Inhibition of eIF2α Dephosphorylation Maximizes Bortezomib Efficiency and Eliminates Quiescent Multiple Myeloma Cells Surviving Proteasome Inhibitor Therapy (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 15.
Abstract
The proteasome inhibitor bortezomib (Velcade) effectively eradicates multiple myeloma (MM) cells, partly by activating endoplasmic reticulum (ER) stress apoptotic signaling. However, MM recurrences in bortezomib-treated patients are invariable. We have shown that ER stress signaling can also induce growth arrest and survival in cancer cells. Thus, we hypothesized that bortezomib therapy could induce quiescence and survival of residual MM cells, contributing to disease recurrence. Here, we report that in MM cells, proteasome inhibition with MG-132 or bortezomib results in a surviving cell fraction that enters a prolonged quiescent state (G0–G1 arrest). Mechanism analysis revealed that bortezomib-surviving quiescent cells attenuate eIF2α phosphorylation and induction of the ER stress proapoptotic gene GADD153. This occurs independently of the eIF2α upstream kinases PERK, GCN2, and PKR. In contrast, the prosurvival ER-chaperone BiP/Grp78 was persistently induced. The bortezomib-surviving quiescent fraction could be eradicated by a simultaneous or sequential combination therapy with salubrinal, an inhibitor of GADD34-PP1C phosphatase complex, and, in consequence, eIF2α dephosphorylation. This effect was mimicked by expression of a phosphorylated mimetic eIF2α-S51D mutant. Our data indicate that bortezomib can induce growth arrest in therapy-surviving MM cells and that attenuation of eIF2α phosphorylation contributes to this survival. Most importantly, this survival mechanism can be blocked by inhibiting eIF2α dephosphorylation. Thus, strategies that maintain eIF2α in a hyperphosphorylated state may be a novel therapeutic approach to maximize bortezomib-induced apoptosis and reduce residual disease and recurrences in this type of cancer.
Introduction
Proteasome inhibitors induce multiple myeloma (MM) cell apoptosis by modulating several pathways, including endoplasmic reticulum (ER) stress signaling (1). In the clinic, bortezomib is now widely used in patients with refractory or relapsed MM and is also being studied as an adjunct to first-line therapies (1). However, the use of prolonged bortezomib therapy has lead to the development of drug resistance (1). Drug resistance has been linked to enhanced ER stress signaling and increased chaperone expression (1, 2), which may be an adaptive capacity of MM cells.
Proteasome inhibitors induce an ER stress response in MM cells contributing to apoptosis (2, 3). However, a primary function of ER stress signaling is also to adapt to exogenous stress and induce growth arrest and survival. This is achieved in part by reducing cyclin D1 levels (4) and inducing protein folding and degradation genes that alleviate the damage caused by unfolded proteins. Thus, it could be anticipated that growth arrest and survival may be a response to proteasome inhibitor therapy.
Depending on the activation intensity and cellular context, eIF2α phosphorylation by upstream kinases, like PERK or GCN2, can induce survival, growth arrest, and/or apoptosis in response to ER stress (5, 6). We showed that eIF2α signaling was linked to the induction and maintenance of HEp3 head and neck squamous carcinoma cell dormancy and survival (5, 7, 8). Growth arrest (i.e., dormancy) is in part due to PERK-dependent phosphorylation of eIF2α, which leads to the down-regulation of cyclins D1/D3 and cyclin-dependent kinase 4 (CDK4). Survival and resistance to chemotherapy, on the other hand, is due to induction of BiP, ATF6 activation, and also eIF2α phosphorylation (5, 8). In other cases, very intense eIF2α phosphorylation can activate apoptotic programs (9). The capacity of eIF2α signaling to decide cell fate in response to stress might be particularly important for MM patients treated with proteasome inhibitors, as different levels of stress may impinge on the cells during this therapy.
We set out to explore the link between proteasome inhibition, activation of ER stress, and growth arrest in MM cells. We hypothesized that proteasome inhibition with MG-132 or bortezomib in clinically relevant concentrations may force cells into a growth arrest/survival program due to an ER stress adaptation response. This could be a potential contributor to therapy resistance and disease recurrence. We further hypothesized that maximizing ER stress signaling could enhance sensitivity to proteasome inhibitors by tipping the balance from signaling for growth arrest/survival to apoptosis. Here, we report that a fraction of MM cells, surviving a single treatment with bortezomib, down-regulate eIF2α phosphorylation and enter a prolonged G0–G1 arrest. More importantly, the quiescent surviving fraction of MM cells with different genetic abnormalities could be almost completely eradicated by enhancing eIF2α phosphorylation with salubrinal (an inhibitor of GADD34-PP1c complex assembly) or by expression of a phosphorylated mimetic eIF2αS51D protein. Our findings reveal that enhancement of ER stress signaling can be exploited as a strategy to maximize efficiency of proteasome inhibitor therapy.
Materials and Methods
Reagents, antibodies, reverse transcription–PCR, cell lines, and tissue culture plasmids
MG-132 and salubrinal were purchased from Calbiochem; bortezomib (Velcade) was kindly provided by Millenium Pharmaceuticals. Proteasome inhibitor treatments were performed using 400 nmol/L MG-132 or 4 nmol/L bortezomib (Velcade) for 24 h. The drug was then removed by serial washes with PBS, and the remaining viable cells were replated. Salubrinal treatments were performed using the drug at 5 or 10 μg/mL, as indicated, for 24 h. RPMI 8226 cells [c-MYC insertion on t(16;22)(q32;q11):der16] were kindly provided by Dr. Douglas Conklin (State University of New York), U266B1 cells [t(11;14)] were from American Type Culture Collection (ATCC). Cells were cultured according to ATCC recommendations. Antibodies used were p-Rb, Rb, p-eIF2α, eIF2α, GCN2, p-GCN2, p-PKR, and PKR from Cell Signaling; p-PERK and PERK from Santa Cruz Biotechnology; BiP from BD; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Calbiochem; and β-tubulin from Abcam. Total RNA was extracted using Trizol (Invitrogen). Primer sequences were published previously (8, 10). The pCLBabe-eIF2αS51D plasmid was kindly provided by Dr. David Schubert (Salk Institute). Transfection of RPMI 8226 cells was performed using Amaxa technology according to the manufacturer’s instructions.
Cell cycle analysis and label retention assay
Cell cycle analysis was performed using propidium iodine (PI)/Rnase staining buffer from BD according to the manufacturer’s instruction. For label retention and viability quantification, cells were treated with proteasome inhibitors or DMSO for 24 h, then stained with 50 nmol/L CFDA/SE (Molecular Probes) for 15 min, and replated. Before analysis, cells were stained using Hoechst-33342 (10 μg/mL) for 15 min at 4°C to determine cell viability. Cells were then fixed using 3% paraformaldehyde for 20 min and analyzed by flow cytometry (LSR-II from BD).
Viability assay and immunoblotting
Cell viability was assessed using trypan blue. Immunoblotting was performed as described previously (7).
Image quantification and statistical analysis
Image semiquantification was performed using ImageJ from NIH. Quantifications of all Western blots and reverse transcription–PCR (RT-PCR) experiments in Figs. 3–5 showing changes in expression levels are given in Supplementary Figs. S1 and S2. Western blots showing protein cleavage were not quantified. Statistical analysis was performed using Prism 4.0 from GraphPad Software and Student’s t test.
Figure 3.
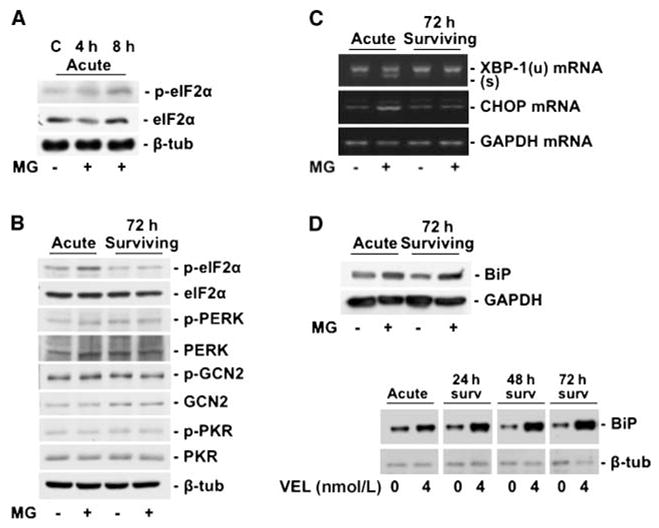
A, Western blots showing phosphorylation of eIF2α at Ser51 acutely at 4 to 8 h. B, Western blots showing phosphorylation of eIF2α at 24 h (left two lanes). EIF2α phosphorylation does not persist after drug washout at 72 h (right two lanes). There was no activation of the upstream kinases PERK, GCN2, and PKR at 24 h nor after drug washout at 72 h. C, RT-PCR showing increased XBP-1 mRNA splicing and CHOP mRNA expression in the acute phase. Note that splicing of XBP-1 and induction of CHOP are not maintained after drug washout at 72 h. D, Western blots showing increased expression of Grp78/BiP in the acute phase of proteasome inhibition with 400 nmol/L MG-132 (top) or 4 nmol/L Velcade (VEL, bottom). As opposed to p-eIF2α, CHOP, and XBP-1, this induction persists after drug washout for both proteasome inhibitors.
Figure 5.
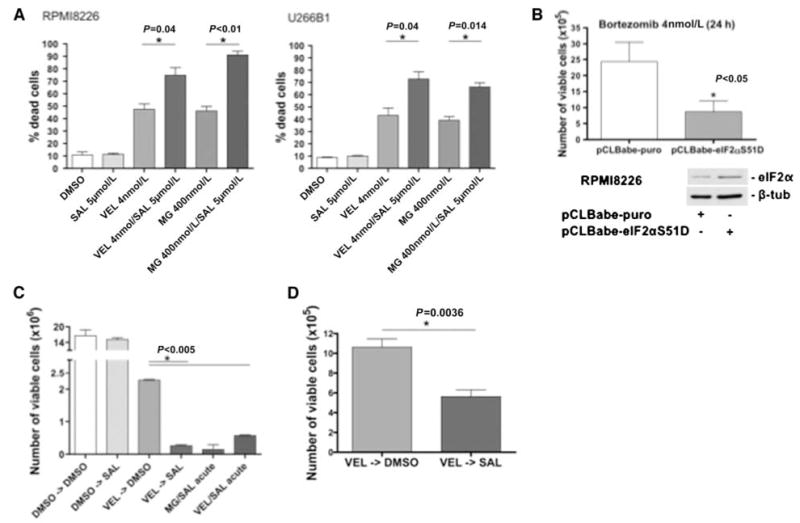
A, percentage of dead cells in RPMI 8226 (left) and U266B1 (right) after 24 h of the indicated treatments with salubrinal (SAL) and/or proteasome inhibitors (MG or Velcade, VEL; left). B, RPMI 8226 cells transfected with a phosphomimetic S51D eIF2α mutant showed enhanced Velcade (VEL) sensitivity compared with the empty vector control. The inset shows increased total protein levels of eIF2α in the cells expressing the construct. C, viability of RPMI 8226 cells after 24 h of the indicated treatments. Cells were treated with either DMSO or Velcade (VEL, 4 nmol/L) for 24 h, then washed, and treated with DMSO or salubrinal (SAL) after that. The right two columns are combination treatments with proteasome inhibitors and salubrinal for 24 h as controls. D, the enhanced sensitivity to salubrinal observed 3 d posttreatment could still be observed at 5 d after proteasome inhibitor washout, suggesting that the sensitivity to salubrinal postproteasome inhibition is long lasting.
Results
Bortezomib induces G0–G1 arrest in a fraction of surviving cells
Treatment of RPMI 8226 MM cells with MG-132 (400 nmol/L) and bortezomib (4 nmol/L) strongly induced cell death (sub-G0 population) in a time-dependent fashion (Fig. 1_A_). This may be due to an initial (4–8 hours) G2-M arrest, as determined by fluorescence-activated cell sorting (data not shown) and in accordance with the data in other systems (11). We next explored the fate of MM cells surviving proteasome inhibitor therapy. RPMI 8226 cells treated with 400 nmol/L MG-132 for 24 hours were washed to remove the drug. We found that the absolute number of viable cells that were then replated remained constant for 7 days. In contrast, vehicle-treated cells initiated exponential growth after 48 hours (Fig. 1_B_). The lack of expansion in the surviving cells was not due to a balance between proliferating and dying cells but to a growth arrest as shown by PI staining followed by flow cytometry (Fig. 1_C_). Cell cycle analysis revealed that the number of cells in G0–G1 increased significantly from ~35% in the control population to ~55% to 65% in the cells surviving proteasome inhibition (Fig. 1_C_). Similar results were obtained using 4 nmol/L bortezomib, a dose that is pharmacodynamically achievable in patients (ref. 12; Fig. 1_D_top). The arrest was due to a failure to transit the G1-S boundary because, 72 hours after washout of proteasome inhibitors, there was an intermediate (Ser780) to strong reduction (Ser795/807/811) in phosphorylated Rb protein levels. These Ser residues are phosphorylated to promote G1-S transition (Fig. 1_D_bottom). Our results show that two proteasome inhibitors are able to induce G0–G1 arrest in RPMI 8226 MM cells after a single treatment.
Figure 1.
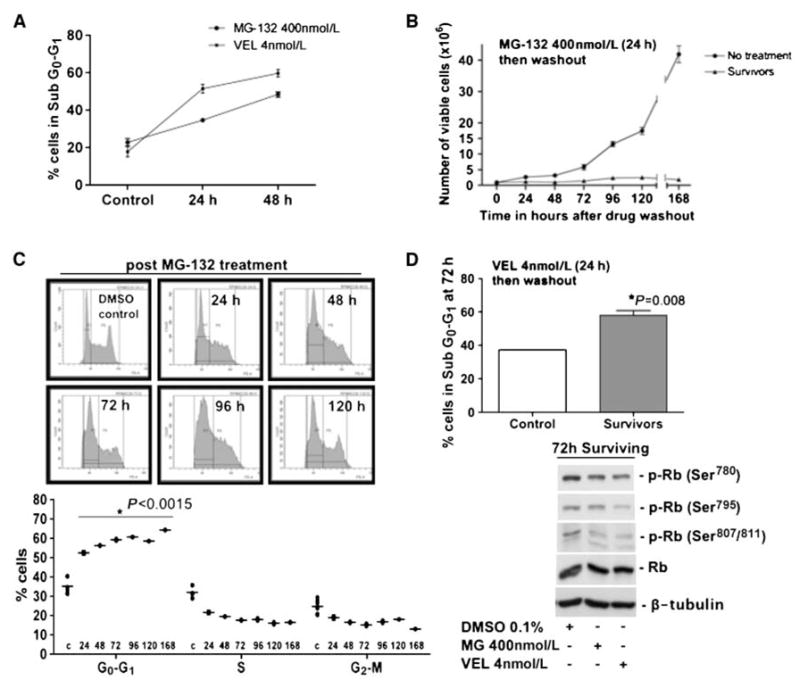
A, RPMI 8226 cells were treated with MG-132 (400 nmol/L) or bortezomib (Velcade, VEL; 4 nmol/L) for 24 to 48 h and stained with PI. The cell cycle was analyzed by flow cytometry. The graph shows the sub-G0–G1 fraction. B, counting of control RPMI 8226 cells and cells surviving proteasome inhibition with 400 nmol/L MG-132 for 24 h using trypan blue exclusion. Note that the cells surviving the treatment do not resume growth for up to a week. C, RPMI 8226 cells surviving proteasome inhibition were stained with PI at the indicated time points after drug washout, and the cell cycle profile was measured by flow cytometry. Cell cycle profile (top) and quantification (bottom). *, P < 0.0015 for all time points, unpaired t test. D, RPMI 8226 cells surviving 4 nmol/L Velcade (VEL) were stained with PI at 72 h after drug washout. The graph (top) shows the G0–G1 fraction. *, P = 0.008. Western blots for phosphorylated (p-Rb ) and total (Rb ) Rb protein in cells surviving proteasome inhibition 72 h after drug washout (bottom). β-Tubulin was used as a loading control.
To corroborate our cell cycle analysis, we performed double labeling with CFSE and Hoechst-33342 to measure viable CFSE retaining cells and to gate out sub-G0 cells. CFSE, due to its irreversible esterification onto cellular proteins and its equal distribution within daughter cells, can be used to monitor cell division (13). We found a highly significant ~4-fold to 7-fold increase in the percentage of CFSE-positive cells at 72 hours in cells surviving both MG-132 and bortezomib (Fig. 2_A_ and B), suggesting that at 72 hours, after the washout of proteasome inhibitors, the surviving cells divide to a lesser extent. These results indicate that, whereas proteasome inhibition can cause acute apoptosis, the surviving MM cells rapidly enter a G0–G1 arrest. Given that pharmacologic treatments do not reach maximal lethal doses in all targeted cells, the induction of a growth-arrested surviving fraction adapting to the treatment may be a possible side effect of proteasome inhibition.
Figure 2.
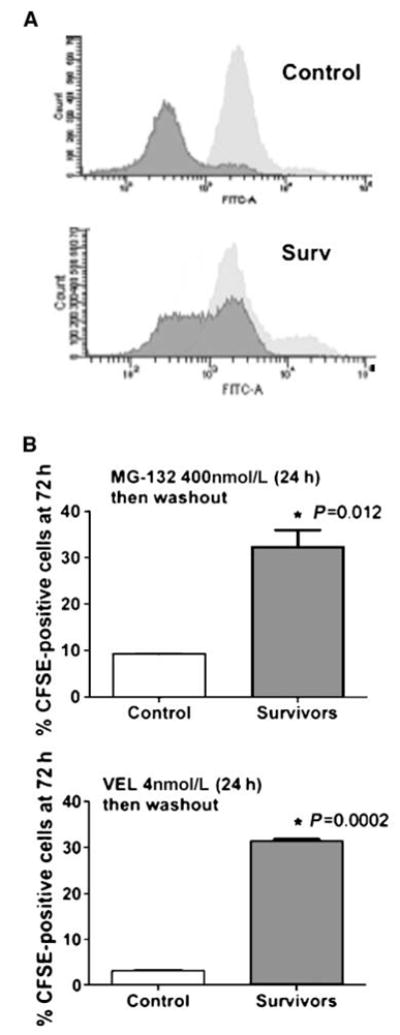
A, RPMI 8226 control or pretreated [MG-132, 400 nmol/L or Velcade (VEL), 4 nmol/L] cells were labeled with CFSE for 15 min, washed, and analyzed by flow cytometry immediately (light gray) or 72 h later (dark gray). Graphs show a representative result for cells surviving MG-132. B, quantification of CFSE labeling. Gates for CFSE-positive were set between 103 and 104 FLH2 (FITC) channel intensity. *, P = 0.012 for MG and P = 0.0002 for Velcade (VEL), unpaired t test.
Bortezomib-surviving cells attenuate eIF2α phosphorylation and uncouple the induction of proapoptotic from prosurvival ER stress genes
We next investigated if the cells surviving proteasome inhibitors undergo any specific changes in ER stress signaling. Proteasome inhibitors are known to induce an unfolded protein response (UPR) in MM cells by inhibition of the ER-associated degradation pathway (2, 14). Furthermore, UPR mediates cell survival and drug resistance in several models, including MM (5, 8, 15). We were interested in the phosphorylation of eIF2α, which attenuates translation and induces a selective gene expression program as an adaptive response to ER stress. We also monitored the expression of the prosurvival chaperone BiP/Grp78 (5, 8), the transcription factor XBP-1 (5), and the proapoptotic transcription factor CHOP (10). MG-132 treatment caused increased eIF2α phosphorylation (Fig. 3_A_ and B), XBP-1 splicing (Fig. 3_C_), BiP (Fig. 3_D_), and CHOP (Fig. 3_C_) up-regulation in the acute phase (8–24 hours). This was not associated with changes in the phosphorylation of the eIF2α upstream kinases PERK, GCN2, and PKR (Fig. 3_B_). With the exception of BiP, which was strongly induced, the expression of CHOP mRNA, spliced XBP-1, or increased eIF2α phosphorylation did not persist in the surviving cells (Fig. 3_A–D_). Phosphorylation of p38 was also activated both by MG-132 and bortezomib, and p38 remained phosphorylated in the surviving quiescent cells (data not shown). This suggests that, as reported (16), p38 activation can have a prosurvival role in MM cells. BiP can induce survival in other cancer cells (5, 17–19), suggesting that these functions might be preserved during quiescence induced by proteasome inhibitors in MM cells.
Interestingly, both CHOP induction and XBP-1 splicing are only observed during the acute phase and both are then down-regulated in the surviving cells. The functional role for the down-regulation of XBP-1 splicing is not clear. However, it is possible that down-regulation of CHOP eliminates the proapoptotic function of this gene (20), further promoting survival of the residual MM cells. Although the levels of phosphorylated eIF2α kinases did not change in acute treatments or after proteasome inhibitor washout, eIF2α phosphorylation was consistently down-regulated. This suggests that other mechanisms may be responsible for this event.
Pharmacologic or genetic enhancement of eIF2α phosphorylation potentiates bortezomib-induced cell death minimizing the surviving cell fraction
Loss of eIF2α phosphorylation and of its downstream target CHOP in the surviving fraction may be associated with an evasion of apoptosis, as enhanced phosphorylation of this protein can induce cell death (21). Thus, we tested whether sustained eIF2αphosphorylation in the surviving cells would limit their survival after bortezomib treatment. To this end, we used salubrinal, an inhibitor of GADD34-PP1c complex assembly and, therefore, eIF2α-dephosphorylation, identified in a screen for small molecules protecting from ER stress (22). We tested whether salubrinal would enhance bortezomib cytotoxicity acutely and, more importantly, in the fraction of MM cells surviving proteasome inhibitor treatment. At a dose of 5 μmol/L, salubrinal had no effect on the basal viability or growth of RPMI 8226 (Fig. 4_A_) despite inducing eIF2α phosphorylation (Fig. 4_B_). Thus, this salubrinal dose was used for treating MM cells in combination with bortezomib or after proteasome inhibitor treatment. This would reveal if maintaining this phosphorylation could potentiate bortezomib-induced killing in the acute phase and also cause death of the surviving cells. As mentioned above, salubrinal alone resulted in increased phosphorylation of eIF2α at 8 hours. However, proteasome inhibition alone caused even higher p-eIF2α levels than salubrinal (Fig. 4_B_). Combining salubrinal and the proteasome inhibitor resulted in the highest levels of p-eIF2α at 8 hours in RPMI 8226 MM cells, suggesting that this combination can potentiate the activation of this pathway (Fig. 4_B_). Treatment with salubrinal, bortezomib, and the combination of both decreased the activation of PERK marginally whereas no effects could be seen on the activation of GCN2, and PKR was only slightly induced by 10 μmol/L salubrinal (Fig. 4_B_ and Supplementary Fig. S2). This suggests that the observed induction of eIF2α phosphorylation by salubrinal, proteasome inhibitors, and the combination of both is mediated for the most part by pathways other than PERK, GCN2, and PKR (Fig. 4_B_). Combination of MG-132 or bortezomib with salubrinal on RPMI 8226 or U266B1 cells for 12 and 24 hours did not further enhance eIF2α phosphorylation but resulted in pronounced cleavage of eIF2α (not observed at 8 hours). This was reported to depend on caspase-3 activation and is indicative of an irreversible cellular commitment to apoptosis (ref. 23; Fig. 4_C_ and D). Consistent with these findings, acute combination of salubrinal with bortezomib also caused a strong induction of the mRNA for the proapoptotic gene CHOP (Fig. 4_D_right), which suggests that the enhancement of bortezomib-induced apoptosis by salubrinal is due to strong proapoptotoic mediators regulated by this transcription factor (24).
Figure 4.
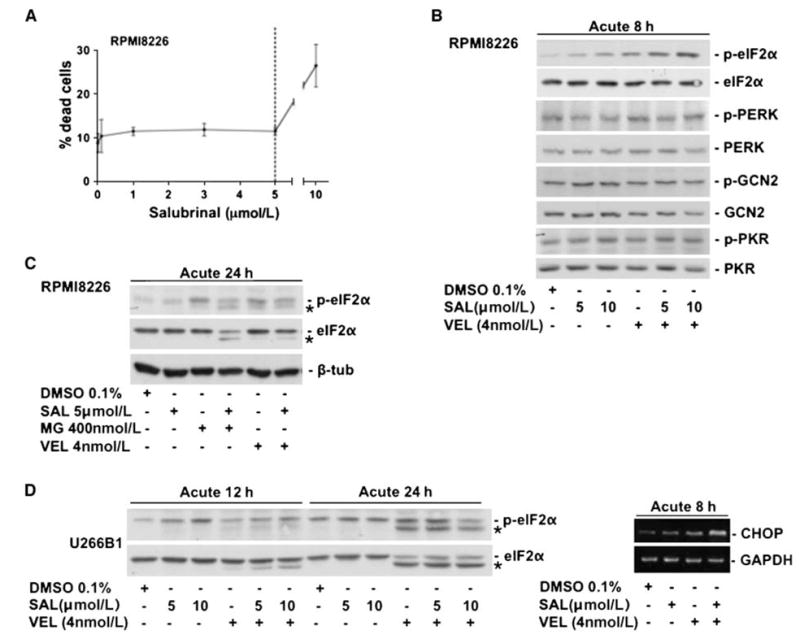
A, viability curve in RPMI 8226 cells in response to salubrinal treatment. Note that 5 μmol/L salubrinal caused no significant induction of cell death (left). B, Western blots for p-eIF2α, eIF2α, p-PERK, PERK, p-GCN2, GCN2, p-PKR, and PKR in RPMI 8226 cells 8 h after treatment with salubrinal, Velcade (VEL), or the combination of both. Note that phosphorylation of eIF2α via salubrinal treatment and Velcade (VEL) is further enhanced by the combination of both drugs at 8 h in RPMI 8226 cells whereas the effect on the upstream kinases PERK, GCN2, and PKR is only marginal. C, Western blots showing that, at 24 h, both MG132 and Velcade (VEL) lead to eIF2α proteolytic processing (*), which was shown to be caspase-3–dependent (23) and is enhanced by the addition of salubrinal. D, Western blots in U266B1 cells showing increased eIF2α phosphorylation with salubrinal, Velcade (VEL), and the combination of both (12 h). Note that, at 12 h, the addition of salubrinal to Velcade (VEL) results in enhanced eIF2α proteolytic processing (*) whereas the effect plateaus at 24 h, wherein similar eIF2α processing with Velcade (VEL) and the Velcade (VEL)/salubrinal combination can be observed (left). RT-PCR showing increased CHOP mRNA levels with 4 nmol/L Velcade (VEL) for 8 h, which are further enhanced by combination of Velcade (VEL) with salubrinal (SAL). GAPDH was used as a loading control (right).
While in both RPMI 8226 and U266B1 cells, MG-132 or bortezomib alone caused roughly 40% to 50% cell death at 24 hours, salubrinal could further augment this effect to roughly 70% to 90% (Fig. 5_A_), which is in agreement with the activation of apoptotic signals. It is unknown if salubrinal can also inhibit other PP1c complexes, but the initial study argues for this compound being highly specific for the GADD34-PP1c complex (22). We cannot rule out that other targets of GADD34-PP1c might contribute to the observed effect. However, our data, obtained by transfecting RPMI 8226 cells with a phosphorylated mimetic eIF2αS51D mutant (25), strongly suggest that the effect is specific for GADD34-PP1c activity on eIF2α (Fig. 5_B_). Transient transfection efficiency in these cells using electroporation is ~50% (data not shown), and cell death was enhanced by around the same magnitude when combining eIF2αS51D overexpression with proteasome inhibition (Fig. 5_B_). Thus, it can be deduced that majority of MM cells expressing the eIF2αS51D mutant were more sensitive to bortezomib.
Finally and most importantly, we tested whether salubrinal treatment of the cells that are arrested but still surviving proteasome inhibition will affect their viability. Treatment of this MM cell population with 5 μmol/L salubrinal, which does not affect the control population, caused ~10-fold reduction in the number of viable cells after 24 hours (Fig. 5_C_). This was comparable with the effect observed using the combination therapy (Fig. 5_C_). The effect could still be observed in quiescent cells 5 days after the bortezomib treatment (Fig. 5_D_). Thus, these cells, while still growth arrested, are in a high ER stress condition, evidenced by the high Grp78 levels, and remain highly sensitive to salubrinal. These results further suggest that the reduction in p-eIF2α observed after bortezomib treatment is indeed a mechanism to evade apoptosis.
Discussion
Exploring mechanisms that might induce quiescence in cancer cells led us to test the hypothesis that therapies known to induce the UPR might be the cause of a protective response in cancer cells (5, 7, 8). We had previously shown that head and neck cancer cells (HNSCC) entered a state of cellular dormancy, which was linked to the activation of ER stress pathways that, in a concomitant fashion, promoted both growth arrest and survival (5, 7, 8). These are the same pathways that can be activated by proteasome inhibitors (1, 2, 14), which led us to reason that proteasome inhibition with MG-132 and the clinically used drug bortezomib (Velcade) might induce quiescence and persistence of cells surviving the therapy.
Several findings are worth discussing. We discovered that, whereas a portion of MM cells treated with proteasome inhibitors die, a significant fraction (30–50% of the cells) could adapt to the stress and survive by entering a protracted state of quiescence. The fact that ~30% to 50% of the cells survive suggests that the G0–G1 arrest is more likely due to treatment adaptation than to selection of a cell population genetically predisposed to undergo quiescence. This response is plausible as one function of ER stress signaling is to allow cells to pause proliferation while unfolding of proteins in the ER is being corrected (6, 26). This ultimately leads to cell survival and, in the context of MM, may allow for recurrence. We cannot rule out that this response may also be operational in MM cells with preexistent mutations that confer resistance to bortezomib (27). If true, this adaptive response, which seems to be functional in a large proportion of cells, may further accelerate the selection of these underrepresented clones. However, such hypothesis needs to be tested.
We also found that survival of the residual quiescent cells hinges on the down-regulation of eIF2α phosphorylation, which allows these cells to silence proapoptotic signals downstream of eIF2α signaling. This was primarily evidenced by the loss of CHOP mRNA expression. The silencing of CHOP might be an important apoptosis escape mechanism, as this transcription factor was shown to repress Bcl-2 expression and at the same time induce expression of BimEL (24), a powerful proapoptotic protein.
First and importantly, we found a potential solution to the problem by combining bortezomib treatment with salubrinal, an inhibitor of GADD34-PP1c complex assembly. Maintaining eIF2α phosphorylation through drugs like salubrinal can potentiate killing of MM cells, and they may serve as an adjuvant therapy to proteasome inhibitors when given simultaneously. Second and more importantly, salubrinal can virtually eliminate the fraction of quiescent MM cells surviving proteasome inhibition. We therefore propose that salubrinal or similar compounds affecting phosphorylation of eIF2α may serve as a potential strategy to maximize proteasome inhibitor efficiency and decrease MM cell resistance to this substance class. The ability of salubrinal to potentiate killing of MM cells by enhancing eIF2α phosphorylation is in accordance with previous models hypothesizing a sigmoidal relationship between stress intensity and eIF2α activation leading to survival at very low and apoptosis at very high levels (9). Thus, the enhancement of eIF2α phosphorylation during MG-132 or bortezomib treatment prevents MM cells to modulate this pathway to adapt to and survive ER stress.
This response was observed in two MM cell lines with different genetic alterations [c-MYC insertion on t(16;22)(q32;q11):der16 in RPMI 8226 and t(11;14) in U266B1], suggesting that it may not be linked to a given genotype. Still, as discussed above, this does not rule out that it may, in the long-term, favor the expansion of clones predisposed to resist bortezomib therapy. We also found that, while diffuse B-cell lymphoma U937 cells were highly sensitive to 4 nmol/L bortezomib and K-562 chronic myeloid leukemia cells were intermediately sensitive, neither of these cell lines were susceptible to an enhancement of bortezomib-induced killing by salubrinal (data not shown). Furthermore, neither bortezomib alone nor its combination with salubrinal were effective in inducing killing of HEp3 HNSCC or Raji Burkitt’s lymphoma cells (data not shown). This suggests that the combination therapy using a proteasome inhibitor and salubrinal may be mostly suited for the treatment of MM.
We hypothesize that the induction of XBP-1 during the acute phase of bortezomib treatment is most likely associated with a survival/adaptation response (5, 15, 28). It is possible that, whereas XBP-1 may function to protect cells during the acute phase, its ability to lead to the prolonged induction of chaperones, such as BiP/grp78, may prevent apoptosis in the cells surviving the treatment. This response may be related to the inherent capacity of the UPR to protect from subsequent insults to the ER (9). Thus, prolonged survival of the quiescent fraction may be linked to the abundant induction of BiP/grp78. Previous studies provided compelling evidence for a prosurvival function for this chaperone in cancer (5, 17–19). Thus, it is possible that, in addition to the inhibition of GADD34-PP1c and enhancement of p-eIF2α signaling, blockade of Grp78 expression and/or function may be an additional strategy to prevent adaptation and survival of MM and other cancer cells to bortezomib treatment. Similar strategies applied to identify HSP90 inhibitors (29, 30) may be successful in identifying small molecule inhibitors specific for Grp78.
Several upstream kinases, such as PERK, PKR, and GCN2, can phosphorylate eIF2α (26). Our study revealed that during the acute phase there was only a marginal modulation of PERK, PKR, and GCN2 activation. Thus, their functional contribution to the growth arrest remains to be elucidated. It is also unclear whether the down-regulation of eIF2α phosphorylation is solely due to GADD34-PP1c expression/activity. Still, our data support that this is the most plausible option. What seems to be evident from our results is that in both acute and post–bortezomib treatments with salubrinal the strong phosphorylation of eIF2α is associated with apoptosis. Studies in HNSCC cells showed that, whereas PERK and eIF2α signaling can contribute to survival, persistent eIF2α signaling is also linked to the quiescent state (5). In the case of MM cells, phosphorylation of eIF2α does not persist posttreatment during the quiescence phase. This suggests that the growth arrest is linked to other mechanisms or that persistent eIF2α phosphorylation is not needed for this effect.
Whereas Rb phosphorylation in sites that are indicative of G1 exit was reduced after proteasome inhibition, we failed to detect significant changes in CDK or cyclin protein levels (data not shown). Only p21 induction was observed in the acute phase but not in the surviving fraction of arrested cells (data not shown). We speculate that this acute induction is sufficient to induce the long-term MM cell arrest and that this or other mechanisms (i.e., p15 and p16 induction) maintain cyclin/CDK complexes inactive for prolonged periods.
In summary, we report that the induction of MM tumor cell quiescence and survival can be an undesirable side effect of proteasome inhibition. We also show that, by blocking eIF2α dephosphorylation, proteasome inhibitor efficiency can be maximized during acute treatment and that residual cells can be eliminated by nontoxic doses of salubrinal as a monotherapy for MM minimal residual disease after proteasome inhibition.
Supplementary Material
Supplementary Figure Legends
Supplementary figures 1-2
Acknowledgments
Grant support: Deutsche Krebshilfe Dr. Mildred-Scheel postdoctoral grant (D.M. Schewe), NIH/National Cancer Institute grant CA109182 (J.A. Aguirre-Ghiso), and a grant from Samuel Waxman Cancer Research Foundation Tumor Dormancy Program.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Chauhan D, Hideshima T, Mitsiades C, Richardson P, Anderson KC. Proteasome inhibitor therapy in multiple myeloma. Mol Cancer Ther. 2005;4:686–92. doi: 10.1158/1535-7163.MCT-04-0338. [DOI] [PubMed] [Google Scholar]
- 2.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chauhan D, Singh A, Brahmandam M, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111:1654–64. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci U S A. 1999;96:8505–10. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, Aguirre-Ghiso JA. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008;68:3260–8. doi: 10.1158/0008-5472.CAN-07-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–95. [PubMed] [Google Scholar]
- 8.Schewe DM, Aguirre-Ghiso JA. ATF6α-rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A. 2008;105:10519–24. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–76. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF, Aguirre-Ghiso JA. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS ONE. 2007;2:e615. doi: 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin D, Zhou H, Kumagai T, et al. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM) Oncogene. 2005;24:344–54. doi: 10.1038/sj.onc.1208225. [DOI] [PubMed] [Google Scholar]
- 12.Papandreou CN, Daliani DD, Nix D, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–21. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 13.Lyons AB, Hasbold J, Hodgkin PD. Flow cytometric analysis of cell division history using dilution of carboxyfluorescein diacetate succinimidyl ester, a stably integrated fluorescent probe. Methods Cell Biol. 2001;63:375–98. doi: 10.1016/s0091-679x(01)63021-8. [DOI] [PubMed] [Google Scholar]
- 14.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695–704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura M, Gotoh T, Okuno Y, et al. Activation of the endoplasmic reticulum stress pathway is associated with survival of myeloma cells. Leuk Lymphoma. 2006;47:531–9. doi: 10.1080/10428190500312196. [DOI] [PubMed] [Google Scholar]
- 16.Navas TA, Nguyen AN, Hideshima T, et al. Inhibition of p38α MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, bcl-X(L), mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia. 2006;20:1017–27. doi: 10.1038/sj.leu.2404200. [DOI] [PubMed] [Google Scholar]
- 17.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 18.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 19.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 20.Zhang K, Kaufman RJ. Chapter twenty identification and characterization of endoplasmic reticulum stress-induced apoptosis in vivo. Methods Enzymol. 2008;442:395–419. doi: 10.1016/S0076-6879(08)01420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cnop M, Ladriere L, Hekerman P, et al. Selective inhibition of eukaryotic translation initiation factor 2 α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J Biol Chem. 2007;282:3989–97. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- 22.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 23.Marissen WE, Guo Y, Thomas AA, Matts RL, Lloyd RE. Identification of caspase 3-mediated cleavage and functional alteration of eukaryotic initiation factor 2α in apoptosis. J Biol Chem. 2000;275:9314–23. doi: 10.1074/jbc.275.13.9314. [DOI] [PubMed] [Google Scholar]
- 24.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 25.Tan S, Somia N, Maher P, Schubert D. Regulation of antioxidant metabolism by translation initiation factor 2α. J Cell Biol. 2001;152:997–1006. doi: 10.1083/jcb.152.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97:12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oerlemans R, Franke NE, Assaraf YG, et al. Molecular basis of bortezomib resistance: proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–99. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 28.Davies MP, Barraclough DL, Stewart C, et al. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int J Cancer. 2008;123:85–8. doi: 10.1002/ijc.23479. [DOI] [PubMed] [Google Scholar]
- 29.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–69. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 30.Davenport EL, Moore HE, Dunlop AS, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends
Supplementary figures 1-2