Patterns of gene expression and copy-number alterations in VHL disease-associated and sporadic clear cell carcinoma of the kidney (original) (raw)
. Author manuscript; available in PMC: 2010 Jun 1.
Abstract
Recent insights into the role of the VHL tumor suppressor gene in hereditary and sporadic clear cell carcinoma of the kidney (ccRCC) have led to new treatments for patients with metastatic ccRCC, although virtually all patients eventually succumb to the disease. We performed an integrated, genome-wide analysis of copy-number changes and gene expression profiles in 90 tumors, including both sporadic and VHL disease-associated tumors, in hopes of identifying new therapeutic targets in ccRCC. We identified 14 regions of nonrandom copy-number change, including 7 regions of amplification (1q, 2q, 5q, 7q, 8q, 12p, and 20q) and 7 regions of deletion (1p, 3p, 4q, 6q, 8p, 9p, and 14q). An analysis aimed at identifying the relevant genes revealed VHL as one of 3 genes in the 3p deletion peak, CDKN2A and CDKN2B as the only genes in the 9p deletion peak, and MYC as the only gene in the 8q amplification peak. An integrated analysis to identify genes in amplification peaks that are consistently overexpressed among amplified samples confirmed MYC as a potential target of 8q amplification and identified candidate oncogenes in the other regions. A comparison of genomic profiles revealed that VHL disease-associated tumors are similar to a subgroup of sporadic tumors, and thus more homogeneous overall. Sporadic tumors without evidence of biallelic VHL inactivation fell into 2 groups: one group with genomic profiles highly dissimilar to the majority of ccRCC, and a second group with genomic profiles that are much more similar to tumors with biallelic inactivation of VHL.
Keywords: Renal cancer, VHL, oncogenes, chromosomal changes, SNP arrays
Introduction
Approximately 54,000 new cases of kidney cancer are diagnosed each year in the United States, with more than one-quarter resulting in death (1). The vast majority of both cases and deaths are represented by clear cell renal cell carcinoma (ccRCC). Until recently, the only effective therapies for patients with metastatic ccRCC have been immunotherapies, which unfortunately have high levels of toxicity and low rates of response (2).
A better understanding of the somatic genetics of ccRCC has led to recent improvements in therapy. The tumor suppressor gene (TSG) VHL, originally identified in families with von-Hippel Lindau (VHL) disease, has been shown to play a major role in the development of both VHL disease-associated and sporadic ccRCC (3-6). Antiangiogenic therapies, which block some of the downstream effects of VHL inactivation, are effective at controlling the growth of metastatic ccRCC in some of patients, but in almost all cases the disease will eventually progress (7-9).
In order to improve the treatments of ccRCC, two major goals should be addressed. First, the underlying heterogeneity of ccRCC that leads to responses in most, but not all, patients needs to be understood (10, 11). This may improve selection of patients for existing therapy. Second, and most importantly, novel therapies are warranted. Given the progress with therapies targeting the VHL pathway, the identification of activated oncogenes and additional inactivated TSGs in ccRCC may provide targets for such novel therapeutics. The current availability of high-throughput platforms to assess genome-wide changes in copy number and gene expression provides an ideal opportunity to achieve these two goals. We performed an integrated analysis of copy-number and expression profiles of ccRCC for both sporadic and VHL-disease associated ccRCC.
Materials and Methods
Renal Cell Carcinoma samples and nucleic acid extraction
De-identified fresh frozen sporadic ccRCC tissues were obtained from the Brigham & Women's Hospital, Beth Israel Deaconess Medical Center and University Hospital in Zurich, Switzerland. De-identified fresh frozen metachronous tumors from patients with VHL disease were obtained from the National Cancer Institute. Genomic DNA was purified by phenol/chloroform extraction and ethanol precipitation. All tumor tissues were needle dissected to ensure at least 70% contribution from tumor cells. In addition, DNA was extracted from 48 paired samples of normal renal cortes and 21 renal cancer cell lines: 769-P, A-704, Caki-2, KC 12, KU 19-20, SK-RC 29, SK-RC 31, SK-RC 38, SK-RC 42, SK-RC 52, SLR 20, SLR 21, SLR 22, SLR 23, SLR 24, SLR 25, SLR 26, SN12-PM6, SW 156, UMRC2, UMRC6.
Total RNA was extracted from 2mm punch biopsies of regions containing at least 70% tumor cells. Frozen tissue fragments were pulverized with a chilled mortar and pestle and homogenized in 1 ml of Trizol reagent (Gibco/BRL). RNA was purified in accordance with the manufacturer's instructions. RNA integrity was assessed by denaturing gel electrophoresis, using the visibility of rRNA bands.
Analysis of VHL
VHL mutational analysis was performed by direct sequencing. Amplification of exons 1-3 (and flanking regions) of VHL was carried out using sets of primers designed with an automated primer selection program (see Supplementary Methods). Bi-directional sequences were generated by the DNA Sequencing Laboratory of the Harvard Partners Genome Center and manually reviewed. The methylation status of the VHL promoter was examined by sodium bisulfite modification and methylation-specific PCR as described previously (12).
Genome-wide copy-number analysis
Genomic DNA was applied to the Sty I (250K) SNP array of the 500K Human Mapping Array set according to manufacturer's instructions (Affymetrix, Inc., Santa Clara, CA). Arrays were scanned using the GeneChip Scanner 3000 7G. Probe-level signal intensities were normalized to a baseline array with median intensity using invariant set normalization (13) and SNP-level signal intensities were obtained using a model-based (PM/MM) method (14). Significant regions of copy-number variation were determined using the Genomic Identification of Significant Targets In Cancer (GISTIC) method (15). SNP, gene, and cytogenetic band locations are based on the hg17 (May 2004) genome build (http://genome.ucsc.edu). Criteria for calling genes oncogenes or TSGs included published evidence of somatic genetic alterations in tumors and functional data supporting the tumorigenic consequences of these alterations. For the comparison between the Euclidean distance distributions among profiles of different subgroups, p-values were calculated by permuting group labels.
Gene expression analysis
RNA was hybridized to HT_HG-U133A High-Throughput Arrays according to the manufacturer's instructions (Affymetrix, Santa Clara, CA) and scanned using the HT Scanner. Signal intensities were determined using RMA (16). Genes were assessed for overexpression among amplified samples or underexpression among deleted samples, compared to normal cortex, by the signal to noise ratio. P-values were calculated by permuting tumor labels. Hierarchical clustering was performed using pairwise complete linkage (WPGMA) on all genes and Euclidean distance as the similarity measure. Gene Set Enrichment Analysis was performed as described (17) with p-values calculated by permuting tag labels.
All SNP and expression array data are available through the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.gov/projects/geo (accession no.GSE14994).
Results
Copy-number profiles from 90 clear cell RCCs
We performed a global survey of amplifications and deletions in 90 ccRCC tumors using oligonucleotide arrays interrogating 238,304 single nucleotide polymorphisms (SNPs) throughout the genome. The dataset comprised 54 sporadic cases (49 primary tumors and 5 metastases) and 36 metachronous tumors from 12 patients with VHL disease (all primaries). All cases were reviewed by a pathologist (S.S.) with expertise in kidney cancer. Tumor cells were enriched by microdissection to ensure >70% purity in each sample. In addition, copy-number changes were assessed for 21 renal cancer cell lines (see Methods).
The ccRCC genome appears complex, with every region of the genome amplified or deleted in at least one tumor (Figure 1). However, the genomes of individual tumors are much simpler, with an average of 5.8 amplifications and 6.8 deletions. The majority of these events are broad gains and losses: 55% of amplifications and 52% of deletions cover most of a chromosome arm. Moreover, recurrent events, such as amplifications of chromosome (chr) 5q and deletions of chr 3p can be readily observed, as noted in prior cytogenetic studies (18, 19). Not surprisingly, the cell lines exhibited many more events, and many more focal events, than the tumors (Figure 1). For this reason, the cell line data were not included in any further analyses below.
Figure 1.
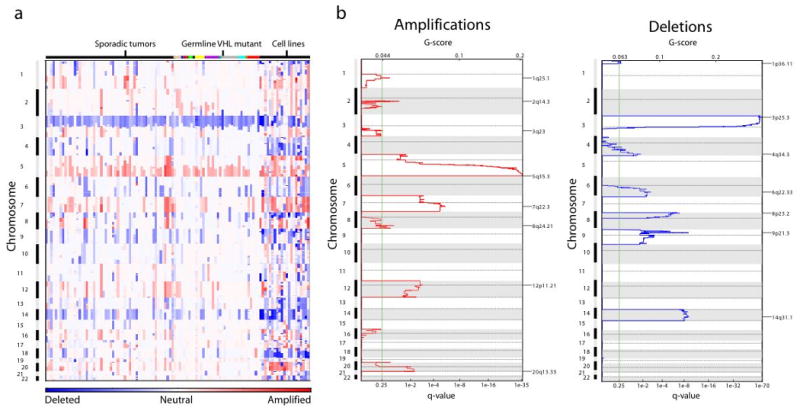
Significant copy-number alterations in ccRCC. (a) Amplifications (red) and deletions (blue), determined by segmentation analysis of normalized signal intensities from 250K SNP arrays (see Methods), are displayed across the genome (chromosome positions, indicated along the y axis, are proportional to marker density) for 54 sporadic tumors, 36 metachronous tumors from 12 VHL patients, and 18 renal cancer cell lines. Metachronous tumors from the same patient are designated by the same color across the top. (b) GISTIC analysis of copy-number changes in ccRCC tumors. The G-score represents the frequency × average amplitude of the aberrations identified in (a). The FDR q-values, representing the statistical significance associated with these scores with correction for multiple hypothesis testing(20), are displayed along the bottom of each panel. Regions with q-values < 0.25 (green lines) were considered significantly altered. Chromosome positions are indicated along the y axis with centromere positions indicated by dotted lines. The locations of the peak regions of maximal copy-number change are indicated to the right of each panel.
Metachronous tumors from the same VHL patient appear no more similar to each other than to tumors from other VHL patients. As can be seen in Figure 1 and Supplementary Figure 1, such metachronous tumors exhibit very different copy-number profiles. We compared each pair of tumors in our dataset using a Euclidean distance metric that takes into account all copy-number differences across the genome. The average distance between tumors in the same patient was 59, not significantly smaller than the average distance between tumors from different VHL patients (61, one-sided p = 0.48). This observation strongly suggests that metachronous tumors from the same patient are not clonal, and therefore do not represent metastatic spread from a common precursor.
Analysis of non-random copy-number events suggests candidate oncogene and TSG targets
We applied the statistical method Genomic Identification of Significant Targets In Cancer (GISTIC) to distinguish non-random copy-number events that may play a role in tumorigenesis (15). Briefly, for both amplifications and deletions, the GISTIC method scores each region according to its combined frequency and amplitude of copy-number changes, and compares these scores to the distribution expected if these copy-number changes resulted from chance alone. The method takes into account multiple hypotheses by applying the False Discovery Rate framework (20), to generate q-values that represent the likelihood each region was affected only by such random events. We considered all events with q-values less than 0.25 to be statistically significant.
We identified 7 significant amplifications and 7 significant deletions in ccRCC (Figure 1b, Table 1). As anticipated, the most significant events are chr 3p loss and chr 5q gain. In addition, we see significant losses of chromosomes 1p, 4q, 6q, 8p, 9p, and 14q, and significant gains of 1q, 2q, 7q, 8q, 12p, and 20q.
Table 1. Peak regions of amplification and deletion.
| Amplification | Cytoband* | Boundaries of peak* | FDR q value | Frequency | # of genes in peak | # of genes in wide peak | Oncogene or TSG in region |
|---|---|---|---|---|---|---|---|
| 1 | 1q25.1 | 172.16-172.25 | 0.14 | 13% | 0 | 45 | |
| 2 | 2q14.3 | 123.81-124.13 | 0.05 | 13% | 0 | 101 | |
| 3 | 5q35.3 | 178.59-180.85 | 1e-35 | 69% | 22 | 62 | |
| 4 | 7q22.3 | 105.27-105.85 | 4e-6 | 30% | 2 | 24 | |
| 5 | 8q24.21 | 128.18-129.00 | 0.09 | 12% | 1 | 38 | MYC |
| 6 | 12p11.21 | 30.84-32.11 | 1e-3 | 24% | 3 | 150 | |
| 7 | 20q13.33 | 61.03-62.44 | 7e-3 | 20% | 27 | 136 | |
| Deletion | |||||||
| 1 | 1p36.11 | 21.42-26.21 | 0.23 | 18% | 51 | 97 | RUNX3 |
| 2 | 3p25.3 | 10.14-10.28 | 1e-69 | 94% | 2 | 24 | VHL |
| 3 | 4q34.3 | 178.38-179.04 | 0.02 | 22% | 1 | 2 | |
| 4 | 6q22.33 | 129.81-129.88 | 3e-3 | 23% | 1 | 53 | |
| 5 | 8p23.2 | 3.06-3.13 | 1e-7 | 32% | 1 | 5 | |
| 6 | 9p21.3 | 21.98-22.01 | 1e-9 | 29% | 2 | 2 | CDKN2A/B |
| 7 | 14q31.1 | 78.81-80.32 | 1e-9 | 42% | 3 | 3 |
Only one gene, VHL, is known to cause ccRCC tumorigenesis through its somatic genetic modification, including its deletion (5). The identification of 13 additional nonrandom copy-number events in our dataset suggests that at least 13 oncogenes and TSGs are targeted by these events and remain to be discovered. For each observed event, we considered genes in the region with the minimum q-value (the peak region), which have the most frequent and high-level aberrations, to be the most likely to contain these gene targets (15) (Table 1 and Supplementary Table 1). This approach does in fact recover VHL as one of 2 genes within the peak region of deletion in chr 3p. This result can be ascribed in part to the presence of a homozygous VHL deletion in a patient with germline hemizygous VHL loss.
In addition to VHL, two other peak regions appear to precisely identify genes that are known to play a role in cancer, though not in ccRCC. In the case of deletions of chr 9p, the presence of focal, homozygous deletions in 2 samples leads the peak region to contain only 2 genes, the known TSGs CDKN2A and CDKN2B. In the case of amplifications of chr 8q, the presence of a focal amplification in a single sample leads the peak region to contain only the known oncogene MYC.
In addition to these 3 peaks, 11 peak regions do not precisely identify genes known to play a role in cancer. In 8 instances, these peak regions each identify fewer than 4 genes, including 3 peaks (deletions of chrs 4q, 6q, and 8p) each containing only 1 gene. This gene is CSMD1 in the case of chr 8p loss. Although functional data demonstrating its cancer-related role are lacking, recurrent deletion of CSMD1 has also been observed in several other tumor types (21). The candidate TSG targets for losses of chr 4q and 6q (AGA and LAMA2 respectively) are poorly characterized in cancer and their role in tumorigenesis remains unknown. The other 3 peak regions (amplification of chrs 5q and 20q and deletions of 1p) each contain more than 20 genes. The deletion peak on chr 1p includes RUNX3, which has been shown to suffer hemizygous loss and hypermethylation in a variety of cancers (22). Functional data, including its genetic inactivation in mice, supports its role as a TSG (23). For all these peak regions, further investigation of the potential gene targets is warranted.
Although these peak regions are the most likely locations of the oncogene and TSG targets of the copy-number changes described above, it is possible that some of them have been displaced by the presence of passenger amplification events that occur next to, but not overlapping, these gene targets. This may be the case for the peak regions of amplification on 1q and 2q, which do not contain any described genes. We therefore accounted for such possible displacements by performing a leave-one-out analysis as previously described (15), in which we identified “wide peak” regions whose boundaries are robust to the removal of any one sample from the dataset (Table 1 and Supplementary Table 1). As expected, these wide peak regions contain many more genes: in the cases of 1q and 2q amplifications, 45 and 101 genes respectively.
Integrated analysis of expression profiles to rank candidate oncogenes and TSGs
Under the assumption that the oncogene targets of these copy-number events are activated by overexpression, we performed an integrated analysis of copy-number and expression data to prioritize the candidate genes in the peak regions of amplification above. Specifically, we obtained genome-wide expression data for 59 of the tumors undergoing SNP array analysis and identified genes consistently overexpressed in amplified tumors relative to normal kidney cortex, using a signal-to-noise ratio metric and calculating p-values by permuting tumor labels. We performed this comparison rather than comparing to tumors without the amplification because we recognized that oncogene expression levels may be modulated in unamplified tumors by factors other than copy-number change.
Among the 47 genes in the peak regions for which probes exist on the expression arrays, 36 are overexpressed among amplified samples. For 23 of these genes, the overexpression among amplified samples was sufficiently consistent as to be statistically significant (p<0.05). These significantly overexpressed genes were found in 5 peak regions: 5q, 7q, 8q, 12p, and 20q (Table 2a). The observation that MYC was consistently overexpressed among tumors with 8q24 amplification relative to normal controls (p < 0.005) suggests these genes may include the target oncogenes in the other peak regions.
Table 2. Overexpressed and underexpressed genes in peak regions of amplification and deletion respectively.
| A) Amplifications | ||
|---|---|---|
| Peak Region | Overexpressed Gene | p-value* |
| 5q | GNB2L1 | <0.0001 |
| MGAT1 | <0.0001 | |
| RUFY1 | <0.0001 | |
| RNF130 | <0.0001 | |
| MAPK9 | <0.0001 | |
| CANX | <0.0001 | |
| CNOT6 | 0.0002 | |
| SQSTM1 | 0.0011 | |
| LTC4S | 0.0029 | |
| TBC1D9B | 0.0092 | |
| HNRPH1 | 0.0373 | |
| FLT4 | 0.0449 | |
| 7q | PBEF1 | <0.0001 |
| 8q | MYC | 0.003 |
| 12p | C12orf35 | <0.0001 |
| 20q | RGS19 | <0.0001 |
| TPD52L2 | 0.0002 | |
| TNFRSF6B | 0.0006 | |
| C20orf11 | 0.0014 | |
| PRPF6 | 0.0104 | |
| BIRC7 | 0.0114 | |
| RTEL1 | 0.0305 | |
| SOX18 | 0.0396 | |
| B) Deletions | ||
| Peak Region | Underexpressed Gene | p-value* |
| 1p | C1QA | <0.0001 |
| RPL11 | <0.0001 | |
| C1QB | 0.0001 | |
| RUNX3 | 0.0003 | |
| HNRPR | 0.0005 | |
| HSPG2 | 0.0029 | |
| IL22RA1 | 0.0209 | |
| TMEM50A | 0.0329 | |
| 4q | NEIL3 | 0.0286 |
| AGA | 0.0297 | |
| 9p | CDKN2A | 0.0002 |
| 14q | NRXN3 | 0.0242 |
A similar analysis of underexpressed genes in deleted regions identifies the known TSG CDKN2A (Table 2b), but not VHL or CDKN2B. This result may reflect one of at least two factors. First, decreases in expression are more difficult to detect than increases in expression by expression arrays, due to noise and high background signal intensities. Second, many deletion events may be expected to inactivate TSGs by leading to loss of heterozygosity in the setting of a mutation in the remaining allele. The overall expression level may not be significantly affected due to overexpression of the remaining (mutated) allele, as previously observed with TP53 (24). For these reasons, prior studies have focused on identifying overexpressed genes in amplified regions (25).
As the peak regions of amplification on 1q and 2q contain no genes (Table 1), the overexpressed genes in the wide peak regions may represent oncogene targets. Therefore, we integrated gene expression data for all the genes in these wide peak regions, and identified 12 genes in the 1q wide peak and 34 genes in the 2q wide peak that are overexpressed (Supplementary Table 1a). Similarly, the wide peak regions for the other significant amplification events contain many significantly overexpressed genes (Supplementary Table 1a), including the genes in the peak regions that were previously identified as most likely to include the oncogene targets (Table 2a). We also performed a similar analysis for deleted regions (Supplementary Table 1b).
Tumors from VHL patients are more homogeneous than sporadic tumors
The presence of tumors from VHL patients (germVHL) in our dataset allowed us to assess the impact of early biallelic inactivation of VHL on the genomic profile of these tumors. Biallelic inactivation of VHL is known to occur in preneoplastic renal lesions in VHL patients (26), but the timing of biallelic VHL inactivation within the sequence of events leading to tumorigenesis in sporadic ccRCC is not known. In addition, a subset of sporadic tumors does not have biallelic VHL inactivation.
We first evaluated the overall level of genetic similarity between germVHL and sporadic tumors by comparing their SNP array profiles. To determine the level of similarity between germVHL and sporadic tumors, we compared the average Euclidean distance amongst tumors within each subset to the average distance between tumors in different subsets. This average distance is significantly smaller amongst germVHL than sporadic tumors (61 vs 89, p < 0.001), suggesting the germVHL tumors constitute a more homogenous group. Although the germVHL tumors tend to have a lower grade than sporadic tumors (p = 0.012), this lower average grade does not appear to explain the level of homogeneity between these tumors. Indeed, the average distance amongst germVHL tumors continues to be smaller than amongst sporadic tumors even after controlling for grade (p = 0.003; see Methods). In addition, the average distance between germVHL and sporadic tumors (77) is smaller than the average distance amongst the sporadic tumors themselves (89), suggesting the copy-number space taken up by these two groups largely overlaps. These interpretations are consistent with the appearance of the spaces occupied by these tumor subsets when mapped to a two-dimensional plane (Figure 2a; see Methods) and imply that sporadic tumors have a more heterogeneous copy-number profile.
Figure 2.
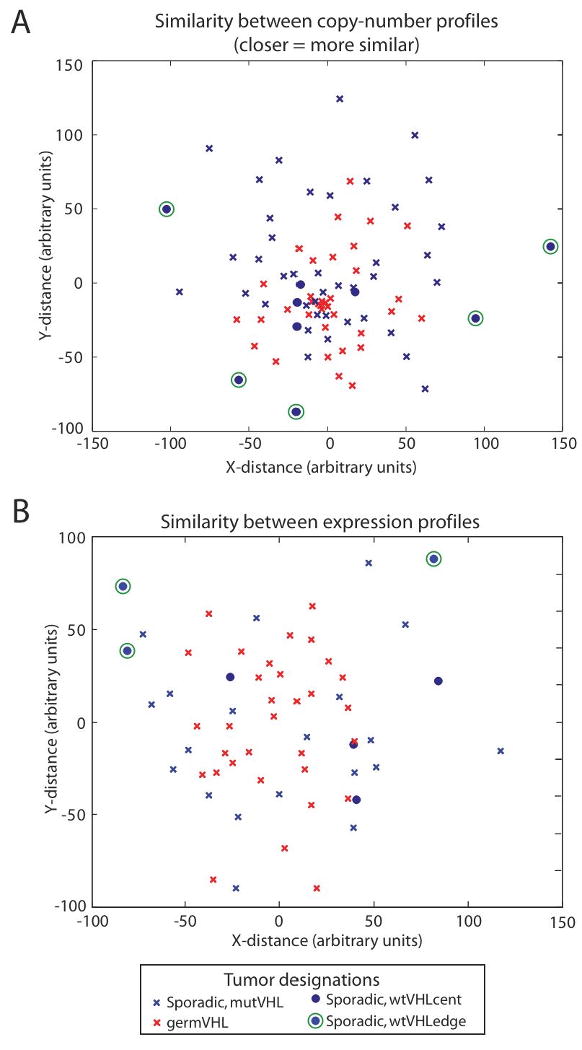
Graphic representation of level of similarity between (a) copy-number and (b) expression profiles of ccRCC samples. Individual tumors, represented in red (germVHL) and blue (sporadic) have been mapped to a 2-dimensional plane to display the Euclidean distances among them. (see Methods). Tumors with biallelic inactivation of VHL (germVHL and mutVHL) are represented by crosses, while those without biallelic inactivation (wtVHL) are represented by dots. Five of the wtVHL tumors, distributed along the periphery of the copy-number space occupied by the tumor set (a), are marked by green circles. The three of these with expression profiles are similarly marked in (b).
The relative homogeneity among germVHL tumors in copy-number space is also observed in gene expression space. The average Euclidean distance amongst expression profiles of germVHL tumors is significantly smaller than the average distance amongst sporadic tumors (75 vs 97, p = 0.001). As seen with their copy-number profiles, the average distance in gene expression space between germVHL and sporadic tumors (89) is smaller than the distance amongst the sporadic tumors themselves (97), consistent with the largely overlapping spaces occupied by these tumor subsets when mapped to a two-dimensional plane (Figure 2b). In both their copy-number and expression profiles, therefore, it appears that germVHL tumors are similar to a subset of sporadic tumors, and therefore lack the same level of heterogeneity overall.
Sporadic tumors exhibit more copy-number events than tumors from VHL patients
The increased level of homogeneity among germVHL tumors reflects the smaller number of copy-number events they exhibit. These tumors have, on average, 4.0 amplifications as compared to 7.0 in sporadic tumors (p = 0.04) and 5.1 deletions as compared to 7.9 deletions in sporadic tumors (p = 0.03). Similar to sporadic tumors, copy-number events in VHLgerm tumors are also typically broad, with 49% of amplifications and 53% of deletions covering the majority of a chromosome arm.
In accordance with the higher average number of alterations in sporadic tumors as compared to germVHL tumors, several of the nonrandom events identified in the GISTIC analysis are also more prevalent among the sporadic tumors. These include amplifications of chr 1q, 7q, and 20q, and deletions of 1p, 4q, and 9p (Supplementary Table 2). To optimize our power to detect nonrandom events that might be restricted to either the sporadic or germVHL tumors, we performed GISTIC independently on each subgroup. This analysis identified an additional significant amplification, of chr 3q28, in the sporadic tumors while no additional significant events were identified in the germVHL subgroup. Amplification of 3q28 was observed in 11/54 (20%) of sporadic tumors, and the boundaries of the peak region (from 188.6-199.5 Mb) include over 50 genes. A focused study of larger numbers of sporadic tumors may be able to limit this region further.
Tumors without biallelic VHL inactivation separate into two groups
We assessed biallelic inactivation of VHL in our sporadic tumors by combined assessment of VHL copy loss and mutation and methylation status (see Methods). We identified mutation events in 35 of the 54 tumors (65%) (Supplementary Table 3). VHL promoter methylation was assessed in 50 tumors and detected in 9 (17%). Three tumors (6%) exhibited both mutation and methylation of VHL. Overall, 41 of the 50 tumors (82%) assessed for both events were observed to have either mutation or methylation of VHL. Loss of chr 3p was observed in 40 of these tumors (mutVHL), for an overall rate of biallelic VHL loss of 80%.
As shown in Figure 2a, tumors without biallelic inactivation (wtVHL) tend to separate into 2 groups: 1) a set (wtVHLcent; represented by blue dots) with copy-number profiles similar to those of the majority of sporadic and VHL disease-associated tumors with biallelic VHL inactivation (seen as residing near the center of the space taken up by the other tumors), and 2) a set of tumors (wtVHLedge; blue dots with green circles) with copy-number profiles that are among the most dissimilar to the other tumors in the dataset (seen as residing at the edges of the space taken up by these tumors). To confirm this finding, we calculated the average distance between the wtVHL tumors and those with biallelic inactivation of VHL. Indeed, unsupervised hierarchical clustering of these distances separated these wtVHL tumors into 2 groups (Supplementary Figure 2a). Although we see that some tumors with biallelic inactivation of VHL also lie at the edge of Figure 2a, the number of “edge” tumors appeared to be enriched among the wtVHL group (p = 0.0002; see Supplementary Methods). As expected, inspection of the copy-number profiles reveals a greater number and diversity of copy-number events in the wtVHLedge tumors (data not shown). In addition, wtVHLedge tumors exhibit chr 3p loss in 3 of 5 samples, as compared to 4 of 4 samples in wtVHLcent tumors. We were able to obtain gene expression profiles for 3 of the 5 wtVHLedge tumors and all 4 wtVHLcent tumors. As shown in Figure 2b, the wtVHL tumors appear to maintain a similar distribution in gene expression space, with the wtVHLedge tumors remaining at the periphery and the wtVHLcenter tumors localized more centrally. Although these results are limited by the small number of wtVHL tumors, these results suggest that wtVHL tumors comprise pathogenetically distinct groups of tumors. Similarly, unsupervised hierarchical clustering of the entire tumor set based upon the expression profiles places the wtVHLedge samples in a separate cluster from wtVHLcenter, mutVHL, and germVHL (Supplementary Figure 2b), whereas the tumors in these latter 3 groups were interspersed.
Because the wtVHLcent tumors have similar copy-number and expression profiles to tumors with biallelic inactivation of VHL, wherease wtVHLedge tumors are more distinct, we hypothesized that the former maintain cryptic biallelic inactivation of VHL or lesions of other genes that lead to similar inactivation of the VHL pathway, whereas the latter do not rely on similar levels of VHL pathway inactivation. To test this hypothesis, we performed Gene Set Enrichment Analysis (17) to compare expression of HIF-1 targets among the 4 groups defined above (Figure 3). We first found that expression of these genes is significantly higher in tumors with biallelic VHL inactivation (mutVHL and germVHL) as compared to wtVHL (p < 0.01). Strikingly, we also found heterogeneity within the wtVHL group, with far lower levels of expression of these genes among wtVHLedge tumors relative to wtVHLcent tumors (p < 0.01) (Figure 3). This result was validated at the protein level by immunohistochemical evaluation of HIF1A, HIF2A, and CA9, the single HIF-1 target most differentially expressed between wtVHL and tumors with biallelic VHL inactivation (data not shown). This heterogeneity among the wtVHL tumors suggested that wtVHLcent tumors might have similar inactivation of the VHL pathway as tumors with biallelic VHL inactivation. Indeed, although wtVHLcent tumors exhibit lower expression levels for VHL pathway members than do tumors with biallelic VHL inactivation (p < 0.01), inspection of these expression levels reveals that these differences are modest (Figure 3).
Figure 3.
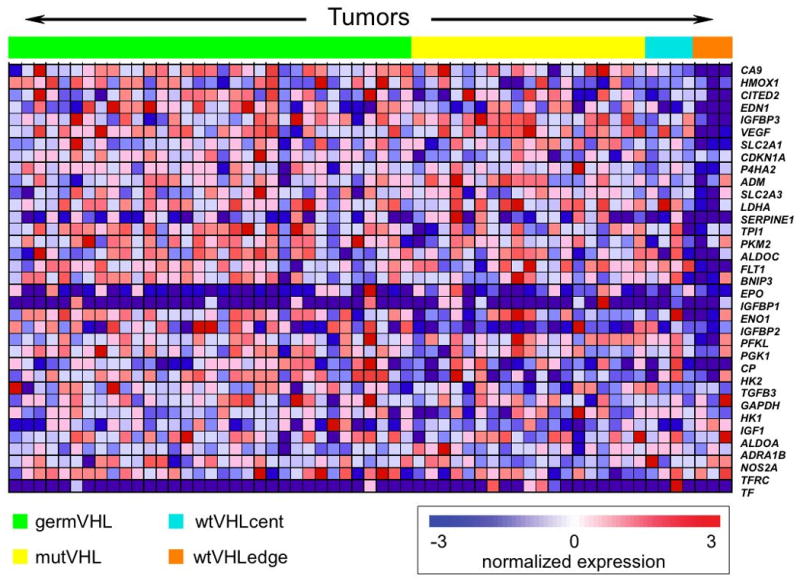
Heat map representing expression levels of HIF-1 targets in ccRCC samples. Tumors (across the top) are divided into germVHL, mutVHL, wtVHLcent, and wtVHLedge groups. Genes (along the right) are ordered from top to bottom according to the degree to which they are differentially expressed between tumors with and without biallelic inactivation of VHL.
The finding that wtVHLedge tumors have such markedly distinct genomic profiles to the vast majority of ccRCC raised the possibility that they represent tumors misclassified as ccRCC. However, all 3 of these tumors exhibit loss of 3p, which is pathognomonic of ccRCC and none have copy-number changes characteristic of other known subtypes of kidney cancer (data not shown).
Discussion
By taking an unbiased, genome-wide approach we have identified 7 regions of the genome that are amplified and 7 that are deleted significantly more often than the background rate across all ccRCC, and an additional region of amplification that is significant only in sporadic ccRCC. The enrichment of these events over the background rate suggests they drive the development and progression of the disease, therefore suggesting the existence of at least 8 amplified oncogenes and 7 deleted TSGs in ccRCC. In addition to recovering VHL as the likely target of 3p loss, we identified MYC as the most likely target of 8q amplicons, and CDKN2A and CDKN2B as the most likely targets of 9p deletions. Although both 8q amplification and 9p loss have been reported previously (19, 27-30), our ability, through an unbiased approach, to precisely locate the likely targets to well-known cancer genes confirms the utility of the method. In addition, further study of the functional relevance of these genes in ccRCC is needed.
An additional region of particular interest is 5q, subject to amplification in ∼70% of tumors. Despite the large number of tumors with the amplification, we find a large region, containing 22 genes, is consistently amplified across all of these samples. Twelve of these genes are also consistently overexpressed among amplified samples relative to normal renal cortex. The consistency with which such a large number of genes are amplified and overexpressed suggests the possibility that more than one gene is the target. For this reason, functional validation of all of these candidate genes is warranted. In addition, copy-number profiles of more tumors should be obtained in an attempt to resolve the minimal region of amplification and more robustly identify of the target(s).
Patients with VHL disease are prone to develop metachronous renal tumors (31, 32). The overall predisposition of these patients to ccRCC and the good prognosis associated with these tumors suggest that they represent separate primaries rather than metastatic spread. In agreement with these observations and a prior study (33), we find that these metachronous tumors are not clonal and therefore developed independently.
The identification of VHL as a TSG in ccRCC from patients with VHL disease led to the recognition of its TSG role in sporadic ccRCC (3). However, whether tumors from patients with VHL disease share additional cooperating mutations with sporadic tumors is not known. By comparing high-resolution copy-number profiles between these VHL disease-associated and sporadic tumors, we have found that their overall profiles are similar, except that VHL disease-associated tumors tend to be more homogenous and have fewer events per tumor, including both likely random (passenger) and nonrandom (driver) events. This smaller number of events may result from at least 3 different reasons. First, the more intense surveillance these VHL patients received enabled their tumors to be diagnosed when they were small, potentially before they acquired additional events. Second, the early biallelic inactivation of VHL documented in patients with VHL disease may occur later in some sporadic tumors, leading to a different set of required coordinating events. Third, because sporadic tumors require an additional driver event (VHL mutation) that for VHL patients is already present in the germline, VHL patients develop tumors in a shorter time and the tumors acquire fewer passenger events before transformation.
Similar to their somatic genetic profiles, the expression profiles of ccRCC from patients with VHL disease are similar to sporadic ccRCC, but more homogenous, suggesting a similar cell of origin and set of dysregulated pathways. Indeed, unsupervised hierarchical clustering fails to distinguish the two groups. The finding of greater heterogeneity among sporadic ccRCC may, in part, result from a lack of biallelic inactivation of VHL, known to occur in a subset of sporadic tumors (34). Indeed, we were unable to identify biallelic inactivation of VHL in ∼20% of sporadic tumors. Surprisingly, while a subset of these tumors have little similarity to the overall ccRCC profile, we found another subset have copy-number and gene expression profiles that overlap with ccRCC with biallelic VHL inactivation. When we specifically assess genes downstream from VHL, we find a clear difference in expression levels between tumors with and without biallelic VHL inactivation. However, this difference was much more pronounced in the subset of tumors with dissimilar copy-number and expression profiles.
The finding that a subset of tumors without biallelic inactivation of VHL has copy-number and expression profiles that are highly dissimilar to the overwhelming majority of ccRCC including expression levels of _HIF_-responsive genes, suggests that these represent a separate group of ccRCC that is not likely to respond to treatments that target the VHL pathway. Therefore, these tumors might require alternative therapeutic strategies. In contrast, the observation that a subset of the tumors without biallelic VHL inactivation has similar copy-number and expression profiles to tumors with such inactivation raises the possibility that the former group have unobserved genetic alterations of the VHL pathway and may respond to treatments targeting this pathway. In this scenario, assessment of the expression of appropriate markers of VHL pathway dysregulation (e.g. CA9) may be more useful in predicting response to therapy than assessment of VHL inactivation status.
Supplementary Material
Supp Fig 1
Supp Fig 2
Supp Methods
Supp Table 1
Supp Table 2
Supp Table 3
Acknowledgments
This work was supported by grants from NIH (Dana-Farber/Harvard Cancer Center Kidney Cancer SPORE and U54CA112962) and Department of Defense (PC050266) and an Award from Istituto Dermopatico dell'Immacolata, Italy, to S.S.; grants from NIH (Dana-Farber/Harvard Cancer Center Prostate Cancer SPORE and K08CA122833) and Department of Defense (PC040638 and PC061642) to R.B.; grants from the Swiss National Science Foundation 3238B0-103145, Sasella Stiftung, Switzerland and from the Zurich Cancer League, Switzerland to H.M.; and by the Doris Duke Charitable Foundation. W.G.K. is recipient of a Doris Duke Clinical Scientist Development Award. A.D.N. is recipient of a Fellowship Award from the Achille Lattuca Foundation, Italy.
References
- 1.http://seer.cancer.gov/csr/1975_2005/, based on November 2007 SEER data submission, posted to the SEER web site, 2008
- 2.McDermott DF, Rini BI. Immunotherapy for metastatic renal cell carcinoma. BJU Int. 2007;99:1282–8. doi: 10.1111/j.1464-410X.2007.06818.x. [DOI] [PubMed] [Google Scholar]
- 3.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 4.Kaelin WG., Jr The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin Cancer Res. 2004;10:6290S–5S. doi: 10.1158/1078-0432.CCR-sup-040025. [DOI] [PubMed] [Google Scholar]
- 5.Linehan WM, Grubb RL, Coleman JA, Zbar B, Walther MM. The genetic basis of cancer of kidney cancer: implications for gene-specific clinical management. BJU Int. 2005;95 2:2–7. doi: 10.1111/j.1464-410X.2005.05189.x. [DOI] [PubMed] [Google Scholar]
- 6.Linehan WM, Lerman MI, Zbar B. Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer Jama. 1995;273:564–70. [PubMed] [Google Scholar]
- 7.Clark PE, Cookson MS. The von Hippel-Lindau gene: turning discovery into therapy. Cancer. 2008;113:1768–78. doi: 10.1002/cncr.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rini BI, Flaherty K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol Oncol. 2008;26:543–9. doi: 10.1016/j.urolonc.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Srinivasan R, Linehan WM. Antiangiogenic therapy in renal cell carcinoma: from concept to reality. Nat Clin Pract Urol. 2007;4:74–5. doi: 10.1038/ncpuro0705. [DOI] [PubMed] [Google Scholar]
- 10.Choueiri TK, Vaziri SA, Jaeger E, et al. von Hippel-Lindau gene status and response to vascular endothelial growth factor targeted therapy for metastatic clear cell renal cell carcinoma. J Urol. 2008;180:860–5. doi: 10.1016/j.juro.2008.05.015. discussion 5-6. [DOI] [PubMed] [Google Scholar]
- 11.Signoretti S, Bratslavsky G, Waldman FM, et al. Tissue-based research in kidney cancer: current challenges and future directions. Clin Cancer Res. 2008;14:3699–705. doi: 10.1158/1078-0432.CCR-07-4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-8-research0032. RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovacs G, Akhtar M, Beckwith BJ, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997;183:131–3. doi: 10.1002/(SICI)1096-9896(199710)183:2<131::AID-PATH931>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 19.Dal Cin P. Genetics in renal cell carcinoma. Curr Opin Urol. 2003;13:463–6. doi: 10.1097/00042307-200311000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc, Ser B. 1995;57:289–300. [Google Scholar]
- 21.Sun PC, Uppaluri R, Schmidt AP, et al. Transcript map of the 8p23 putative tumor suppressor region. Genomics. 2001;75:17–25. doi: 10.1006/geno.2001.6587. [DOI] [PubMed] [Google Scholar]
- 22.Vogiatzi P, De Falco G, Claudio PP, Giordano A. How does the human RUNX3 gene induce apoptosis in gastric cancer? Latest data, reflections and reactions. Cancer Biol Ther. 2006;5:371–4. doi: 10.4161/cbt.5.4.2748. [DOI] [PubMed] [Google Scholar]
- 23.Li QL, Ito K, Sakakura C, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–24. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 24.Thompson AM, Steel CM, Chetty U, et al. p53 gene mRNA expression and chromosome 17p allele loss in breast cancer. Br J Cancer. 1990;61:74–8. doi: 10.1038/bjc.1990.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrasco DR, Tonon G, Huang Y, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–25. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Mandriota SJ, Turner KJ, Davies DR, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–68. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 27.Gronwald J, Storkel S, Holtgreve-Grez H, et al. Comparison of DNA gains and losses in primary renal clear cell carcinomas and metastatic sites: importance of 1q and 3p copy number changes in metastatic events. Cancer Res. 1997;57:481–7. [PubMed] [Google Scholar]
- 28.Junker K, Weirich G, Amin MB, Moravek P, Hindermann W, Schubert J. Genetic subtyping of renal cell carcinoma by comparative genomic hybridization. Recent Results Cancer Res. 2003;162:169–75. doi: 10.1007/978-3-642-59349-9_15. [DOI] [PubMed] [Google Scholar]
- 29.Moch H, Presti JC, Jr, Sauter G, et al. Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res. 1996;56:27–30. [PubMed] [Google Scholar]
- 30.Schraml P, Struckmann K, Bednar R, et al. CDKNA2A mutation analysis, protein expression, and deletion mapping of chromosome 9p in conventional clear-cell renal carcinomas: evidence for a second tumor suppressor gene proximal to CDKN2A. Am J Pathol. 2001;158:593–601. doi: 10.1016/s0002-9440(10)64001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 32.Vira MA, Novakovic KR, Pinto PA, Linehan WM. Genetic basis of kidney cancer: a model for developing molecular-targeted therapies. BJU Int. 2007;99:1223–9. doi: 10.1111/j.1464-410X.2007.06814.x. [DOI] [PubMed] [Google Scholar]
- 33.Phillips JL, Ghadimi BM, Wangsa D, et al. Molecular cytogenetic characterization of early and late renal cell carcinomas in von Hippel-Lindau disease. Genes Chromosomes Cancer. 2001;31:1–9. doi: 10.1002/gcc.1111. [DOI] [PubMed] [Google Scholar]
- 34.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Fig 1
Supp Fig 2
Supp Methods
Supp Table 1
Supp Table 2
Supp Table 3