proliferator-activated receptor-β/δ in epithelial cell growth and differentiation. Cell. Signal. 2006;18:9–20. doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Desvergne B, et al. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 4.Grimaldi PA. Regulatory role of peroxisome proliferator-activated receptor δ (PPARδ) in muscle metabolism. A new target for metabolic syndrome treatment? Biochimie. 2005;87:5–8. doi: 10.1016/j.biochi.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Lee CH, et al. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 6.Peraza MA, et al. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR) Toxicol. Sci. 2006;90:269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- 7.Braissant O, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 8.Braissant O, et al. Differential expression of peroxisome proliferator-activated receptor-α, -β, and -γ during rat embryonic development. Endocrinology. 1998;139:2748–2754. doi: 10.1210/endo.139.6.6049. [DOI] [PubMed] [Google Scholar]
- 9.Girroir EE, et al. Quantitative expression patterns of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) protein in mice. Biochem. Biophys. Res. Commun. 2008;371:456–461. doi: 10.1016/j.bbrc.2008.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forman BM, et al. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc. Natl Acad. Sci. USA. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westergaard M, et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J. Invest. Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- 12.Sznaidman ML, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)-synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- 13.Berger J, et al. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARδ ligands produce distinct biological effects. J. Biol. Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 14.Leibowitz MD, et al. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- 15.Oliver WR, Jr, et al. A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc. Natl Acad. Sci. USA. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holst D, et al. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta. 2003;1633:43–50. doi: 10.1016/s1388-1981(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 17.Luquet S, et al. Roles of peroxisome proliferator-activated receptor δ (PPARδ) in the control of fatty acid catabolism. A new target for the treatment of metabolic syndrome. Biochimie. 2004;86:833–837. doi: 10.1016/j.biochi.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl Acad. Sci. USA. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang YX, et al. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 20.Kitz Kramer D, et al. Role of AMP kinase and PPARδ in the regulation of lipid and glucose metabolism in human skeletal muscle. J. Biol. Chem. 2007;282:19313–19320. doi: 10.1074/jbc.M702329200. [DOI] [PubMed] [Google Scholar]
- 21.Lee CH, et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl Acad. Sci. USA. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sprecher DL, et al. Triglyceride: high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor δ agonist. Arterioscler. Thromb. Vasc. Biol. 2007;27:359–365. doi: 10.1161/01.ATV.0000252790.70572.0c. [DOI] [PubMed] [Google Scholar]
- 23.Kilgore KS, et al. PPARβ/δ ligands as modulators of the inflammatory response. Curr. Opin. Investig. Drugs. 2008;9:463–469. [PubMed] [Google Scholar]
- 24.Peters JM, et al. Role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clin. Sci. 2008;115:107–127. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollingshead HE, et al. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. doi: 10.1093/carcin/bgm183. [DOI] [PubMed] [Google Scholar]
- 26.Girroir EE, et al. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology. 2008;243:236–243. doi: 10.1016/j.tox.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalik L, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J. Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peters JM, et al. Growth, adipose, brain and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ) Mol. Cell. Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim DJ, et al. Peroxisome proliferator-activated receptor β (δ)-dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J. Biol. Chem. 2004;279:23719–23727. doi: 10.1074/jbc.M312063200. [DOI] [PubMed] [Google Scholar]
- 30.Kim DJ, et al. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006;13:53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- 31.Schmuth M, et al. Peroxisome proliferator-activated receptor (PPAR)-β/δ stimulates differentiation and lipid accumulation in keratinocytes. J. Invest. Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- 32.Tan NS, et al. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strickland JE, et al. Development of murine epidermal cell lines which contain an activated rasHa oncogene and form papillomas in skin grafts on athymic nude mouse hosts. Cancer Res. 1988;48:165–169. [PubMed] [Google Scholar]
- 34.Yuspa SH, et al. Mouse skin cells resistant to terminal differentiation associated with initiation of carcinogenesis. Nature. 1981;293:72–74. doi: 10.1038/293072a0. [DOI] [PubMed] [Google Scholar]
- 35.Yuspa SH, et al. A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Res. 1980;40:4694–4703. [PubMed] [Google Scholar]
- 36.Kulesz-Martin MF, et al. Quantitative assay for carcinogen altered differentiation in mouse epidermal cells. Carcinogenesis. 1980;1:995–1006. doi: 10.1093/carcin/1.12.995. [DOI] [PubMed] [Google Scholar]
- 37.Yuspa SH, et al. Cultivation and characterization of cells derived from mouse skin papillomas induced by an initiation-promotion protocol. Carcinogenesis. 1986;7:949–958. doi: 10.1093/carcin/7.6.949. [DOI] [PubMed] [Google Scholar]
- 38.Hough-Monroe L, et al. Quantitation of cross-linked protein: an alternative to counting cornified envelopes as an index of keratinocyte differentiation. Anal. Biochem. 1991;199:25–28. doi: 10.1016/0003-2697(91)90264-t. [DOI] [PubMed] [Google Scholar]
- 39.Hennings H, et al. Development of an in vitro analogue of initiated mouse epidermis to study tumor promoters and antipromoters. Cancer Res. 1990;50:4794–4800. [PubMed] [Google Scholar]
- 40.Burdick AD, et al. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits cell growth of human N/TERT-1 keratinocytes. Cell. Signal. 2007;19:1163–1171. doi: 10.1016/j.cellsig.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Man MQ, et al. Deficiency of PPARβ/δ in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J. Invest. Dermatol. 2007;128:370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- 42.Knutsen GL, et al. Gross and microscopic lesions in the female SENCAR mouse skin and lung in tumor initiation and promotion studies. Environ. Health Perspect. 1986;68:91–104. doi: 10.1289/ehp.866891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiano HF, et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002;62:3395–3401. [PubMed] [Google Scholar]
- 44.Kim DJ, et al. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits epidermal cell proliferation by down-regulation of kinase activity. J. Biol. Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 45.Lippens S, et al. Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death Differ. 2000;7:1218–1224. doi: 10.1038/sj.cdd.4400785. [DOI] [PubMed] [Google Scholar]
- 46.Weil M, et al. Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr. Biol. 1999;9:361–364. doi: 10.1016/s0960-9822(99)80162-6. [DOI] [PubMed] [Google Scholar]
- 47.Gandarillas A, et al. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp. Dermatol. 1999;8:71–79. doi: 10.1111/j.1600-0625.1999.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 48.Polakowska RR, et al. Apoptosis in human skin development: morphogenesis, periderm, and stem cells. Dev. Dyn. 1994;199:176–188. doi: 10.1002/aja.1001990303. [DOI] [PubMed] [Google Scholar]
- 49.Singh RP, et al. Silymarin inhibits growth and causes regression of established skin tumors in SENCAR mice via modulation of mitogen-activated protein kinases and induction of apoptosis. Carcinogenesis. 2002;23:499–510. doi: 10.1093/carcin/23.3.499. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh M, et al. COX-2 suppresses tissue factor expression via endocannabinoid-directed PPARδ activation. J. Exp. Med. 2007;204:2053–2061. doi: 10.1084/jem.20070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez PM, et al. Tissue factor and fibrin in tumor angiogenesis. Semin. Thromb. Hemost. 2004;30:31–44. doi: 10.1055/s-2004-822969. [DOI] [PubMed] [Google Scholar]
- 52.Mousa SA. Anti-thrombotics in thrombosis and cancer. Future Oncol. 2005;1:395–403. doi: 10.1517/14796694.1.3.395. [DOI] [PubMed] [Google Scholar]
- 53.Mousa SA. Antithrombotics in thrombosis and cancer. Hamostaseologie. 2005;25:380–386. [PubMed] [Google Scholar]
- 54.Rickles FR. Mechanisms of cancer-induced thrombosis in cancer. Pathophysiol. Haemost. Thromb. 2006;35:103–110. doi: 10.1159/000093551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Data]