Structure of mammalian protein geranylgeranyltransferase type-I (original) (raw)
Abstract
Protein geranylgeranyltransferase type-I (GGTase-I), one of two CaaX prenyltransferases, is an essential enzyme in eukaryotes. GGTase-I catalyzes C-terminal lipidation of >100 proteins, including many GTP- binding regulatory proteins. We present the first structural information for mammalian GGTase-I, including a series of substrate and product complexes that delineate the path of the chemical reaction. These structures reveal that all protein prenyltransferases share a common reaction mechanism and identify specific residues that play a dominant role in determining prenyl group specificity. This hypothesis was confirmed by converting farnesyltransferase (15-C prenyl substrate) into GGTase-I (20-C prenyl substrate) with a single point mutation. GGTase-I discriminates against farnesyl diphosphate (FPP) at the product turnover step through the inability of a 15-C FPP to displace the 20-C prenyl-peptide product. Understanding these key features of specificity is expected to contribute to optimization of anti-cancer and anti-parasite drugs.
Keywords: crystal structure/G proteins/lipid modification/protein prenylation/signal transduction
Introduction
Over 100 proteins important for cell growth, differentiation and morphology, including many GTP-binding regulatory proteins (G proteins), require post-translational modification by covalent attachment of an isoprenoid lipid (prenylation) for proper function (Tamanoi and Sigman, 2001). The three known enzymes that catalyze protein prenylation are the two CaaX prenyltransferases, protein geranylgeranyltransferase type-I (GGTase-I) and protein farnesyltransferase (FTase), and a third enzyme, protein geranylgeranyltransferase type-II (RabGGTase) (Casey and Seabra, 1996). GGTase-I modifies most monomeric G proteins in the Rho, Rac and Rap subfamilies, and nine of the 12 heterotrimeric G protein γ-subunits. Loss of GGTase-I function has dramatic biological effects, blocking the cell cycle at the G1 to S phase transition and promoting apoptosis (Vogt et al., 1996; Li et al., 2002). Since the demonstration that inhibition of FTase causes tumor regression in mice (Kohl et al., 1995), the prenyltransferase enzyme family has been studied in increasing detail. Drug design efforts have produced a number of CaaX prenyltransferase inhibitors that are now in advanced clinical trials as anti-cancer treatments (Johnston, 2001). Although the majority of these drug discovery efforts have focused on FTase inhibition, GGTase-I is increasingly of interest as a drug target. GGTase-I inhibitors have demonstrated efficacy in pre-clinical models of tumor progression (Sebti and Hamilton, 2000) and show promise in the treatment of smooth muscle hyperplasia (Stark et al., 1998). Recently, GGTase-I inhibitors were shown to attenuate clinical signs of disease in animal models of multiple sclerosis (Walters et al., 2002). GGTase-I has also been proposed as a target for countering parasitic infections such as malaria by selective inhibition of the parasite enzyme (Chakrabarti et al., 1998).
GGTase-I and FTase catalyze the transfer of a 20-carbon and a 15-carbon isoprenoid, respectively, from geranylgeranyl diphosphate (GGPP) or farnesyl diphosphate (FPP) to a protein, or short peptide, with a C-terminal CaaX sequence recognition motif. The CaaX box is defined by the cysteine (C), two typically aliphatic residues (aa) and the C-terminal residue (X) that contributes to substrate specificity (Reiss et al., 1990; Casey et al., 1991; Moores et al., 1991; Yokoyama et al., 1991). The steady-state kinetic parameters of GGTase-I are similar to those of FTase, although the GGTase-I reaction has not been characterized in as much detail, and indicate that the enzyme binds substrates by an ordered mechanism (Yokoyama et al., 1995; Stirtan and Poulter, 1997). For both GGTase-I and FTase, product release is the slow step in the reaction. In FTase, this step is accelerated by the binding of additional substrate (Tschantz et al., 1997). Despite these similarities, the two enzymes differ in cofactor requirements: unlike FTase, GGTase-I does not require magnesium for activity (Zhang and Casey, 1996). Three-dimensional structures of mammalian FTase with substrates, products and inhibitors have been determined (Park et al., 1997; Dunten et al., 1998; Long et al., 1998, 2000, 2001, 2002; Strickland et al., 1998). Structural information is available only for the apo RabGGTase (Zhang et al., 2000); RabGGTase is more specialized than the CaaX enzymes, exclusively modifying members of the Rab subfamily of G proteins (Seabra, 1998). RabGGTase processively transfers geranylgeranyl groups to both cysteine residues of CC- or CxC-containing Rab proteins (Farnsworth et al., 1994; Thoma et al., 2001). RabGGTase also requires an escort protein (REP) to present the Rab substrate for modification (Andres et al., 1993). Unlike the CaaX enzymes, short peptides are not substrates for RabGGTase (Seabra et al., 1992).
Here we present the first structural information for a GGTase-I, including a series of structures that represent the major steps along the reaction coordinate, from binding of substrates to product formation and release. We conclude that the main mechanistic features of the catalytic cycle are common to all three protein prenyltransferases. Comparative analysis of the structures exposes features unique to GGTase-I. In particular, determinants within the protein prenyltransferase family that dominate substrate specificity are revealed. We have confirmed the importance of these features by mutagenesis. The GGTase-I structures also provide further insight into the double prenylation mechanism of RabGGTase and its substrate selection. The structures of the GGTase-I complexes are likely to facilitate design of inhibitors that are selective for one type of prenyltransferase over another. Furthermore, the product complexes suggest a mechanism for the transportation of newly prenylated proteins within the cell.
Results and discussion
The overall structure of GGTase-I
The overall structure of GGTase-I is shown in Figure 2. Four ligand complexes are presented: (1) binary complex with GGPP; (2) ternary complex with a non-hydrolyzable GGPP analog and CaaX peptide; (3) binary complex with prenyl-peptide product; and (4) ternary complex with prenylated product and GGPP. The differences between the structures are confined primarily to the conformation of the ligands (backbone r.m.s.d. value of ∼0.2 Å) and do not involve significant structural rearrangements within the enzyme during the course of the reaction cycle (Figure 3).
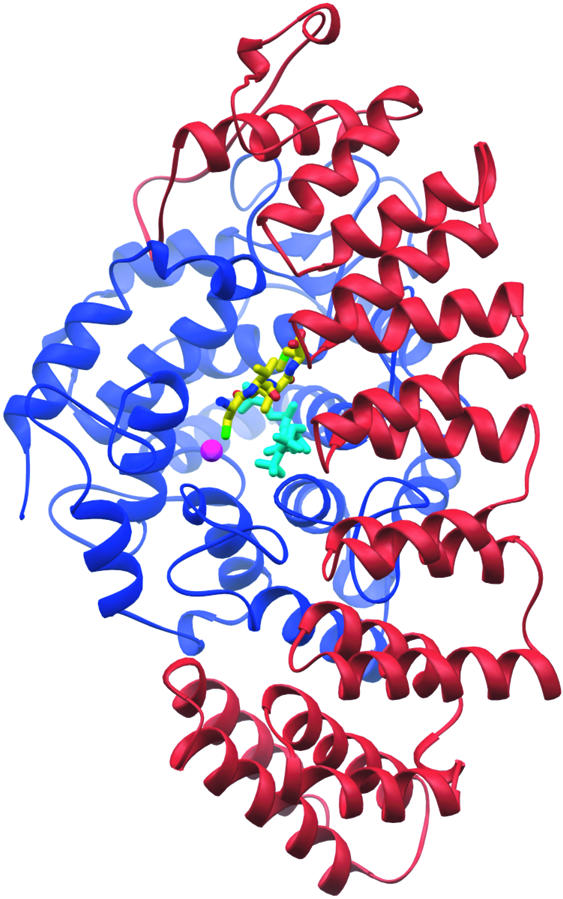
Fig. 2. GGTase-I ternary substrate complex. The GGTase-I heterodimer consists of a 48 kDa α-subunit (red) and a 43 kDa β-subunit (blue). The non-reactive isoprenoid analog 3′azaGGPP (cyan) binds similarly to GGPP in the active site. The CaaX portion of the KKKSKTKCVIL peptide substrate (yellow) binds against the isoprenoid with the cysteine sulfur coordinating the catalytic zinc ion (magenta).

Fig. 3. Structures of the GGTase-I reaction cycle. The GGTase-I active site is shown in stereo as a molecular surface, with the α-subunit colored red, the β-subunit blue, and the exit groove highlighted in cyan. Complex 1, the enzyme with bound GGPP. Complex 2, ternary complex with 3′azaGGPP analog and CaaX peptide substrate (only CVIL shown). Complex 3, prenylated peptide product complex. Complex 4, displaced prenylated peptide product and GGPP.
GGTase-I crystals belong to the _I_222 space group, with three complete 91 kDa heterodimers in the asymmetric unit (Table I). Phases were determined by single isomorphous replacement with anomalous scattering (SIRAS). A second crystal form was obtained that diffracted to higher resolution. The molecular packing of the two crystal forms was nearly identical, but a slight shift reduced the symmetry of the second crystal form to space group _C_2, with six heterodimers in the asymmetric unit. In both crystal forms, the structures of the individual heterodimers within the crystallographic asymmetric units are identical, except for a few side chains in crystal contacts. Therefore, only one representative molecule from the asymmetric unit is considered for discussion.
Table I. Data collection and refinement statistics.
| | Derivative λ1 | Derivative λ2 | Native (ternary) | Binary (1) | Ternary (2) | Product (3) | Displaced product (4) | | | -------------------------------- | ------------- | ---------------- | -------------- | --------------- | --------------- | ------------------------- | ------------- | | Data collection (all data) | | | | | | | | | Beamline | NSLS X12B | NSLS X12B | APS 14BMC | APS 14BMC | APS 14BMC | NSLS X12B | NSLS X25 | | Wavelength, Å | 1.071416 | 1.070676 | 1.00000 | 0.900000 | 1.00000 | 1.00008 | 1.00000 | | Resolution, Å | 40–3.5 | 40–3.5 | 50–2.7 | 30–2.65 | 30–2.4 | 30.0–2.8 | 40–2.6 | | Outer shell, Å | 3.63–3.5 | 3.63–3.5 | 2.8–2.7 | 2.74–2.65 | 2.49–2.4 | 2.9–2.8 | 2.69–2.6 | | No. of reflections | | | | | | | | | Unique | 125 705 | 126 788 | 139 616 | 286 863 | 355 317 | 240 519 | 300 000 | | Total | 393 410 | 337 591 | 745 344 | 1 038 571 | 997 257 | 785 048 | 1 011 902 | | Mean I/σIa | 8.6 (1.7) | 9.2 (2.0) | 18.8 (2.4) | 14.0 (2.2) | 15.1 (2.1) | 13.0 (2.3) | 13.6 (2.6) | | Completeness % | 98.0 (97.1) | 98.8 (97.4) | 99.6 (99.6) | 98.7 (91.5) | 93.0 (87.1) | 99.4 (98.0) | 99.7 (100) | | _R_sym %a | 9.2 (58.0) | 8.5 (65.4) | 7.8 (66.3) | 7.7 (50.0) | 5.6 (35.2) | 7.4 (43.2) | 6.9 (43.5) | | _R_iso/_R_anom % | 22.1/7.9 | 22.2/8.3 | | | | | | | Space group | _I_222 | _I_222 | _I_222 | _C_2 | _C_2 | _C_2 | _C_2 | | Unit cell dimensions, Å | a = 185.25 | a = 185.25 | a = 185.05 | a = 272.34 | a = 271.05 | a = 272.07 | a = 271.12 | | | b = 204.34 | b = 204.34 | b = 204.31 | b = 271.57 | b = 268.03 | b = 268.80 | b = 268.43 | | | | c = 273.01 | c = 273.01 | c = 269.22 | c = 185.42 | c = 184.97 | c = 185.31 | c = 184.82 | | | | β = 90° | β = 90° | β = 90° | β = 131.56° | β = 131.72° | β = 131.55° | β = 131.68° | | | Refinement (F ≥ σF) | | | | | | | | | Completeness, %a | | | 98.2 (97.3) | 98.2 (90.5) | 92.8 (86.8) | 99.1 (97.4) | 99.2 (97.3) | | _R_cryst, %a | | | 20.7 (31.5) | 20.4 (34.4) | 21.4 (33.4) | 19.8 (31.4) | 19.4 (29.3) | | _R_free, %a | | | 23.0 (33.3) | 22.8 (36.9) | 23.4 (35.6) | 21.7 (32.9) | 21.4 (31.5) | | Non-hydrogen atoms | | | | | | | | | Total | | | 16 344 | 33 444 | 33 546 | 33 247 | 33 926 | | Solvent | | | 260 | 1024 | 1117 | 708 | 1298 | | Ramachandran plot | | | | | | | | | Most favored regions, % | | | 88.8 | 89.0 | 89.2 | 88.0 | 89.0 | | Allowed regions | | | 11.2 | 11.0 | 10.8 | 12.0 | 11.0 | | R.m.s.d. from ideal geometry | | | | | | | | | Bond lengths, Å | | | 0.01 | 0.007 | 0.006 | 0.007 | 0.006 | | Bond angles, ° | | | 1.45 | 1.18 | 1.19 | 1.18 | 1.17 | | Average isotropic _B_-value, Å2 | | | 59.5 | 53.7 | 58.3 | 54.2 | 54.0 |
The helices of the GGTase-I α-subunit are arranged in α-helical hairpin pairs, forming a crescent that wraps around the β-subunit. Although identical in sequence to the FTase α-subunit, the curvature of the crescent-shaped α-subunit in GGTase-I is slightly different (1.5 Å r.m.s.d.) because the rat GGTase-I β-subunit is smaller than the rat FTase β-subunit (377 residues versus 437). The interface between the α- and β-subunits is extensive, covering >3300 Å2. The β-subunit forms a compact, globular, α–α barrel domain with a central cavity. The substrate-binding site opens into the α–β subunit interface and extends into the central funnel-shaped cavity of the β-subunit, which is lined with hydrophobic residues. A zinc ion is bound at the top of this active site funnel. The zinc ion is required for catalytic activity (Zhang and Casey, 1996); it can be removed using chelating agents, however, without significantly altering the structure (data not shown). Because the α-subunit is common with FTase, functional differences are the result of differences in the β-subunits, where sequence identity is just 25%.
The GGTase-I structure facilitates a structure-based sequence alignment of the β-subunits of all three protein prenyltransferases which shows that despite nearly identical topology, there is only 32% sequence similarity with FTase and RabGGTase (Figure 4). The solvent-accessible surfaces of the GGTase-I and FTase β-subunits differ not only in side chain identities, but also in the lengths of several loops. The FTase β-subunit has an additional 53 residues at the N-terminus and two insertions near the C-terminus of 13 and 16 residues. In GGTase-I, residues 79β–121β form a loop connecting helix 3β and helix 4β that has an insertion of 26 residues relative to FTase and RabGGTase (Figure 4). This loop terminates at helix 4β, which makes up part of the CaaX-binding site, and thus a shift in its position could influence the enzyme’s CaaX specificity (see below). These loops are located primarily on the surface of the molecule and, while the differences may simply be the result of divergent evolution with no functional consequence, the differences in the molecular surfaces of the enzymes could be important for the specificity of protein–protein interactions.

Fig. 4. β-Subunit alignment. Top: superposition of β-subunits from the three protein prenyltransferases (Cα atoms only). FTase (blue) and RabGGTase (green) are aligned with GGTase-I (red). Bottom: structure-based sequence alignment highlights residues that contact the zinc (blue), peptide substrate (red) or isoprenoid (green).
Determinants of isoprenoid specificity
One of the key functional differences between the three protein prenyltransferases is the selective binding of either GGPP or FPP. The critical feature that distinguishes GGPP and FPP is the length of the lipid (Figure 1). The complex of GGTase-I with the isoprenoid diphosphate substrate GGPP (Figure 3A) clearly suggests how lipid length is used to selectively bind the correct substrate. The GGPP binds with its diphosphate moiety placed at the α–β subunit interface and the lipid inserted into the central cavity of the β-subunit (Figure 5A). The first three isoprene units are arranged along a straight line. The fourth isoprene unit is turned ∼90° relative to this axis. This geometry of the bound lipid is quite different from that of the other known structures with bound isoprenoids, i.e. FTase (Long et al., 1998), RhoGDI (Hoffman et al., 2000) and the phosducin–Gtβγ complex (Loew et al., 1998), in which the isoprenoid is completely extended. Alignment of GGTase-I and FTase complexes by Cα atom superposition of the heterodimers shows a slight divergence in the position of the first isoprene unit, shifting the diphosphate moiety of GGPP ∼1.0 Å toward Asn199α, while the second and third isoprene units are in essentially identical positions. The position of the fourth isoprene unit of the GGPP indicates that Thr49β is the primary determinant of lipid length discrimination. In FTase, a tryptophan residue fills the space where the fourth isoprene unit binds in GGTase-I (Figure 5B), accounting for the inability of FTase to bind GGPP productively. In GGTase-I, Phe324β is also positioned near the fourth isoprene unit; the hydroxyl group on the corresponding FTase tyrosine residue may also help discriminate against GGPP binding in FTase. In a sequence alignment, the identity of the residues corresponding to 49β and 324β in GGTase-I is highly correlated with the enzyme type, providing a strong indication of whether an unknown sequence is a GGTase or FTase (Figure 4). Across many species, residue 49β is always a small amino acid such as threonine, serine, valine or alanine in GGTase-I and RabGGTase, whereas in FTase it is always tryptophan.

Fig. 1. Comparison of the chemical structures of isoprenoid diphosphates and a non-reactive analog. Geranylgeranyl diphosphate (GGPP) has four isoprene units; farnesyl diphosphate (FPP) has three. The non-reactive GGPP analog (3′azaGGPP) was used to form ternary substrate complex 2.

Fig. 5. Substrate-binding site. (A) Stereo view of the active site, in approximately the same orientation as Figure 3, showing the CaaX portion of the KKKSKTKCVIL peptide (yellow) and 3′azaGGPP (purple). The CaaX peptide is bound in an extended conformation with the cysteine thiolate coordinated by the zinc ion (magenta). Carbonyl oxygens and the C-terminus of the CaaX sequence make water-mediated (cyan) and direct hydrogen bonds with conserved side chains in both the α- (red) and β- (blue) subunits. (B) Comparison of isoprenoid binding in FTase and GGTase-I. In FTase (red), the larger tryptophan fills the space where the fourth isoprene binds in GGTase-I (blue) and is one of the primary determinants of isoprenoid specificity.
To test directly the importance of the threonine/tryptophan identity in the determination of isoprenoid substrate specificity, residue Trp102β of human FTase (corresponding to Thr49β in GGTase-I) was mutated to a threonine. The mutant was cloned, overexpressed in Escherichia coli, purified (Long et al., 2001), and its substrate specificity characterized by enzymatic assays (Zhang et al., 1994b). As predicted, the resulting mutant FTase has acquired the substrate specificity of a GGTase, creating an FTase enzyme greatly preferring GGPP over FPP as its isoprenoid substrate without significantly altering CaaX sequence specificity (Figure 6).

Fig. 6. Altered substrate specificity of a protein prenyltransferase. Prenylation reactions assayed the activity of human wild-type FTase, wild-type GGTase-I, and the W102T FTase mutant with four substrate combinations: FPP + Ras-CVLS (blue), FPP + Ras-CVLL (green), GGPP + Ras-CVLS (yellow) and GGPP + Ras-CVLL (red). The activities are shown as percentages of activity with optimal isoprenoid and protein substrates for each of the enzymes tested (FPP and Ras-CVLS, FTase; GGPP and Ras-CVLS, W102T FTase; GGPP and Ras-CVLL, GGTase-I). Turnover numbers under the optimal substrate concentrations were 1.0, 0.59 and 0.65/min for FTase, W102T FTase and GGTase-I, respectively.
Steric hindrance plays a dominant role in FTase in selecting the 15-C farnesyl in preference to the 20-C geranylgeranyl substrate. A different mechanism must operate in GGTase to prevent the shorter farnesyl group from functioning as a substrate because there is no steric block to prevent GGTase-I from binding FPP in a productive conformation. Indeed, FPP is a weak substrate for GGTase-I (Yokoyama et al., 1995). As described below, the product release step in the GGTase-I reaction cycle is the dominant mechanism by which GGTase-I selects for GGPP over FPP.
Metal cofactors
All of the GGTase-I complexes contain a zinc ion bound at full occupancy with a _B_-factor comparable with the surrounding protein residues. The zinc ion is coordinated by three residues, Asp269β, Cys271β and His321β, that are strictly conserved across all the protein prenyltransferases. In the ternary complex 2 and product complex 3, the zinc ion is also coordinated by the CaaX cysteine. A water molecule occupies an analogous position in complexes 1 and 4. Spectroscopic observations suggest that the CaaX cysteine sulfur is in the deprotonated thiolate form when coordinated by the zinc ion and is directly involved in the chemical step (Hightower et al., 1998). GGTase-I requires only zinc, whereas FTase requires both zinc and millimolar levels of magnesium for optimal activity (Zhang and Casey, 1996). The GGTase-I structure suggests an explanation for this difference. Residue 311β is lysine in GGTase-I, whereas it is aspartic acid in FTase and RabGGTase. In GGTase-I, the amine group of Lys311β is observed near where the Mg2+ is located in the FTase transition state model (Long et al., 2002). Hence, the GGTase-I Lys311β amine group may substitute for the positively charged magnesium ion in FTase, permitting magnesium-independent catalysis by GGTase-I (Figure 7A).

Fig. 7. Model for the transition state of the prenylation reaction. (A) The scissile phosphoether bond between the diphosphate and geranylgeranyl group, and the nascent thioether bond between the cysteine and geranylgeranyl group are shown as dotted lines. Hydrogen bonds observed in complexes 1, 2 and 3 are colored blue, while those predicted to stabilize the transition state are shown in red. The amine of Lys311β replaces the magnesium ion that is required for catalysis by FTase (see text). (B) Stereo view of the two substrates, GGPP and CVIL peptide (gray) superimposed on the prenylated peptide product (yellow). Before catalysis, the zinc-coordinated cysteine thiolate of the CVIL peptide and the C1 of the GGPP are separated by 8.2 Å (dotted line). Rotation of the first two isoprene units brings the substrate into the product conformation (black arrow).
Determinants of protein substrate specificity
The GGTase-I ternary complex 2 shows the 3′azaGGPP (Steiger et al., 1992) bound at the same location as the GGPP in 1, and a peptide substrate with the terminal sequence –CVIL (the sequence of GTPase Rap2B) bound adjacent in an extended conformation (Figures 3B and 5A). The CaaX peptide buries a total of 140 Å2 of the isoprenoid from the solvent. This large contact area between the two substrates is the likely origin for the observed ordered substrate binding. In the absence of GGPP, the peptide binds non-productively and must dissociate before the reaction can proceed (Yokoyama et al., 1995; Stirtan and Poulter, 1997). The cysteine of the CaaX peptide coordinates the catalytic zinc ion located at the top of the funnel-shaped central cavity. The C-terminus of the CaaX peptide is anchored to the bottom of this cavity by a hydrogen bond to Gln167α, and water-mediated hydrogen bonds to His121β, Glu169β and Arg173β. One additional hydrogen bond is present between the carbonyl oxygen of the a2 residue (isoleucine) and Arg173β. These four protein residues, as well as the position of the water molecule, are conserved in FTase (Long et al., 2000). GGTase-I preferentially binds CaaX protein substrates with leucine in the X position, whereas FTase accepts substrates with methionine, serine, alanine or glutamine (Casey et al., 1991; Yokoyama et al., 1991).
A comparison of the GGTase-I and FTase ternary complexes reveals the structural determinants for this selectivity. Specificity at the X position is determined by surface complementarity between the X residue and the ‘specificity pocket’ in which the X side chain binds (Figure 5A). The hydrophobic specificity pocket discriminates against polar side chains; the shape and volume further restrict the identity of the X residue, which for GGTase-I must be leucine. Other amino acids with van der Waals volumes similar to leucine are either polar (histidine and glutamine) or are actually weak substrates for GGTase-I (phenylalanine and methionine). CaaX sequences ending in glycine or alanine are worse GGTase-I substrates (Moores et al., 1991) than would be expected from simply not filling the pocket, indicating that these sequences may bind in an alternative and unproductive conformation. Some of the same structural features in GGTase-I that allow it to bind the longer GGPP isoprenoid also shape the pocket to fit leucine. A threonine at residue 49β (tryptophan in FTase) provides space for the Cδ1 atom of the leucine side chain. Additionally, a steric clash between the leucine Cδ2 atom and Ala123β (Ala151β in FTase) is avoided through a small shift in the backbone of helix 4β. There is little or no sequence constraint on the a1 residue, as its side chain projects into the solvent and makes no direct contact with the protein. In GGTase-I, the a2 side chain makes extensive hydrophobic contacts with Phe53β and Leu320β as well as the fourth isoprene unit. All three of these a2 contacts are different in FTase: Phe53β is tryptophan, Leu320β is tyrosine and the isoprenoid contact is replaced in FTase by Trp102β. These changes may result in altered sequence preference at the a2 position and provide additional steric differences that can be taken advantage of in the design of specific inhibitors.
Product complexes
The structures of GGTase-I product complexes reveal a change in the isoprenoid conformation and a secondary product-binding site, thus generalizing for the family of protein prenyltransferases recent observations for FTase (Long et al., 2002). Crystals of the GGTase-I product complexes show that the product remains bound in the active site at full occupancy many weeks after crystallization (Figure 3C). In this complex, the active site is occupied by the prenylated peptide, and the characteristic electron density of the diphosphate leaving group is not observed. The peptide portion of the product is in the identical position to that in the substrate complex 2, with the sulfur atom, now part of a thioether, still coordinating the catalytic zinc. However, the GGPP markedly changes conformation during the reaction. The third and fourth isoprene units are in the same location as in 2, but atoms C1–C10 are in a distinct conformation, with the C1 atom now covalently bonded to the cysteine of the CaaX peptide. The conformational change includes an ∼160° rotation about the C8–C9 bond in the second isoprene unit and additional rotations in the first isoprene unit. We see no evidence of any conformational change in the protein backbone during the reaction.
A fourth complex, in which the product is displaced to a secondary binding site, was created when crystals of 3 were soaked in a solution containing additional GGPP (Figure 3D). The new GGPP binds in the same location as in 1, but the product is not completely released. Instead, to make space for the new GGPP, the isoprenoid moiety of the prenylated product is displaced to a solvent-accessible groove that in 1–3 is solvated and runs from the active site to the rim of the β subunit α–α barrel, the ‘exit groove’. In this displaced product complex, 4, the first three isoprene units of the prenyl product lie in the exit groove in an extended conformation, while the fourth unit is turned ∼90° towards the solvent. As a result of the rearrangement, the cysteine of the prenylated product no longer interacts with the zinc ion. The a2 and X residues (isoleucine and leucine) are displaced by ∼0.9 Å but are otherwise in a conformation similar to that seen in 2 and 3. The a1 (valine) residue is rotated 120° about its ψ Ramachandran backbone angle, which reorients the side chain and brings the prenyl-cysteine residue to a position where the carbonyl oxygen can hydrogen-bond with the backbone nitrogen of the a2 residue (isoleucine), forming a type-1 β-turn. A type-1 β-turn can accommodate any residue including proline at the a1 position, but precludes proline from the a2 position. A CaaX peptide with proline in the a2 position can adopt the conformation required to be a substrate, but there are no known prenylated proteins that end in CaPX, suggesting that the formation of the type-1 β-turn in the exit complex is important.
Upon moving to the exit groove, the product can be fully displaced from the enzyme by soaking new peptide substrate into the crystals or simply by waiting several days. In vivo, product release may require an interaction with a membrane or other protein, or could be triggered by binding of a new CaaX substrate. Similar complexes with a product in the exit groove have been observed in FTase (Long et al., 2002), and the RabGGTase also has an exit groove at the same location. The GGTase-I structures confirm that the exit groove region is a functionally conserved structural feature, despite little sequence conservation between the three enzymes.
The protein prenyltransferase reaction cycle
Based on a similar series of substrate and product complexes, an unusual reaction cycle was proposed for FTase (Long et al., 2002). Here we show that the critical aspects of the proposed cycle are also retained in GGTase-I, suggesting that the reaction mechanism is general for all protein prenyltransferases. The GGTase-I structures capture four different states in a reaction cycle that begins with the binding of GGPP (Figure 3; an animation of the GGTase-I reaction cycle can be found in the Supplementary data available at The EMBO Journal Online). Once charged with GGPP, 1, the enzyme binds a protein with a C-terminal CaaX motif 2. The observed ordered substrate binding is consistent with the CaaX binding mode seen in 2, where the isoprenoid in complex 1 forms much of the CaaX-binding site in complex 2. These two steps are identical to those seen in FTase, with the exception of the substrate specificity differences, the structural origins for which have been discussed above. The formation of 2 is followed by a conformational change in the isoprenoid diphosphate substrate, repositioning it for catalysis while maintaining the conformation of the peptide substrate position and keeping the protein rigid (Figure 7B). The conformational change in the GGPP reorients the diphosphate moiety and the first two isoprene units relative to their positions in 2. The β-phosphate of the leaving diphosphate group stays in roughly the same position, but the α-phosphate is repositioned to interact with Lys164α, Lys311β and Tyr272β, bringing the C1 atom close to the cysteine thiolate. The CaaX peptide substrate remains in place, with the zinc and the Cβ–Sγ bond of the cysteine thiolate orienting a free pair of electrons for nucleophilic attack on C1. Synthesis of all available data on the mechanism, including the comparison of the structures of complexes 2 and 3, allows a model of the transition state to be constructed (Figure 7A), as was done for FTase (Long et al., 2002). The proposed structure retains elements of both the postulated electrophilic and nucleophilic components of the reaction mechanism (Yokoyama et al., 1997; Clausen et al., 2001). In this model, Lys311β in GGTase-I fulfills the role of Mg2+ in FTase (see previous section). The enzyme then releases pyrophosphate forming 3, a stable product complex. Binding of a new GGPP molecule then shifts the product to the exit site 4, where binding the next protein substrate or an interaction with the next enzyme in the pathway facilitates its release (see below). These product complexes again confirm observations of the FTase reaction cycle in all critical aspects, including that changes in substrate conformation during the reaction are not correlated with conformational changes in the enzyme. Indeed, the structures of these four GGTase-I complexes, determined using two crystal forms, have the same protein conformations.
GGTase-I couples product release to lipid substrate specificity
The nature of the GGTase-I reaction cycle suggests that lipid substrate specificity can be coupled to product release, resolving the apparent paradox of how GGTase-I selects GGPP over FPP. The four structures presented here indicate that the GGTase-I lipid-binding site is always occupied, either by GGPP or by the geranylgeranyl moiety of the prenyl-peptide product. Reaction cycle progression requires the binding of fresh isoprenoid diphosphate to displace the product from the active site. FPP cannot displace GGPP from the active site, and we find that only GGPP can displace the prenyl-peptide product. Thus, there is no opportunity for FPP to bind to GGTase-I during the reaction cycle. Repeated attempts to displace a geranylgeranyl-peptide product from GGTase-I by soaking product crystals with FPP failed (soak times of up to 1 week, results not shown), suggesting that the stability of the enzyme–product complex contributes towards selectivity of GGPP over FPP. RabGGTase-I probably shares the same mechanism. To our knowledge, the protein prenyltransferase enzymes are unique in that they couple product release to lipid substrate specificity.
Implications of substrate-mediated product release
After lipidation, the prenyl-protein product is processed further first by Rce1, a protease that cleaves the ‘aaX’ portion from the CaaX motif, followed by Icmt, which methylates the C-terminus (Tamanoi and Sigman, 2001). The CaaX motif is the main determinant for directing proteins to this next processing step (Choy et al., 1999). We propose that the unusual substrate-mediated product release provides a mechanism for the regulated handover of the prenyl-protein product to the next step in this processing pathway (Figure 8). In the CaaX prenyltransferases, the prenylated product remains tightly bound and is therefore shielded from the cytoplasm, preventing aggregation or association with an incorrect membrane compartment. Only upon binding of an additional GGPP molecule is the product isoprenoid displaced into the exit groove, presenting the product for delivery to the next processing step. The product can then be either directly delivered to the membrane bilayer or passed to another protein such as the Rce1 protease, the next enzyme in the prenylation pathway. The mechanism for this handover is analogous to one of the roles of REP, which escorts RabGGTase products to their final destination (Alexandrov et al., 1994), and similar to that of the GDI proteins, which extract prenylated G proteins such as Rho from membranes by binding the isoprenoid moiety in a hydrophobic pocket and transporting them throughout the cell (Hoffman et al., 2000). Consistent with this hypothesis of a role for CaaX prenyltransferases in product delivery, in vivo studies of Rce1 knock-out fibroblasts have shown that Ras, although prenylated normally, becomes mislocalized (Kim et al., 1999; Bergo et al., 2000).

Fig. 8. Regulated handover of product. GGPP-charged GGTase-I (orange) (1) binds a protein substrate with a CaaX motif (green) (2) and catalyzes the addition of a geranylgeranyl group in the cytosol. The enzyme retains the prenylated product (3) until binding of a new GGPP (blue highlight) displaces the product into the exit groove (4). The CaaX prenyltransferase hands over the prenylated product to the next enzyme in the prenylation pathway, the membrane-bound Rce1 CaaX protease (yellow) (see text). After handover of the prenylated protein to RceI, the GGPP-charged GGTase-I is ready to bind a new protein substrate.
Molecular mechanisms of RabGGTase
Insights gained from the GGTase-I and FTase structures can be applied to understand the mechanism of RabGGTase. There is a high degree of structural similarity between GGTase-I and RabGGTase at the active site (Figure 4). The 24 residues surrounding the GGPP molecule in the GGTase-I complexes are identical or structurally conservative substitutions in RabGGTase. Consequently, a single GGPP can be modeled into the apo structure of RabGGTase in the same conformation as seen in GGTase-I. Additionally, there is insufficient room to bind two GGPP molecules, supporting biochemical studies showing that RabGGTase binds only a single GGPP at a time (Desnoyers and Seabra, 1998; Thoma et al., 2000). The isoprenoid specificity residues identified in the GGTase-I structures, Thr49β and Phe324β, are identical in RabGGTase, suggesting that a general mechanism is used for isoprenoid selection by all of the protein prenyltransferases. In contrast, the selection of protein substrates in RabGGTase must differ from the CaaX prenyltransferases. In CaaX protein substrates, the distance between the cysteine and the C-terminus is invariant. However, RabGGTase must accommodate variations in this distance both when moving from one cysteine to the next during the processive reaction and from variation within the Rab substrates (i.e. –CC or –CxC). Comparison of the structures suggests that in RabGGTase, the region analogous to the C-terminal CaaX peptide-binding site has a negative charge and the peptide specificity pocket is blocked. Thus, unlike the CaaX prenyltransferases, RabGGTase cannot anchor the C-terminus of Rab substrates. Although it remains to be seen how RabGGTase recognizes its protein substrates, we note that REP is required for this to occur and presumably contributes determinants of the interaction (Andres et al., 1993).
The reaction cycle proposed here for CaaX prenyltransferases may be relevant to understanding the processive addition of isoprenoids by RabGGTase (Thoma et al., 2001). Many processive enzymes feature two product-binding sites analogous to those observed for CaaX prenyltransferases. A notable example is the ribosome with its product (P) and exit (E) sites. In RabGGTase, a translation of the product would allow the unmodified cysteine to interact with the zinc ion in preparation for the second catalytic reaction (Long et al., 2002). The exit groove observed in CaaX prenyltransferases is a conserved structural feature of RabGGTase. Additionally, RabGGTase has a tunnel that may serve to stabilize the mono-prenylated product during the processive reaction.
Conclusions
The structural snapshots of the GGTase-I reaction cycle observed in this study are consistent with the unusual reaction mechanism proposed for FTase (Long et al., 2002), thereby indicating that this cycle is a common feature of the protein prenyltransferase family. A unique feature of the mechanism is that these enzymes may regulate handover of their products to the next step in the post-translational processing pathway. Consequently, the reaction mechanism intimately combines the requirements of chemistry with cellular physiology. Furthermore, the ability to conduct detailed structural comparisons of the active sites of CaaX prenyltransferases reveals the dominant structural features that account for their distinct substrate specificities.
Protein prenyltransferases are promising targets for chemotherapeutics, but their exploitation is likely to require the design of drugs that are highly selective for one enzyme. This work, along with the earlier work on FTase, provides the basis for the structure-based design and characterization of drugs specific to a particular CaaX prenyltransferase. The features that can be exploited to create specific inhibitors include the difference between tryptophan or threonine at 49β, providing discrimination for binding both isoprenoids and peptides, the altered binding surface near Phe53β and Leu320β that contacts the a2 residue of the CaaX motif, the aspartic acid for lysine substitution at 311β, and the arginine for proline substitution at 317β in the exit groove.
Materials and methods
Expression, purification, crystallization and data collection
Rat GGTase-I was expressed in Sf9 insect cells and purified as previously described (Zhang et al., 1994a) with two additions. The purified enzyme was incubated with GGPP (Sigma) before application to a 26/10 phenyl Sepharose FPLC column (Pharmacia), followed by a 16/60 Superdex 200 FPLC column (Pharmacia). GGTase-I was concentrated to ∼15 mg/ml and stored at –80°C.
GGTase-I crystals were grown at 17°C in hanging drops using equal volumes of protein and reservoir solution [1.3 M NH4SO4, 175 mM Na3citrate pH 6.5, 20 mM dithiothreitol (DTT) and 100 mM MES pH 6.3]. Micro-seeds were added after equilibration to control nucleation. Seeding solution was produced by crushing GGTase-I crystals in stabilizing solution (1.5 M NH4SO4, 175 mM Na3citrate pH 6.5, 5 µM ZnCl2, 20 mM DTT and 100 mM MES pH 6.3). Initial protein batches produced orthorhombic crystals (I222, three molecules per asymmetric unit), but subsequent protein preparations yielded monoclinic crystals (C2, six molecules per asymmetric unit). Complex 2 was formed prior to crystallization by equilibrating GGTase-I first with 3′azaGGPP followed by CaaX peptide, KKKSKTKCVIL (Genosys, >95% purity), at a molar ratio of 1:2:2. Product complex 3 was formed similarly, with GGPP instead of 3′azaGGPP, at a molar ratio of 1:1:2. Displaced product complex 4 was obtained by soaking product crystals 3 in stabilization solution containing 0.1 mM GGPP for 18 h. Binary complex 1 was obtained by extending the soak time to 1 week. For data collection, crystals were transferred stepwise into cryosolvent [30% (w/v) sucrose, 1.8 M NH4SO4, 5 µM ZnCl2, 10 mM TCEP and 100 mM MES pH 6.3] before flash-freezing in liquid nitrogen. Derivative crystals were soaked for 18 h in cryosolvent containing 1 mM di-µ-iodobis (ethlyenediamine) diplatinum (II) nitrate (PIP) and no TCEP.
Diffraction data were collected at 100°K using beamlines X12B and X25 at NSLS, Brookhaven National Laboratories (BNL) and beamline 14-BMC at APS, Argonne National Laboratories (ANL). Data were integrated and scaled using DENZO and SCALEPACK.
Phasing, model building and refinement
Phases for the I222 diffraction data were determined using SIRAS. SOLVE (Terwilliger and Berendzen, 1999) was used to locate 18 diplatinum sites and to make phase calculations. At 3.7 Å resolution, the isomorphous phasing power was 1.45/1.01 (acentric/centric), anomalous phasing power was 0.45 and the mean figure of merit was 0.52. Initial maps revealed three molecules in the asymmetric unit and a 73% solvent content. Experimental phases were improved using maximum-likelihood density modification in RESOLVE (Terwilliger, 2000).
An initial model was constructed using the experimental phases and, as refinement progressed, these were combined with partial model phases using the σA weighting scheme in CNS v1.0 (Brünger et al., 1998). Iterative cycles of manual building using O followed by simulated annealing, minimization, _B_-factor refinement and phase extension techniques in CNS v1.0 continued until the _R_free converged at the full resolution limit (2.7 Å). Strict non-crystallographic symmetry (NCS) was enforced until the _R_-factors dropped below 25%. All included waters had a 3σ peak in omit _F_o – _F_c maps, with density recapitulated in 2_F_o – _F_c maps.
Phases for the C2 diffraction data were determined by molecular replacement using all three molecules in the I222 structure as the probe. Structure refinement was carried out as described above. The larger size of the C2 model required NCS restraints in order to minimize _R_free. Restraints were chosen empirically by monitoring refinement statistics: moderate restraints (150 kcal/mol/Å2) were applied to the protein backbone and internal residues, while solvent-accessible residues in the N- and C-termini were given weak restraints (20 kcal/mol/Å2).
In the C2 and I222 structures, the first 54 and last nine amino acids of the α-subunit, the first 17 and last 15 amino acids of the β-subunit, and the five N-terminal residues of the KKKSKTKCVIL peptide were not seen in the electron density. The first 54 residues in the α-subunit, which include 14 proline residues, are disordered in all FTase and GGTase-I structures determined to date. The atomic coordinates have been submitted to the Protein Data Bank, identification codes 1N4P, 1N4Q, 1N4R and 1N4S.
Preparation, expression, purification and kinetic characterization of FTase mutant
Mutagenesis of the human FTase sequence (Long et al., 2001) was performed using the QuikChange site-directed mutagenesis system from Stratagene, converting the W102β residue to a threonine (W102T mutant). Mutant and wild-type FTase were expressed in E.coli and purified as previously described for the wild-type FTase enzyme (Long et al., 2001). Prenylation reactions were conducted and processed essentially as previously described (Zhang et al., 1994b), with substrate concentrations of 500 nM [3H]FPP or [3H]GGPP and 1 µM Ras-CVLS or Ras–CVLL, and 50 ng of either FTase (wild-type or W102T mutant) or GGTase-I.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The 3′azaGGPP substrate analog was a kind gift from Robert M.Coates. We would like to acknowledge Stephen Long, Joel Tuttle and Carolyn Weinbaum for assistance in the expression and purification of rat GGTase-I, the APS and NSLS staff for their help with data collection, and Homme Hellinga for helpful discussions. Research carried out (in whole or in part) at the National Synchrotron Light Source is supported by the US Department of Energy, Division of Materials Sciences and Division of Chemical Sciences, under contract No. DE-AC02-98CH10886. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. Use of the BioCARS Sector 14 was supported by the National Institutes of Health (NIH), National Center for Research Resources, under grant number RR07707. This work was supported by grants from the NIH to L.S.B. (GM52382) and P.J.C. (GM46372), and a postdoctoral fellowship to J.S.T. from the American Heart Association.
References
- Alexandrov K., Horiuchi,H., Steele-Mortimer,O., Seabra,M.C. and Zerial,M. (1994) Rab escort protein-1 is a multifunctional protein that accompanies newly prenylated rab proteins to their target membranes. EMBO J., 13, 5262–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres D.A., Seabra,M.C., Brown,M.S., Armstrong,S.A., Smeland,T.E., Cremers,F.P.M. and Goldstein,J.L. (1993) cDNA cloning of component A of rab geranylgeranyl transferase and demonstration of its role as a rab escort protein. Cell, 73, 1091–1099. [DOI] [PubMed] [Google Scholar]
- Bergo M.O., Leung,G.K., Ambroziak,P., Otto,J.C., Casey,P.J. and Young,S.G. (2000) Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem., 275, 17605–17610. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Casey P.J. and Seabra,M.C. (1996) Protein prenyltransferases. J. Biol. Chem., 271, 5289–5292. [DOI] [PubMed] [Google Scholar]
- Casey P.J., Thissen,J.A. and Moomaw,J.F. (1991) Enzymatic modification of proteins with a geranylgeranyl isoprenoid. Proc. Natl Acad. Sci. USA, 88, 8631–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti D., Azam,T., DelVecchio,C., Qiu,L., Park,Y.I. and Allen,C.M. (1998) Protein prenyl transferase activities of Plasmodium falciparum. Mol. Biochem. Parasitol., 94, 175–184. [DOI] [PubMed] [Google Scholar]
- Choy E., Chiu,V.K., Silletti,J., Feoktistov,M., Morimoto,T., Michaelson,D., Ivanov,I.E. and Philips,M.R. (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell, 98, 69–80. [DOI] [PubMed] [Google Scholar]
- Clausen V.A., Edelstein,R.L. and Distefano,M.D. (2001) Stereochemical analysis of the reaction catalyzed by human protein geranylgeranyl transferase. Biochemistry, 40, 3920–3930. [DOI] [PubMed] [Google Scholar]
- Desnoyers L. and Seabra,M.C. (1998) Single prenyl-binding site on protein prenyl transferases. Proc. Natl Acad. Sci. USA, 95, 12266–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunten P., Kammlott,U., Crowther,R., Weber,D., Palermo,R. and Birktoft,J. (1998) Protein farnesyltransferase: structure and implications for substrate binding. Biochemistry, 37, 7907–7912. [DOI] [PubMed] [Google Scholar]
- Farnsworth C.C., Seabra,M.C., Ericsson,L.H., Gelb,M.H. and Glomset,J.A. (1994) Rab geranylgeranyl transferase catalyzes the geranylgeranylation of adjacent cysteines in the small GTPases, Rab1A, Rab3A and Rab5A. Proc. Natl Acad. Sci. USA, 91, 11963–11967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hightower K.E., Huang,C.-C., Casey,P.J. and Fierke,C.A. (1998) H-ras peptide and protein substrates bind protein farnesyltransferase as an ionized thiolate. Biochemistry, 37, 15555–15562. [DOI] [PubMed] [Google Scholar]
- Hoffman G.R., Nassar,N. and Cerione,R.A. (2000) Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell, 100, 345–356. [DOI] [PubMed] [Google Scholar]
- Johnston S.R.D. (2001) Farnesyl transferase inhibitors: a novel targeted therapy for cancer. Lancet Oncol., 2, 18–26. [DOI] [PubMed] [Google Scholar]
- Kim E., Ambroziak,P., Otto,J.C., Taylor,B., Ashby,M., Shannon,K., Casey,P.J. and Young,S.G. (1999) Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J. Biol. Chem., 274, 8383–8390. [DOI] [PubMed] [Google Scholar]
- Kohl N.E. et al. (1995) Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat. Med., 1, 792–797. [DOI] [PubMed] [Google Scholar]
- Li X., Liu,L., Tupper,J.C., Bannerman,D.D., Winn,R.K., Sebti,S.M., Hamilton,A.D. and Harlan,J.M. (2002) Inhibition of protein geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis in human endothelial cells. J. Biol. Chem., 277, 15309–15316. [DOI] [PubMed] [Google Scholar]
- Loew A., Ho,Y.K., Blundell,T. and Bax,B. (1998) Phosducin induces a structural change in transducin βγ. Structure, 6, 1007–1019. [DOI] [PubMed] [Google Scholar]
- Long S.B., Casey,P.J. and Beese,L.S. (1998) Co-crystal structure of protein farnesyltransferase with a farnesyl diphosphate substrate. Biochemistry, 37, 9612–9618. [DOI] [PubMed] [Google Scholar]
- Long S.B., Casey,P.J. and Beese,L.S. (2000) The basis for K-Ras4B binding specificity to protein farnesyltransferase revealed by 2 Å resolution ternary complex structures. Structure, 8, 209–222. [DOI] [PubMed] [Google Scholar]
- Long S.B., Hancock,P.J., Kral,A.M., Hellinga,H.W. and Beese,L.S. (2001) The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics. Proc. Natl Acad. Sci. USA, 98, 12948–12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.B., Casey,P.J. and Beese,L.S. (2002) Reaction path of protein farnesyltransferase at atomic resolution. Nature, 419, 645–650. [DOI] [PubMed] [Google Scholar]
- Moores S.L., Schaber,M.D., Mosser,S.D., Rands,E., O’Hara,M.B., Garsky,V.M., Marshall,M.S., Pompliano,D.L. and Gibbs,J.B. (1991) Sequence dependence of protein isoprenylation. J. Biol. Chem., 266, 14603–14610. [PubMed] [Google Scholar]
- Park H.-W., Boduluri,S.R., Moomaw,J.F., Casey,P.J. and Beese,L.S. (1997) Crystal structure of protein farnesyltransferase at 2.25 Å resolution. Science, 275, 1800–1804. [DOI] [PubMed] [Google Scholar]
- Reiss Y., Goldstein,J.L., Seabra,M.C., Casey,P.J. and Brown,M.S. (1990) Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell, 62, 81–88. [DOI] [PubMed] [Google Scholar]
- Seabra M.C. (1998) Membrane association and targeting of prenylated Ras-like GTPases. Cell Signal., 10, 167–172. [DOI] [PubMed] [Google Scholar]
- Seabra M.C., Goldstein,J.L., Sudhof,T.C. and Brown,M.S. (1992) Rab geranylgeranyltransferase: a multisubunit enzyme that prenylates GTP-binding proteins terminating in Cys–Cys or Cys–X–Cys. J. Biol. Chem., 267, 14497–14503. [PubMed] [Google Scholar]
- Sebti S.M. and Hamilton,A.D. (2000) Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene, 19, 6584–6593. [DOI] [PubMed] [Google Scholar]
- Stark W.W. Jr, Blaskovich,M.A., Johnson,B.A., Qian,Y., Vasudevan,A., Pitt,B., Hamilton,A.D., Sebti,S.M. and Davies,P. (1998) Inhibiting geranylgeranylation blocks growth and promotes apoptosis in pulmonary vascular smooth muscle cells. Am. J. Physiol., 275, L55–L63. [DOI] [PubMed] [Google Scholar]
- Steiger A., Pyun,H.-J. and Coates,R.M. (1992) Synthesis and characterization of aza analogue inhibitors of squalene and geranylgeranyl diphosphate synthases. J. Org. Chem., 57, 3444–3449. [Google Scholar]
- Stirtan W.G. and Poulter,C.D. (1997) Yeast protein geranylgeranyltransferase type-I: steady-state kinetics and substrate binding. Biochemistry, 36, 4452–4557. [DOI] [PubMed] [Google Scholar]
- Strickland C.L. et al. (1998) Crystal structure of farnesyl protein transferase complexed with a caax peptide and farnesyl diphosphate analogue. Biochemistry, 37, 16601–16611. [DOI] [PubMed] [Google Scholar]
- Tamanoi F. and Sigman,D.S. (eds) (2001) Protein Lipidation. Academic Press, San Diego, CA. [Google Scholar]
- Terwilliger T.C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D, 56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma N.H., Iakovenko,A., Owen,D., Scheidig,A.S., Waldmann,H., Goody,R.S. and Alexandrov,K. (2000) Phosphoisoprenoid binding specificity of geranylgeranyltransferase type II. Biochemistry, 39, 12043–12052. [DOI] [PubMed] [Google Scholar]
- Thoma N.H., Niculae,A., Goody,R.S. and Alexandrov,K. (2001) Double prenylation by RabGGTase can proceed without dissociation of the mono-prenylated intermediate. J. Biol. Chem., 276, 48631–48636. [DOI] [PubMed] [Google Scholar]
- Tschantz W.R., Furfine,E.S. and Casey,P.J. (1997) Substrate binding is required for release of product from mammalian protein farnesyltransferase. J. Biol. Chem., 272, 9989–9993. [DOI] [PubMed] [Google Scholar]
- Vogt A., Qian,Y., McGuire,T.F., Hamilton,A.D. and Sebti,S.M. (1996) Protein geranylgeranylation, not farnesylation, is required for the G1 to S phase transition in mouse fibroblasts. Oncogene, 13, 1991–1999. [PubMed] [Google Scholar]
- Walters C.E., Pryce,G., Hankey,D.J., Sebti,S.M., Hamilton,A.D., Baker,D., Greenwood,J. and Adamson,P. (2002) Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J. Immunol., 168, 4087–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K., Goodwin,G.W., Ghomashchi,F., Glomset,J.A. and Gelb,M.H. (1991) A protein geranylgeranyltransferase from bovine brain: implications for protein prenylation specificity. Proc. Natl Acad. Sci. USA, 88, 5302–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K., McGeady,P. and Gelb,M.H. (1995) Mammalian protein geranylgeranyltransferase-I: substrate specificity, kinetic mechanism, metal requirements and affinity labeling. Biochemistry, 34, 1344–1354. [DOI] [PubMed] [Google Scholar]
- Yokoyama K., Zimmerman,K., Scholten,J. and Gelb,M.H. (1997) Differential prenyl pyrophosphate binding to mammalian protein geranylgeranyltransferase-I and protein farnesyltransferase and its consequences on the specificity of protein prenylation. J. Biol. Chem., 272, 3944–3952. [DOI] [PubMed] [Google Scholar]
- Zhang F.L. and Casey,P.J. (1996) Influence of metal ions on substrate binding and catalytic activity of mammalian protein geranylgeranyltransferase type-I. Biochem. J., 320, 925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.L., Diehl,R.E., Kohl,N.E., Gibbs,J.B., Giros,B., Casey,P.J. and Omer,C.A. (1994a) cDNA cloning and expression of rat and human protein geranylgeranyltransferase Type-I. J. Biol. Chem., 269, 3175–3180. [PubMed] [Google Scholar]
- Zhang F.L., Moomaw,J.F. and Casey,P.J. (1994b) Properties and kinetic mechanism of recombinant mammalian protein geranylgeranyl transferase type I. J. Biol. Chem., 269, 23465–23470. [PubMed] [Google Scholar]
- Zhang H., Seabra,M.C. and Deisenhofer,J. (2000) Crystal structure of Rab geranylgeranyltransferase at 2.0 Å resolution. Structure, 8, 241–251. [DOI] [PubMed] [Google Scholar]