IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates (original) (raw)
. Author manuscript; available in PMC: 2010 Aug 7.
SUMMARY
During endoplasmic reticulum (ER) stress, homeostatic signaling through the unfolded protein response (UPR) augments ER protein-folding capacity. If homeostasis is not restored, the UPR triggers apoptosis. We found that the ER transmembrane kinase/endoribonuclease (RNase) IRE1α is a key component of this apoptotic switch ER stress induces IRE1α kinase autophosphorylation, activating the RNase to splice XBP1 mRNA and produce the homeostatic transcription factor, XBP1s. Under ER stress—or forced autophosphorylation—IRE1α’s RNase also causes endonucleolytic decay of many ER-localized mRNAs, including those encoding chaperones, as early events culminating in apoptosis. Using chemical-genetics, we show that kinase inhibitors bypass autophosphorylation to activate the RNase by an alternate mode that enforces XBP1 splicing, and averts mRNA decay and apoptosis. Alternate RNase activation by kinase-inhibited IRE1α can be reconstituted in vitro. We propose that divergent cell fates during ER stress hinge on a balance between IRE1α RNase outputs that can be tilted with kinase inhibitors to favor survival.
INTRODUCTION
Chaperones and other enzymatic activities residing in the endoplasmic reticulum (ER) promote folding of secretory proteins. Demand on ER protein folding often outpaces capacity, causing unfolded proteins to accumulate. During these instances of ER stress, intracellular signaling pathways termed the unfolded protein response (UPR) become active. Mammalian cells contain three unfolded protein sensors—IRE1α, PERK, and ATF6—that initiate UPR activation. Signaling by these sensors upregulates transcription of genes encoding ER chaperones, oxidoreductases, and ER-associated degradation (ERAD) components (Travers et al., 2000). The UPR also exerts a translational block during ER stress (Harding et al., 2001). These outputs are adaptive because they reduce ER protein load, augment ER protein folding capacity, and promote degradation of unfolded proteins (Ron and Walter, 2007). If ER stress is successfully reduced, negative feedback causes UPR signaling to wane as homeostasis becomes restored (Merksamer et al., 2008).
Failure to adapt to ER stress causes the UPR to trigger apoptosis. Chemicals that inhibit ER protein glycosylation or deplete ER calcium can be used to impose ER stress at unremediable levels, deterministically triggering apoptosis. Also, mutations in many genes encoding secretory proteins cause these proteins to fold improperly, thereby generating chronic ER stress and leading to cell degenerative diseases. For example, unoxidizable proinsulin mutants cause rare forms of diabetes in humans and rodents (Oyadomari et al., 2002; Stoy et al., 2007). Removal of ER-stressed cells through UPR-mediated apoptosis may occur physiologically, but widespread apoptosis becomes pathological when it leads to the loss of large numbers of cells in vital organs (Kaufman, 2002).
Paradoxically, UPR signaling promotes opposite cell fates—adaptation/survival versus death—thereby acting as a switch. Underlying mechanisms of this switching process remain unclear. Because UPR signaling exhibits heavy crosstalk, individual signaling events are unlikely to completely determine cell fate. However, it is reasonable to expect that the three unfolded protein sensors—IRE1α, PERK, and ATF6—influence the life-death decision. Inability of UPR outputs to restore homeostasis may generate continuous signaling from these sensors, tipping the balance in favor of apoptosis. To explore this possibility, we studied the most ancient ER unfolded protein sensor, IRE1α.
IRE1α is an ER transmembrane protein containing two enzymatic activities, a kinase and an endoribonuclease (RNase), both residing on its cytosolic face (Wang et al., 1998). IRE1α senses ER unfolded proteins through an ER lumenal domain that becomes oligomerized during ER stress (Aragon et al., 2009; Credle et al., 2005; Zhou et al., 2006). Oligomerization juxtaposes the kinase domains, which consequently _trans_-autophosphorylate. Autophosphorylation activates the RNase activity to cleave XBP1 mRNA at specific sites to excise an intron. Religation of IRE1α-cleaved XBP1 mRNA shifts the open reading frame; translation of spliced XBP1 mRNA produces a potent transcription factor called XBP1s (s=spliced) (Calfon et al., 2002; Yoshida et al., 2001). XBP1s’s target genes encode protein products that enhance ER protein folding capacity and quality control (Lee et al., 2003). Thus, IRE1α promotes adaptation via XBP1s.
IRE1α may have functions independent of XBP1 mRNA splicing. IRE1 was implicated in the decay of ER-localized mRNAs in D. melanogaster (Hollien and Weissman, 2006). However, it remains unclear if ER-localized mRNA decay occurs in mammalian cells, if IRE1α’s RNase directly mediates such mRNA decay, and whether the physiological consequences of mRNA decay are adaptive or destructive. IRE1α was also linked to pro-apoptotic c-Jun kinase (JNK) signaling (Urano et al., 2000). Together, existing data support direct and indirect roles for IRE1α in both adaptive and destructive processes, but mechanistic details remain unclear.
Kinase activation in signal transduction pathways is often transitory if downstream effects restore homeostasis. We hypothesized that persistent activation of IRE1α’s kinase could signal an inability to adapt to ER stress and trigger a switch into apoptosis. To test this hypothesis we employed small molecules to forcibly activate IRE1α and key mutants, allowing us to assign physiological functions to the catalytic activities. We learned that alternate outputs from IRE1α’s RNase activity govern opposing cell fate outcomes during ER stress, and that these outputs can be modulated with inhibitors targeting the kinase domain.
RESULTS
IRE1α RNase Activity Can Be Controlled Through Two Distinct Routes
We began studies on UPR-mediated apoptosis using INS-1 insulinoma cells, which are differentiated insulin-producing cells derived from pancreatic islet β-cells. In INS-1 cells undergoing ER stress from exposure to the ER calcium pump inhibitor thapsigargin (Tg), IRE1α autophosphorylates and splices XBP1 mRNA. Both IRE1α-catalyzed events persist throughout Tg treatment as cells undergo apoptosis (Figure S1). To inquire whether IRE1α activation affects cell fate, we designed tools to forcibly trigger its two catalytic activities—together or separately—without imposing upstream ER stress.
Unfolded proteins in the ER promote oligomerization of IRE1α’s ER lumenal domain as the initial activating event, but oligomerization can also be driven through mass action (Shamu and Walter, 1996). We reasoned that by acutely elevating levels of IRE1α its ER lumenal domains should spontaneously oligomerize, juxtaposing the kinase domains to autophosphorylate and activate the RNase. To this end, we introduced tetracycline-inducible expression constructs driving wild-type (WT) IRE1α or various mutants, into INS-1 cells that stably express Tet repressor (Figure 1A and S2) (Thomas et al., 2004). All expression constructs were stably integrated at the same chromosomal FRT docking site so as to cancel out position effects and reliably ascribe differences in downstream physiological effects to the IRE1α variants. To ensure that physiological effects of IRE1α signaling are general, and not cell-type specific, we repeated the strategy using another cell line, T-REx 293.
Figure 1. Conditional Tools to Forcibly Trigger IRE1α Catalytic Activities.

(A) IRE1α mutants in this study, and the chemical structure of 1NM-PP1. (B) Anti-Myc immunoblot of IRE1α transgenic proteins induced in INS-1 stable lines with doxycycline (Dox). (C) Confocal fluorescence micrographs of INS-1 cells showing co-localization of eGFP-WT IRE1α and eGFP-IRE1α (I642G) with transiently transfected ER-DsRED. Time course immunoblots of transgenic IRE1α proteins induced with Dox in INS-1 cells (anti-Myc and anti-phospho IRE1α). (D) Anti-total and anti-phospho IRE1α immunoblots of WT IRE1α, I642G, and K907A proteins produced from constructs transfected into Ire1 α−/− MEFs, +/− 1NM-PP1. (E) Immunoprecipitation (i.p) of Myc-tagged IRE1α proteins from T-REx 293 cells. (F) in vitro autophosphorylation of i.p.-ed Myc-tagged IRE1α and mutants, +/−1NM-PP1.
In these systems, transgenic WT IRE1α is tightly inducible with doxycyline (Dox) (Figure 1B, lanes 1, 2). Induced WT IRE1α proteins become properly ER-targeted, and spontaneously autophosphorylate as they accumulate (Figure 1C and D). Autophosphorylation activates the RNase, which converts essentially all cellular XBP1 mRNA to the spliced form (96+/− 3%) (Figure 2A, lanes 1, 2).
Figure 2. Two Distinct Modes to Produce XBP1s Transcription Factor.

(A) EtBr-stained agarose gel of XBP1 cDNA amplicons after induction of IRE1α variants for 8 hrs, followed by 5µM 1NM-PP1 (or DMSO) for 4 hrs. The cDNA amplicon of unspliced XBP1 mRNA is cleaved by a PstI site within a 26 nucleotide intron to give 2U and 3U. IRE1α-mediated cleavage of the intron and re-ligation in vivo removes the PstI site to give the 1S (spliced) amplicon. *is a spliced/unspliced XBP1 hybrid amplicon. The ratio of spliced over (spliced + unspliced) amplicons—1S/(1S+2U+3U)—is reported as % spliced XBP1 amplicons in histograms. Three independent biological samples were used. Data are means +/− SD. P-values: ** <0.01 and *** <0.002. ns=not significant. (B) % spliced XBP1 amplicons in IRE1α (I642G)-expressing INS-1 cells as a function of [1NM-PP1]. (C) Curve fitting to triplicate 1NM-PP1 concentration-dependent splicing assays in IRE1α (I642G)-expressing T-REx 293 cells. (D) Time course of XBP1 mRNA splicing under “phosphotransfer activation”—defined as provision of Dox (0.1µg/ml) and 1NM-PP1 (5µM) to stable WT IRE1α-expressing cells. 1NM-PP1 is inert for WT IRE1α—Figure 2A, lane 3—but always added to maintain consistency with pseudokinase activation. (E) ER stress-mediated splicing of XBP1 mRNA in INS-1 CAT cells using tunicamycin—Tm—(5µg/ml) for 2 hrs. (F) Time course of XBP1 mRNA splicing under “pseudokinase activation”, defined as provision of Dox (0.1µg/ml) and 1NM-PP1 (5µM) to stable IRE1α (I642G)-expressing cells. Time courses shown used T-REx 293 cells and were identical in INS-1 cells (not shown). (G) Immunoblot of XBP1s transcription factor under phosphotransfer or pseudokinase activation in INS-1 cells, and INS-1 CAT cells under Tm.
We then added a second layer of conditionality to test effects of IRE1α kinase inhibition. We mutated IRE1α at Ile642 to either Ala or Gly to create enlarged kinase pockets (Figure 1A). I642A and I642G mutants are severely compromised for autophosphorylation in vitro, indistinguishably from the kinase-dead mutant K599A (Figure 1F, lanes 1, 3, 5, 7) (Tirasophon et al., 1998). IRE1α (I642A) retains partial XBP1 mRNA splicing upon induction (57+/− 9% splicing), but IRE1α (I642G) is completely crippled for splicing (Figure 2A, lanes 5, 6, 9, 10). IRE1α (I642G) accumulates to levels comparable to transgenic WT IRE1α, localizes properly to the ER, but does not autophosphorylate (Figure 1B, lanes 2, 6; Figure 1C; Figure 1D, lanes 5,6).
Due to their enlarged kinase pockets, I642A and I642G selectively bind 1NM-PP1, a cell-permeable adenosine nucleotide mimic with a bulky chemical head group (Figure 1A) (Shah et al., 1997). As 1NM-PP1 cannot bind the WT IRE1α kinase domain, autophosphorylation and RNase activity are not affected (Figure 1F, lanes 1, 2; Figure 2A, lanes 2, 4). However, for both I642 mutants, 1NM-PP1 completely corrects the defect in XBP1 mRNA splicing (Figure 2A, lanes 2, 6, 8,10,12).
IRE1α (I642A) and IRE1α (I642G) behave as pseudokinases, a class of kinases that lost phosphotransfer activity through evolution and instead regulate functions in attached domains (Boudeau et al., 2006). 1NM-PP1 does not restore autophosphorylation to these mutants (Figure 1F, lanes 4, 6), but instead allosterically activates the attached RNase through binding the pseudokinase.
IRE1α (I642G) was utilized henceforth because of its large dynamic range: 1.8 +/− 0.5% splicing at baseline, to 97.6 +/− 2.5 % when saturated with 1NM-PP1 (Figure 2A, lanes 10, 12). 1NM-PP1 activation of splicing by IRE1α (I642G) is finely tunable, exhibiting an EC50 of 887nM and a Hill coefficient of 1.86 (Figure 2B, C). 1NM-PP1 stimulates splicing in cis: when IRE1α (I642G) is combined with the RNase active site mutation, K907A (Figure 1A) (Tirasophon et al., 1998), splicing becomes unresponsive to 1NM-PP1 (Figure 2A, lanes 12, 16).
Thus, by using Dox to induce expression of either WT IRE1α or IRE1α (I642G), and also adding 1NM-PP1 which only activates IRE1α (I642G), XBP1 mRNA splicing can be forcibly triggered and sustained for many hours (Figure 2D, F). Depending on the cell line we use (those expressing WT IRE1α or IRE1α (I642G), the two different activation modes are referred to as phosphotransfer activation or pseudokinase activation respectively. Both modes trigger XBP1 mRNA splicing with similar kinetics, causing XBP1s protein to accumulate to levels mimicking those that occur during ER stress (Figure 2E, 2G). However, neither activation mode causes ER stress per se, as evidenced by lack of eIF2α phosphorylation over 8 hrs (Figure S3).
Cell Fate is Determined by Distinct Mechanisms of IRE1α RNase Activation
By forcibly activating IRE1α through two different modes without relying on pleiotropic ER stress agents that activate all three UPR arms, we could dissect effects of IRE1α’s catalytic activities on cell fate. We find that phosphotransfer activation triggers apoptosis in INS-1 cells starting at 36 hrs (Figure 3A, B). In contrast, pseudokinase-activated INS-1 cells do not undergo apoptosis. These divergent fates are replicated in phosphotransfer- and pseudokinase-activated T-REx 293 cells, indicating that they are general (Figure S5, Movie S1-Movie S4).
Figure 3. Divergent Cell Fates under Phosphotransfer and Pseudokinase Activation of IRE1α RNase.

INS-1 cells under phosphotransfer or pseudokinase activation, activation of kinase-active/RNase-dead mutants K907A and N906A, and expression of XBP1s: (A) Time course of percent cells staining positive for Annexin V. (B) Time course immunoblots of full-length and cleaved caspase 3, and Myc-IRE1α proteins. NT=not treated (C) Percent Ire1 α−/− and Ire1 α+/+ MEFs staining positive for Annexin V during ER stress induced by brefeldin A (BFA) (0.5µg/mL), Tm (0.5µg/mL) or Tg (0.1µM). (D) Percent cells staining positive for Annexin V 24 hrs after transfection of expression vectors encoding WT IRE1α, I642G, K907A, or N906A into Ire1 α−/− or _Xbp1_−/− MEFs. 5µM 1NM-PP1 (or DMSO) was added at the time of transfection. (E) Percent cells staining positive for Annexin V 24 hrs after transfection of WT IRE1α expression vector (WT) or empty vector (vector) in _Bax_−/− _Bak_−/− MEFs. (F) Percent cells staining positive for Annexin V 24 hrs after provision of varying concentrations of Tm in Ire1 α−/− or _Xbp1_−/− MEFs. All cells were previously transfected with 1µg of expression vectors bearing WT, I642G, K907A, or N906A for 8 hrs, treated with 1NM-PP1 (or DMSO) overnight and then treated for 24 hrs with Tm. Data are means +/− SD. P-values: **<0.02 and ***<0.002. Figure S3 shows equivalent transgenic IRE1α protein levels. For all experiments, three independent biological samples were measured.
Because both activation modes cause complete XBP1 mRNA splicing to produce equivalent amounts of XBP1s, the divergent fates of survival versus death cannot be ascribed to outputs of this transcription factor. Indeed, conditional expression of XBP1s is itself not pro-apoptotic (Figure 3A and B), suggesting instead that phosphotransfer-activated IRE1α transmits pro-apoptotic signals independent of XBP1 mRNA splicing.
A plausible explanation holds that pro-apoptotic outputs of WT IRE1α proceed directly from phosphotransfer activity, which IRE1α (I642G) lacks. If true, one would predict that kinase-active/RNase-dead IRE1α mutants should be as cytotoxic as WT IRE1α, if not more so, since the adaptive benefits derived from XBP1s would also be lost. To test this notion, we conditionally expressed two different kinase-active/RNase-dead mutants, K907A and N906A, in INS-1 cells. Surprisingly, despite retaining full phosphotransfer activity (Figure 1F, lane 1, 9, also Figure S6), K907A and N906A do not induce apoptosis (Figure 3A and B).
Endogenous IRE1α is also pro-apoptotic during unremediable ER stress: _Ire1_−/− mouse embryonic fibroblasts (MEFs) demonstrate greater survival than Ire1+/+ MEFs under toxic doses of various ER stress agents (Figure 3C). In _Ire1_−/− MEFs, apoptosis is spontaneously triggered through transient expression of WT IRE1α, but not through the I642G, K907A, or N906A mutants (Figure 3D). Similarly, apoptosis is triggered when WT IRE1α (but no other mutant) is expressed in _Xbp1_−/− MEFs, strengthening the notion that XBP1s is unnecessary for IRE1α-mediated apoptosis (Figure 3D). WT IRE1α triggers mitochondrial (intrinsic) apoptosis because it requires Bax and Bak genes (Figure 3E) (Wei et al., 2001).
Our results clearly argue that apoptotic signals from WT IRE1α require an active RNase. Yet, pseudokinase activation of the same RNase through 1NM-PP1 does not trigger apoptosis. Furthermore, pre-treatment of cells through pseudokinase activation reduces apoptosis when cells are exposed to Tm (Figure 3F). Cytoprotection requires three components: a catalytically-active RNase in IRE1α (I642G), the XBP1 gene, and 1NM-PP1, indicating that it proceeds from a pre-conditioned state brought about through pre-emptive XBP1s production.
These paradoxical results clearly focus attention on the RNase activity as the immediate downstream effector of both life and death outputs emanating from IRE1α. To reconcile these findings, we hypothesized that for IRE1α the specific kinase activation mode—phosphotransfer vs. pseudokinase—must exert two mechanistically distinct outputs from the RNase that cause opposite effects on cell fate. To illuminate the mechanistic basis of these alternate RNAse outputs we employed gene expression profiling.
Phosphotransfer-Activation of IRE1α RNase Causes ER-Localized mRNA Decay
Using DNA microarrays, we compared cellular mRNAs in phosphotransfer- and pseudokinase-activated T-REx cells at two time points: an early time point of 8 hrs when induction of transgenic IRE1α proteins (and autophosphorylation) and XBP splicing have all reached new steady-state levels, and a later time point of 24 hrs when divergent cell fate outcomes start to emerge between the two activation modes.
Pairwise expression profiling was conducted using untreated cells as controls. Clustering analysis indicates excellent overlap of genes induced more than two-fold between both activation schemes, especially at 24 hrs (Figure 4A). The induced genes have been previously implicated in secretory processes of protein translocation (Travers et al., 2000), ER-to-Golgi trafficking and retrieval (Higashio and Kohno, 2002), and ER protein folding (Table S1).
Figure 4. Gene Expression Profiling Reveals ER-localized mRNA Decay under IRE1α Phosphotransfer Activation and ER Stress.
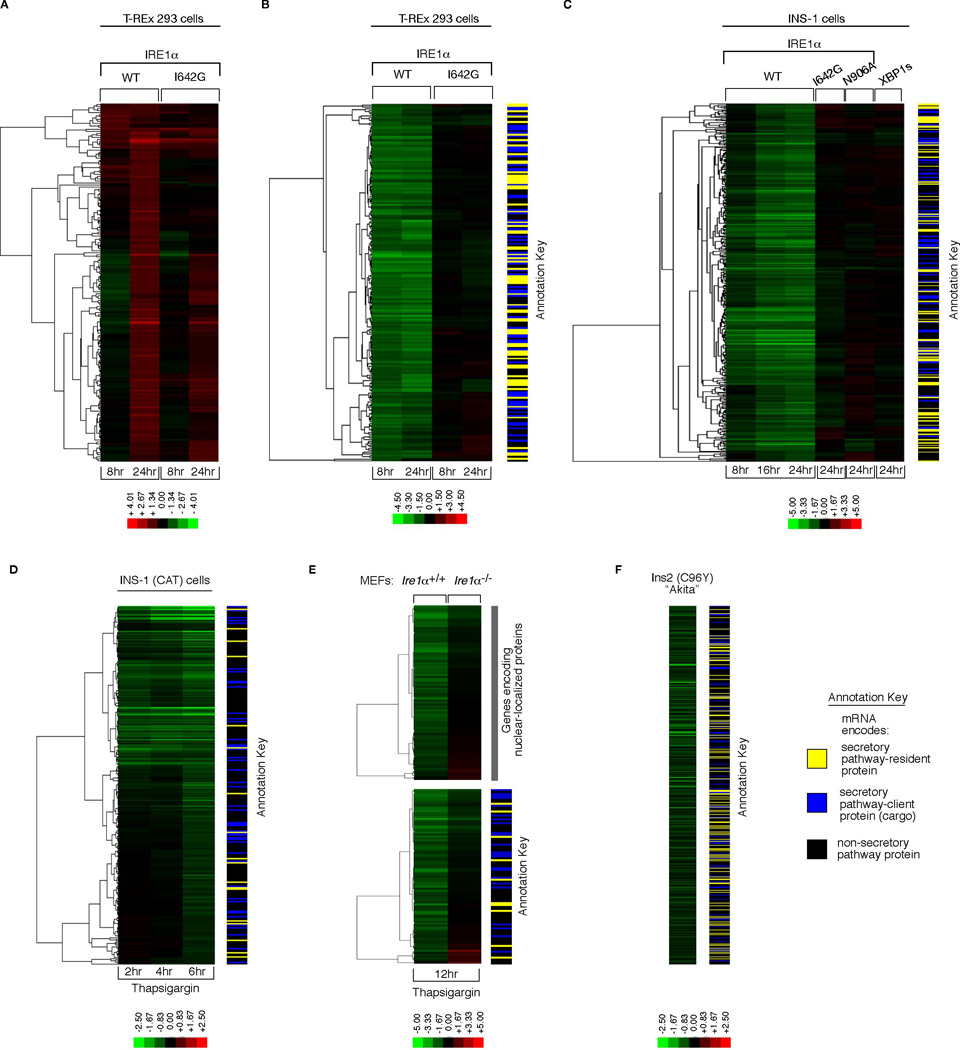
Hierarchical clustering analysis of gene expression changes from DNA microarrays. (A) Genes whose expression is 2-fold or more increased under either phosphotransfer activation or pseudokinase activation in T-REx 293 cells. (B) Genes whose expression is significantly reduced 2-fold or more at 8 hrs in phosphotransfer-activated T-REx 293 cells (vs. untreated) (q-value < 0.05), and significantly different between phosphotransfer activation and pseudokinase activation at 8 hrs (q-value<0.05). (C) Genes whose expression is significantly reduced 2-fold or more at 24 hrs in phosphotransfer-activated INS-1 cells (vs. untreated) (q-value < 0.05), and significantly different between phosphotransfer activation, pseudokinase activation, and phosphotransfer activation of the RNase-dead mutant N906A at 24 hrs (q-value < 0.05)— expression changes for these genes at 8 and 16 hrs under phosphotransfer activation and at 24 hrs in XBP1s-expressing cells are displayed in the same row (D) Genes that are significantly downregulated at 6 hrs under 1µM Tg in INS-1 CAT cells— expression changes for these genes at 2 and 4 hrs under 1µM Tg are displayed in the same row (q-value < 0.05). (E) Genes that are significantly reduced 2-fold or more under 1µM Tg in Ire1 α+/+ MEFs (q-value < 0.05), and significantly different between 1µM Tg-treated WT MEFs (vs. untreated) and 1µM Tg-treated Ire1 α−/− MEFs (vs. untreated) (q-value < 0.05). (F) Genes that are significantly reduced in normally growing Ins2 (C96Y) Akita cells compared to isogenic WT controls (q-value < 0.05). Annotation keys were used to divide the predicted function of down-regulated genes into SPR: Secretory-pathway ER-resident protein, SPC: Secretory-pathway client protein (cargo), or NSP: Non-secretory pathway protein. A set of IRE1α-dependent decreasing mRNAs encoding nuclear localized genes was noted in (E). Colorbars indicate upregulated genes in red, and downregulated genes in green (_log_2). For all experiments, the mean of three or four independent biological samples is shown. See Table S1–Table S6 for gene identities, _log_2 expression changes, and statistics.
However, when comparing genes whose expression is significantly reduced by at least two-fold, large differences are found between the two activation modes. By 8 hrs the expression levels of two hundred different mRNAs are reduced under phosphotransfer activation, but not under pseudokinase activation (Figure 4B) (Table S2). Reduction of these mRNAs is evident as early as 4 hrs after phosphotransfer activation, and levels continue to decline over several hours (Figure S5A). By contrast, XBP1 mRNA splicing is already maximal by 4 hrs. Reduction of these mRNAs precedes entry of phosphotransfer-activated cells into apoptosis by many hours, indicating that it is not a consequence of cell death (Figure 3A, 3B, S5, and movie S1-movie S4). The pattern of mRNA reduction is general, as it also occurs in phosphotransfer-activated INS-1 cells (Figure 4C and S5). mRNA reduction does not occur under pseudokinase-activation, upon expression of kinase-active/RNase-dead mutant (N906A), or expression of XBP1s (Figure 4C).
Using annotation databases, we find that genes encoding secretory pathway proteins are strongly enriched in the target set of reduced expression (see annotation key in Figure 4B and C, and Table S2 and S3 for statistics). mRNAs of these genes are predicted to be localized near the ER membrane during translocational translation of their protein products, in close proximity to IRE1α which could promote their endonucleolytic decay. Inability of the kinase-active/RNase-dead mutant (N906A) to cause decay of these mRNAs is consistent with the notion that the IRE1α RNase catalytic activity is directly responsible for promoting the mRNA decay in vivo.
The profile of ER-localized mRNA decay under IRE1α phosphotransfer activation is mimicked in cells undergoing ER stress. Three examples follow: First, a time course of parent INS-1 CAT cells treated with Tg shows ER mRNA decay evolving over time (Figure 4D and Table S4). Second, Ire1 α−/− MEFs treated with Tg show significantly reduced ER mRNA decay as compared to Ire1 α+/+ MEFs, indicating dependence of decay on endogenous IRE1α (Figure 4E and Table S5). Third, pancreatic β-derived cells carrying a mutation in the insulin-encoding gene, Ins2(C96Y), display significant reductions in ER-localized mRNAs even during normal growth when compared to isogenic WT cells (Figure 4F and Table S6). These so-called “Akita” variants prevent ER oxidative folding of the encoded proinsulin and cause chronic ER stress in β-cells and diabetes in mice (Oyadomari et al., 2002).
Many hundreds of the decaying mRNAs encode proteins that transit through the ER en route to the cell surface, intra-cellular organelles, or the cell exterior. These secretory pathway cargo proteins—“SPC”—include insulin, HMG-CoA reductase, β2 microglobulin, and numerous cell surface receptors (Table S2–Table S6). We validated that IRE1α phosphotransfer activation is sufficient to cause insulin (Ins1) mRNA decay (Figure 5A), or an ER-targeted but not a cytosolic-targeted GFP mRNA (Figure S14).
Figure 5. Validation of Cargo and Chaperone mRNA/Protein Decay under IRE1α Phosphotransfer Activation or ER Stress.
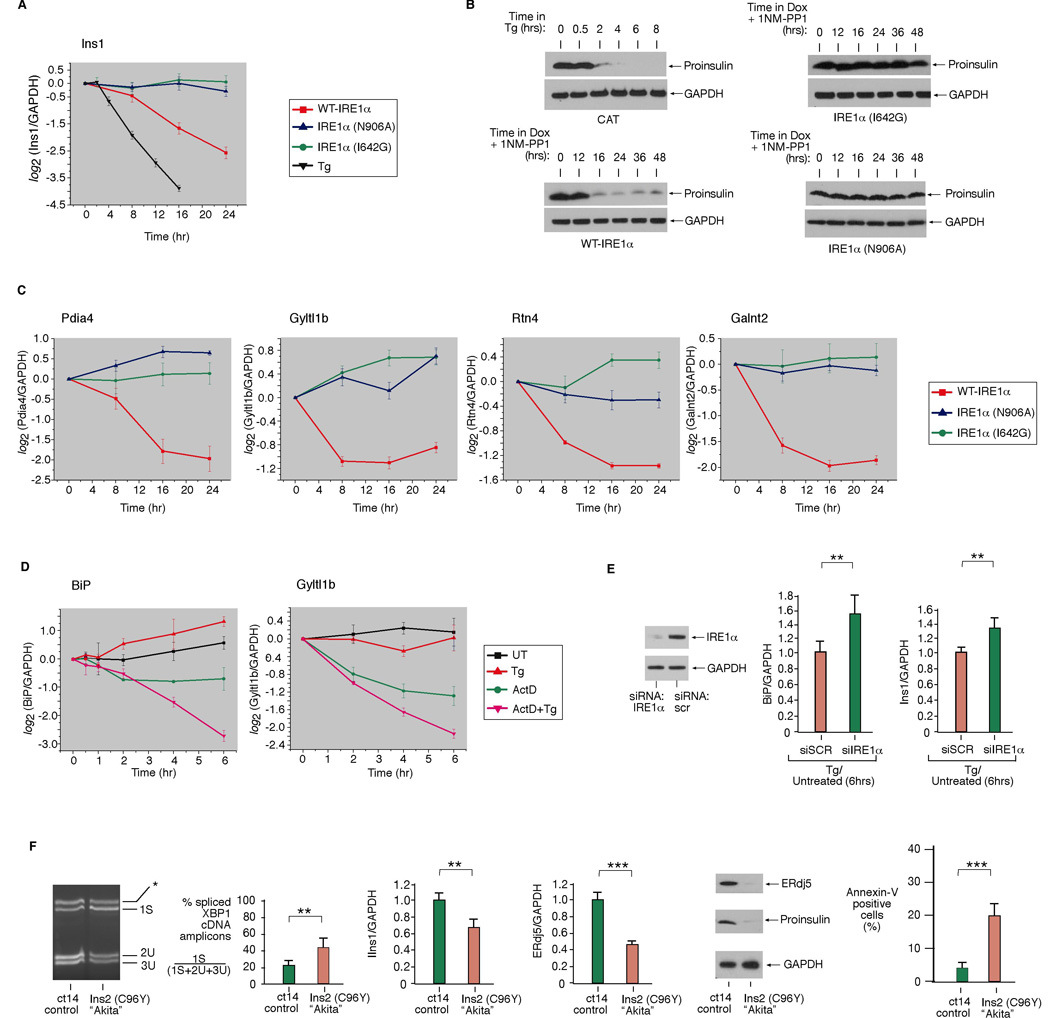
(A) Time course analysis of Ins1 mRNA expression (normalized to GAPDH) during ER stress (1µM Tg), WT IRE1α, I642G, or N906A activation in INS-1 cells by quantitative real-time PCR (Q-PCR). (B) Immunoblot of proinsulin during ER stress (1µM Tg), WT IRE1α, I642G, or N906A activation in INS-1 cells. (C) Time course analysis of the expression of mRNAs encoding ER-resident activities—Pdia4, Gyltl1b, Rtn4, Galnt2—during WT IRE1α, I642G, or N906A activation in INS-1 cells by Q-PCR. (D) Time course analysis of BiP or Gyltl1b mRNA levels during ER stress (1µM Tg) by Q-PCR. To attenuate transcription, cells were pretreated with 5 µg/mL Actinomycin D (or DMSO) for 1 hr, then left untreated or treated for the indicated times with 1µM Tg. (E) Anti-IRE1α immunoblot of INS-1 CAT cells electroporated with IRE1α siRNA or scramble siRNA control (SCR). (F) Baseline XBP1 mRNA splicing in Akita and isogenic WT cells (ct14). Q-PCR analysis of Ins1 or ERdj5 mRNA levels in Akita and ct14 cells. Immunoblot analysis of ERdj5 and proinsulin in Akita and ct14 cells. Annexin V positive staining in Akita and ct14 cells. Three independent biological samples were used for Q-PCR and XBP1 splicing experiments. Data are means +/− SD. P-values: ** <0.02 and *** <0.005.
Phosphotransfer activation causes insulin mRNA to decay rapidly, as was also shown to occur during ER stress (Lipson et al., 2008); insulin mRNA decay precedes large reductions in proinsulin protein (Figure 5A, 5B and S7). In contrast, pseudokinase activation, or expression of the N906A mutant, preserves both insulin mRNA and proinsulin levels (Figure 5A, B). siRNA knockdown of endogenous IRE1α in INS-1 parent cells increases Ins1 mRNA levels during ER stress, confirming that IRE1α is also necessary for Ins1 mRNA decay (Figure 5E, S12).
The mRNAs that we find decaying under phosphotransfer activation or under ER stress not only encode secretory cargo proteins, but also encode many ER and downstream secretory organelle-resident activities. These are annotated as “SPR” for secretory pathway-resident (Figure 4B–F). Validation of four such targets is shown in Figure 5C: mRNAs encoding Golgi-localized glycosylating enzymes Galnt2 and Gyltl1b, ER-localized co-chaperone Pdia4, and the ER membrane structural protein Rtn4. Because many of these mRNAs are replenished by UPR-mediated transcription, the competition through IRE1α-mediated decay may determine their steady-state levels. Therefore, down-regulation of many UPR targets may have even evaded detection in arrays. To identify representative targets, we inhibited transcription with Actinomycin D (ActD) before adding Tg, and find that mRNAs encoding the abundant ER chaperone BiP, as well as Gyltl1b, decay more rapidly during ER stress (Figure 5D). Remarkably, BiP mRNA decay depends on IRE1α because reducing endogenous IRE1α through RNAi increases BiP mRNA levels during ER stress (Figure 5E).
Basal splicing of XBP1 mRNA in normally-growing Akita cells is 42%— twice the level of isogenic WT counterparts (ct14 cells) (Figure 5F). This increased baseline in IRE1α activity correlates with significantly decreased steady-state levels of many ER-localized mRNAs in the Akita cells (Figure 4F). Akita cells have lower insulin mRNA and proinsulin protein content than ct14. While the Akita mutation is in the Ins2 gene, Ins1 mRNA is also reduced, implying a _trans_-dominant effect. Akita cells have significantly decreased levels of many UPR target mRNAs and their protein products, such as ERdj5, and exhibit a threefold higher spontaneous apoptosis rate than ct14 (Figure 5F).
We investigated whether we could uncouple endogenous IRE1α’s XBP1 splicing activity from its mRNA decay activity by varying the degree of ER stress with Tg. Cells cultured in low (5nM) Tg underwent mild XBP1 splicing (26%), proliferated slightly better than untreated cells, and accumulated Ins1 and BiP mRNAs over 24 hrs (Figure S8). However, cells treated with higher Tg (10nM) had larger increases in XBP1 mRNA splicing (to 40%) and IRE1α autophosphorylation, and displayed drop-offs in Ins1 mRNA, and in BiP mRNA after an initial period during which BiP mRNA climbed 3.5 fold. These cells stopped proliferating and underwent apoptosis, indicating that they may have crossed a threshold of unremediable ER stress. These results argue that under low ER stress conditions, IRE1α preferentially splices XPB1, while at higher stress it also causes ER-localized mRNA decay.
Time courses confirm that protein products encoded by many decaying mRNAs decline under ER stress. Figure S9 shows decaying IGFBP1, site 1 and 2 proteases (needed to activate ATF6), BiP, and ERdj5 proteins. Taken together, our data show that under conditions of either overwhelming ER stress induction (e.g. 1µM Tg), or chronic low-level stress (expression of Akita insulin or 10nM Tg), mRNAs encoding ER-resident activities such as chaperones start to decay, as also occurs during phosphotransfer activation. Decay of ER-resident activities under phosphotransfer activation (Figure S10) may result in increased ER stress. Consistent with this notion, phosphorylation of eIF2α (Figure S10) and c-Jun kinases (JNK) (Figure S11) increases at late time points, as also occurs under ER stress. Reciprocally, chaperone levels rise under pseudokinase activation, and baseline levels of phospho-eIF2α and phospho-JNK decline, providing a mechanistic explanation for how 1NM-PP1 could enhance ER functions to pre-condition cells to resist apoptosis during ER stress.
Kinase Inhibitors Constrain IRE1α to Cleave XBP1 and Spare Insulin mRNA
Taken together, we show that IRE1α has two functions in vivo, both requiring its RNase: (1) IRE1α initiates XBP1 mRNA splicing through site-specific cleavage, and (2) is necessary and sufficient for ER-localized mRNA decay. To determine whether the second function proceeds directly from endonucleolytic activity, we reconstituted cleavage against XBP1 and mouse Ins2 RNAs in a cell-free system. We immunoprecipitated three versions of IRE1α: WT, N906A, and I642G, and incubated equivalent protein amounts with in vitro transcribed Ins2 and XBP1 RNA. WT IRE1α cleaves XBP1 RNA at previously identified sites in two loops flanking the intron (Figure 6A) (Calfon et al., 2002). We find that WT IRE1α also cleaves mouse Ins2 RNA in vitro (Figure 6A). Several specific Ins2 RNA cleavage sites are identified that are highly reproducible using WT IRE1α proteins isolated on different occasions. Rat Ins1 RNA was similarly cleaved at specific sites (Figure S13). We mapped the cleavage sites on IRE1α-treated Ins1 and 2 RNA products by primer extension and found sequence similarity to the scission site in XBP1 RNA (Figure 6D).
Figure 6. Reconstitution of Insulin RNA Cleavage by IRE1α in vitro.
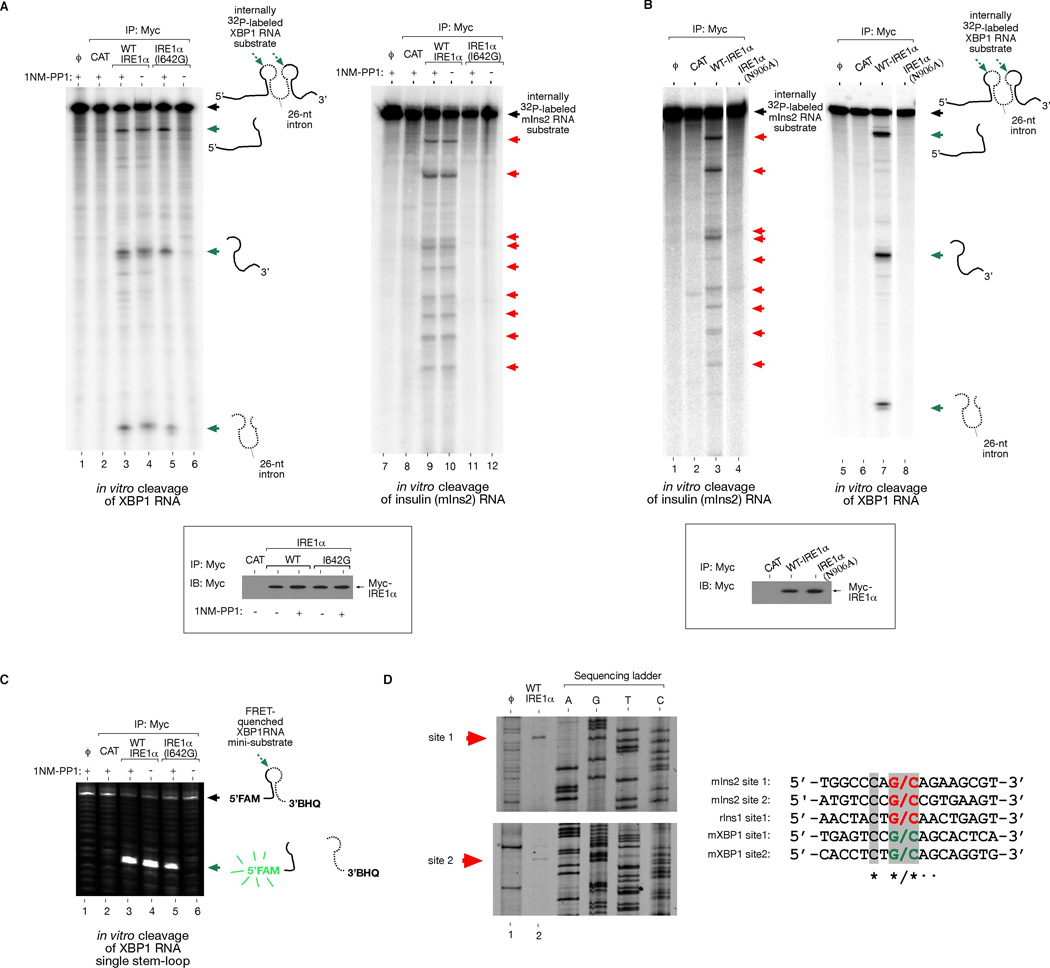
(A) In vitro cleavage of a 352 nucleotide (nt) XBP1 RNA encompassing the 26-nt intron, and a full-length 503-nt mouse Ins2 RNA by immunoprecipitated (i.p.) WT IRE1α and I642G proteins from T-REx 293 cells. ϕ=substrate alone; CAT=mock i.p. from T-REx 293 CAT cells. (B) Control reactions using kinase-active RNase-dead IRE1α (N906A) mutant. Anti-Myc immunoblot analysis show amounts of immunoprecipitated proteins used for in vitro cleavage reactions. (C) In vitro cleavage of 5’FAM -3’BHQ labeled XBP1 single stem-loop mini-substrate by WT IRE1α or I642G proteins +/− 1NM-PP1. ϕ=substrate alone; CAT= mock i.p. from T-REx 293 CAT cells. (D) Mapping of IRE1α cleavage sites in mouse Ins2 RNA, and sequence alignment with the cleavage sites of mouse XBP1 and rat Ins1 shows conservation. An asterisk * indicates complete nucleotide conservation, whereas ● indicates 4 out of 5 conserved nucleotides. See Figure S13 for cleavage site mapping of rat Ins1 RNA.
IRE1α (N906A) cannot cleave either RNA, arguing against the possibility that RNases co-purifying with phosphorylated IRE1α proteins non-specifically degraded Ins1 and Ins2 RNA (Figure 6B and S13). IRE1α (I642G) proteins can cleave XBP1 RNA in vitro, but only under 1NM-PP1 (Figure 6A). Using an internally FRET-quenched XBP1 RNA substrate, 1NM-PP1 activated IRE1α (I642G) is also able to cleave even a single XBP1 RNA stem-loop, similar to WT (Figure 6C). However, IRE1α (I642G) spares Ins2 and Ins1 RNA, even under 1NM-PP1 (Figure 6A and S13), as it does in vivo. Thus, for IRE1α (I642G), 1NM-PP1 uncouples endonucleolytic cleavage of XBP1 RNA from cleavage of insulin RNAs.
To identify kinase inhibitors that similarly activate WT IRE1α RNase, we implemented a screening campaign using the FRET-quenched XBP1 RNA substrate (Figure 6C) in a high-throughput format (Han et al., manuscript in preparation). Several small molecule activators of murine IRE1α including 5-methyl-1H-pyrazol-3-yl-2-phenylquinazolin-4-yl amine (H6) and anti-cancer drugs imatinib and sunitinib (Figure S15A), were identified along with another compound called APY29, recently reported along with sunitinib to activate yeast IRE1 (Korennykh et al., 2009). All these compounds inhibit murine WT IRE1α _trans_-autophosphorylation in vitro to varying degrees (APY29>sunitinib>H6>imatinib). In INS-1 cells, APY29 and H6 increase XBP1 splicing in the absence of ER stress. Moreover, all these compounds allow XBP1 mRNA splicing to proceed fully under ER stress. Under Tg-induced ER stress, APY29 strikingly rescues decline of Ins2 mRNA in a dose-responsive manner, and H6 and imatinib have similar (albeit modest) effects. These results suggest that kinase inhibitors may uncouple the two endonucleolytic output modes of WT IRE1α just as 1NM-PP1 does when acting on the I642G mutant.
DISCUSSION
Mechanistic Basis of Binary Signaling through IRE1α during ER Stress
Our findings focus attention on apoptotic outputs of IRE1α that have received little attention to date. Classically IRE1α is considered a homeostat in the UPR. Here we found that apart from these adaptive roles, IRE1α is also a potent executioner. Thus IRE1α outputs are “double-edged”. Binary outputs are consistent with those expected of a signaling switch, and IRE1α’s divergent signaling may be an important component in the UPR’s homeostatic-apoptotic switching network.
We traced divergent signaling by IRE1α to alternate outputs of its RNase. The adaptive role played by the RNase is well understood: by splicing XBP1 mRNA it produces the cytoprotective XBP1s transcription factor. IRE1α also displays pro-apoptotic signaling (Urano et al., 2000), yet underlying mechanisms are unclear. It was previously reported that IRE1 is needed in D. melanogaster to mediate rapid decay of ER-localized mRNAs (Hollien and Weissman, 2006). Here we showed for the first time that ER-localized mRNA decay also occurs in many mammalian cells undergoing ER stress. Early in the UPR, such decay may provide some immediate respite by reducing protein translocational load. This resembles early outputs of PERK kinase, which temporarily suppresses translation under ER stress. If these rapid measures succeed, reduction of unfolded proteins should cause UPR signaling to decay as homeostasis becomes restored. XBP1s downstream effects may take longer to manifest since they require de novo protein synthesis; these outputs may help pre-condition the ER to combat future ER stress.
Alternatively, under unremediably-high levels of ER stress, sustained increases in XBP1 mRNA splicing are accompanied by decay in many ER-localized mRNAs. These mRNAs encode secretory cargo proteins, and secretory pathway resident-activities that promote folding of cargo. Continued decay of these mRNAs under unmitigated ER stress may deplete crucial cell-surface signaling proteins, and perhaps more importantly, may destabilize ER protein folding as ER-resident protein folding activities become reduced.
Without relying on pleiotropic ER stress-promoting agents that activate all three proximal arms of the UPR simultaneously, our tools allow us to link ER-localized mRNA decay directly to the IRE1α RNase. Since IRE1α activation occurs through self-association of its lumenal domains under ER stress, we simulated this regulated event through a step increase in transgenic IRE1α proteins. Driven from the same locus in isogenic cell lines, equivalent dynamic increases occurred for all IRE1α mutants; thus divergent physiological effects are directly traceable to the varying alleles. Using these tools, we found that phosphotransfer activation is the only mode that suffices to trigger ER-localized mRNA decay, and that IRE1α must contain a catalytically active RNase to cause decay.
Several lines of evidence argue that IRE1α-mediated ER mRNA decay is not confined to situations of controlled overexpression. ER-localized mRNA decay during ER stress is reduced when the Ire1 α gene is deleted or the protein reduced through RNAi. Therefore, IRE1α is both necessary and sufficient for ER mRNA decay under stress. Finally, using cell-free systems we identified insulin RNA as the first direct extra-XBP1 endonucleolytic substrate for IRE1α. We predict that many of the hundreds of decaying mRNAs we identified may also be direct IRE1α endonucleolytic substrates.
In all our experiments, ER mRNA decay correlated positively with apoptosis. A parsimonious interpretation holds that IRE1α-mediated ER mRNA decay contributes to apoptosis by reducing secretory pathway-resident activities that mediate protein folding, and perhaps important cargo substrates, to insufficiently low levels. Downstream JNK phoshorylation also becomes triggered under phosphotransfer activation of the IRE1α RNase, as it does under ER stress (Figure S11 A–C)—this may serve to amplify pro-apoptotic signaling. Thus, beyond some threshold of ER stress, IRE1α outputs may morph from homeostatic feedback loops (XBP1s-driven), into positive feedback pro-apoptotic loops, ushering in a terminal UPR that actively drives cells towards apoptosis.
IRE1α-triggered ER mRNA decay may have previously gone unnoticed in mammalian cells because it causes cell death. In our experience, constitutive overproduction of WT IRE1α in stable lines eventually fails, since surviving cells invariably lose or silence the transgene (data not shown). Therefore, tight conditional systems to produce IRE1α upon demand are necessary to prevent adaptation. Recent reports that IRE1α solely promotes cytoprotection utilized lines constitutively overproducing only kinase-dead IRE1α (Lin et al., 2007; Lin et al., 2009). Without kinase-active controls, those studies concluded that IRE1α outputs are solely adaptive, in contrast to what we found.
Model of How Kinase Inhibitors Suppress IRE1α–mediated ER mRNA Decay
We found excitingly that kinase inhibitors can modulate IRE1α’s divergent RNase outputs. This was first revealed using 1NM-PP1-sensitized IRE1α as proof-of-concept. 1NM-PP1 forcibly activates XBP1 mRNA splicing by I642G in cis. This pharmacological maneuver produces XBP1s in the absence of ER stress, without simultaneously causing decay of ER mRNAs. Preconditioning through XBP1s targets may help cells resist apoptosis when ER stress is subsequently encountered.
How are XBP1 mRNA splicing and ER-localized mRNA destruction separable in IRE1α (I642G), but not in WT? We propose a model: When IRE1α (I642G) is overproduced, its lumenal domains spontaneously oligomerize in the ER membrane. This juxtaposes the cytosolic kinases, but because the space-creating I642G substitution in the kinase pocket kills catalytic activity, the mutant cannot autophosphorylate. Therefore the RNase remains quiescent unless the ligand 1NM-PP1 is provided to bind the excavated kinase pocket and activate the RNase, (Figure 7A). This unusual mechanism of RNase activation through allostery was previously described for yeast IRE1 (Papa et al., 2003), and appears to have been preserved through evolution to mammals (Han et al., 2008).
Figure 7. Model For Divergent IRE1α RNase Outputs and their Modulation by Kinase Inhibitors.

(A) Pseudokinase IRE1α activation. Overproduction of IRE1α (I642G) causes it to cluster in the ER, allowing 1NM-PP1 to allosterically activate the RNase when it binds the engineered kinase pocket. While these two steps are depicted as separable, when provided 1NM-PP1 during IRE1α (I642G) overproduction, XBP1 mRNA splicing is identical to that occurring during overproduction of WT IRE1α—the phosphotransfer activation mode, (B). Oligomerization of the kinase/RNase domains in phosphorylated IRE1α is depicted as higher-order than in IRE1α (I642G) under 1NM-PP1. Relaxed specificity of the RNase pocket in phosphorylated IRE1α promotes ER-localized mRNA decay. IRE1α (I642G) under 1NM-PP1 is maintained in a tight conformation that restricts activity to XBP1 splicing, thus averting ER mRNA decay, (A). (C) KIRAs: kinase-inhibiting RNase attenuators of IRE1α. KIRAs reduce IRE1α autophosphorylation, thus reducing kinase/RNase oligomerization and tempering ER-localized mRNA decay; KIRAs permit, and even enhance XBP1 mRNA splicing, because they still satisfy the kinase ligand requirement in endogenous IRE1α.
A recent crystal structure model sheds light on this ligand requirement (Lee et al., 2008). Occupancy of ADP in the yeast IRE1 kinase active site stabilizes an “open” conformation, promotes formation of an extended interface between IRE1 monomers leading to dimerization, and orients the RNase domains for catalysis. For IRE1α (I642G), 1NM-PP1 appears to satisfy a similar ligand function as ADP does for WT, but without an autophosphorylation prerequisite: working as a ligand, 1NM-PP1 also helps promote an open kinase conformation causing RNase activation.
Phosphotransfer activation of the RNase appears to occur by an alternate mechanism. Upon induction, transgenic WT IRE1α also spontaneously oligomerizes though the lumenal domains, but simultaneously _trans_-autophosphorylates as the kinases are juxtaposed (Figure 7B). Another recent crystal structure shows that yeast IRE1 forms higher-order oligomers through salt-bridges between phosphorylated activation segments of adjacent kinase/RNases dimers (Korennykh et al., 2009). The RNase catalytic pocket in oligomeric IRE1 is larger than in dimeric IRE1, and exhibits catalytic rates that are orders of magnitude higher than that of dimers (Korennykh et al., 2009). We speculate that these phosphorylated oligomers may be “promiscuous” in their RNase activity, and could cause endonucleolytic degradation of ER-localized mRNAs as well as promoting XBP1 splicing; indeed the recognized sequences for Ins1 and Ins2 RNAs are identical to that of XBP1 at the scission sites, but diverge at the flanks (Figure 6D).
Importantly, higher-order oligomerization of kinase/RNase domains should not be available to IRE1α (I642G), since the mutation kills phosphotransfer. Indeed, the in vivo Hill coefficient of 1.8 suggests that active IRE1α (I642G) kinase/RNase splicing units may be lower-order species that form cooperatively upon 1NM-PP1 binding—in the model they are shown speculatively as dimers. The RNase activity of I642G under 1NM-PP1 is nevertheless sufficient to fully splice XBP1 mRNA, which may have evolved structural and/or ER targeting features that make it a highly efficient substrate (Aragon et al., 2009). Indeed, other mutants that abrogate phosphotransfer such as I642A, or that cannot be phosphorylated on the activation segment, still cause significant XBP1 mRNA splicing when oligomerized (Figure 2A, lane 6), but do not promote mRNA decay or apoptosis (data not shown). These results suggest that autophosphorylation may be formally unnecessary for low-level XBP1 mRNA splicing; IRE1α autophosphorylation during high ER stress increases RNase activity but may also carry the risk of increasing ER mRNA decay as higher-order oligomers form.
New approaches are needed to intervene in the apoptotic process to treat ER stress-related diseases. Here we showed that kinase inhibitors can uncouple IRE1α productive XBP1 mRNA splicing from destructive endonucleolytic events, and this may have important therapeutic ramifications. We followed proof-of-concept with 1NM-PP1 with preliminary demonstrations that other small molecule kinase inhibitors of WT IRE1α can reduce insulin mRNA decay in vivo during ER stress. We designate such small molecules kinase-inhibiting RNase attenuators—“KIRAs.” KIRAs may temper RNase-mediated ER mRNA decay by reducing IRE1α kinase/RNase oligomerization, while permitting (or even enhancing) XBP1 mRNA splicing because they still satisfy the ligand requirement (Figure 7C).
Recent provocative reports show that several FDA-approved kinase inhibitors used to treat cancer also reverse type 1 diabetes in rodents (Louvet et al., 2008). Due to the plethora of structurally-related kinases, many kinase inhibitors have effects not confined to intended targets. It remains to be determined if anti-diabetic effects of these agents, which have modest activity against IRE1α are due in part to “off-target” KIRA effects in β-cells. In turn, off-target IRE1α KIRA activity may hamper chemotherapeutic effects, not necessarily because homeostatic XBP1 splicing is increased, but because apoptotic RNase-mediated mRNA decay is reduced. The development of selective and potent KIRAs will provide the necessary tools to further investigate the therapeutic potential of this novel mode of enzyme modulation.
Note added in proof: ER-localized mRNA decay through WT IRE1α, but not (I642G) under 1NM-PP1, is also being reported by Hollien and colleagues (Hollien et al.).
EXPERIMENTAL PROCEDURES
Creation of stable cell lines
INS-1/FRT/TO cells (#5-3.19) (Thomas et al., 2004) were grown in RPMI, 10% fetal calf serum, 1mM sodium pyruvate, 10mM HEPES, 2mM glutamine, 50µM beta-mercaptoethanol, 10µg/ml blasticidin. Cells were grown in 200µg/ml zeocin, then cotransfected with 1µg pcDNA5/FRT/TO::IRE1α constructs and 1µg FLP recombinase (pOG44) using Lipofectamine (Invitrogen). 4 hrs later, cells were switched to zeocin-free media, trypsinized 48 hrs later, then plated in media containing hygromycin (150µg/ml), which was replaced every 3 days until colonies appeared. IRE1α expression vectors were similarly integrated in Flp-In T-REx 293 cells (Invitrogen).
Microarray Analysis
RNA was harvested using Trizol, quantified by Nanodrop, and integrity verified by Bioanalyzer. 15µg RNA was used to synthesize cDNAs, which were purified (Qiagen MinElute Purification Kit) and coupled to Cy3 or Cy5 (Amersham). See supplementary methods for details of array-specific protocols and statistical analysis.
Supplementary Material
01
10
11
02
03
04
05
06
07
08
09
ACKNOWLEDGEMENTS
We thank G. Ryffel for INS-1/FRT/TO cells, L. Glimcher for _Xbp1_−/− MEFs, D. Ron for Ire1 α−/− MEFs, F. Urano for anti-IRE1α antibodies, and C. Zhang and K. Shokat for 1NM-PP1. We thank J. Bluestone and members of the Oakes and Papa labs for comments. This work was supported by NIH: Director’s New Innovator Award DP2 OD001925 (F.R.P), K08 DK065671 (F.R.P.), K08 AI054650 (S.A.O.), RO1 CA136577 (S.A.O.), RO1 DK080955 (F.R.P); HHMI Physician-Scientist Early Career Award (S.A.O.), Steward Trust Foundation (S.A.O.), Sandler Program in Basic Sciences (S.A.O. and F.R.P.), Burroughs Wellcome Foundation (F.R.P.), Hillblom Foundation (F.R.P.), Juvenile Diabetes Research Foundation (F.R.P), Partnership for Cures (F.R.P.), and NIGMS-IMSD R25 GM56847 (A.G.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. Inaugural Article: On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Upton JP, Hagen A, Callahan J, Oakes SA, Papa FR. A kinase inhibitor activates the IRE1alpha RNase to confer cytoprotection against ER stress. Biochem Biophys Res Commun. 2008;365:777–783. doi: 10.1016/j.bbrc.2007.11.040. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Bertolotti A, Zeng H, Zhang Y, Urano F, Jousse C, Ron D. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb Symp Quant Biol. 2001;66:499–508. doi: 10.1101/sqb.2001.66.499. [DOI] [PubMed] [Google Scholar]
- Higashio H, Kohno K. A genetic link between the unfolded protein response and vesicle formation from the endoplasmic reticulum. Biochem Biophys Res Commun. 2002;296:568–574. doi: 10.1016/s0006-291x(02)00923-3. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin J, Li H, Stevens N, Walter P, Weissman J. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. Journal of Cell Biology. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008;132:89–100. doi: 10.1016/j.cell.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE. 2009;4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KL, Ghosh R, Urano F. The role of IRE1α n the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, Bluestone JA. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008;105:18895–18900. doi: 10.1073/pnas.0810246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-Time Redox Measurements during Endoplasmic Reticulum Stress Reveal Interlinked Protein Folding Functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Shah K, Liu Y, Deirmengian C, Shokat KM. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc Natl Acad Sci U S A. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15:3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas H, Senkel S, Erdmann S, Arndt T, Turan G, Klein-Hitpass L, Ryffel GU. Pattern of genes influenced by conditional expression of the transcription factors HNF6, HNF4alpha and HNF1beta in a pancreatic beta-cell line. Nucleic Acids Res. 2004;32:e150. doi: 10.1093/nar/gnh144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes and Development. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. CELL. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
10
11
02
03
04
05
06
07
08
09