The PAR-6 Polarity Protein Controls Dendritic Spine Density Through p190 RhoGAP and the Rho GTPase (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 27.
Summary
The majority of excitatory synaptic transmission in the brain occurs at dendritic spines, which are actin-rich protrusions on the dendrites. The polarized nature of these structures suggests that proteins regulating cell polarity might be involved in their formation. Indeed, the polarity protein PAR-3 is required for normal spine morphogenesis. However, this function is independent of association with atypical protein kinase C (aPKC) and PAR-6. Here we show that PAR-6 together with aPKC plays a distinct but essential role in spine morphogenesis. Knockdown of PAR-6 inhibits spine morphogenesis, whereas over-expression of PAR-6 increases spine density, and these effects are mediated by aPKC. Using a FRET biosensor, we further show that p190 RhoGAP and RhoA act downstream of the PAR-6/aPKC complex. These results define a novel function for PAR-6 and aPKC in dendritic spine biogenesis and maintenance, and reveal an unexpected link between the PAR-6/aPKC complex and RhoA activity.
Introduction
Dendritic spines are small protrusions on neurons that receive the majority of the excitatory synaptic inputs in the brain (Goda and Davis, 2003; Hering and Sheng, 2001; Sheng and Hoogenraad, 2007). The formation of these structures is essential for cognitive functions, as spine abnormalities are associated with various forms of mental retardation (Fiala et al., 2002). In addition, neurodegenerative diseases like Alzheimer’s disease usually begin with spine and synaptic loss, which is closely associated with the pure memory impairment observed in their earliest clinical phases (Selkoe, 2002). Therefore, elucidating the mechanisms of dendritic spine morphogenesis is crucial to understanding how the brain processes and stores information, and how this fails in a diseased state.
During development, the formation of spines begins with the extension of highly dynamic, filopodia-like protrusions on the dendrites. These protrusions are believed to be actively searching for their presynaptic interacting partners and, once a contact has formed, the protrusions stabilize and mature into dendritic spines (Matus, 2005). In contrast to the microtubule-based dendritic shaft, spines are highly enriched in actin. A number of proteins that control actin dynamics, including Rho GTPases and their regulators, have been implicated in spine morphogenesis (Govek et al., 2005).
The synapse forms on the top of the spine head, where a membrane thickening called the postsynaptic density (PSD) occurs. Various neurotransmitter and adhesion receptors, structural and signaling molecules cluster within the PSD. The synapse is an asymmetric adhesive contact that bears structural and functional similarities to the apical junction complex in epithelial cells, and to the immunological synapse between T-cells and antigen-presenting cells (Yamada and Nelson, 2007). Thus, a dendritic spine is a highly polarized structure, which lends credence to the notion that molecules involved in establishing epithelial polarity might also be important in the formation of dendritic spines.
The PAR proteins were first discovered in a screen for genes in C. elegans required for the initial asymmetric cell division of the zygote. Several of the partition-defective (par) gene products - PAR-3, PAR-6, and PKC-3 – co-localize to the anterior end of the zygote, and the mammalian orthologs of these proteins can form a physical complex, along with CDC42, which binds to PAR-6. A conserved signaling pathway containing CDC42, PAR-3, PAR-6, and aPKC is essential for cell polarization in different contexts ranging from asymmetric cell division, epithelial polarity and directional migration to axon specification in neurons (Macara, 2004; Suzuki and Ohno, 2006). However, in some circumstances PAR-3 localizes separately from the PAR-6/aPKC complex, and performs distinct functions.
Previously, we discovered that PAR-3 is necessary for normal dendritic spine morphogenesis in hippocampal neurons (Zhang and Macara, 2006). PAR-3 spatially restricts the Rac guanine nucleotide exchange factor (GEF) TIAM1 to dendritic spines, thereby prevents inappropriate Rac activation. Silencing of PAR-3 by RNA interference (RNAi) results in the formation of dendritic filopodia that do not form mature into spines or form synapses. Interestingly, we found that PAR-3 acts in this pathway independently of its association with aPKC (Zhang and Macara, 2006). A similar situation occurs during tight junction assembly in epithelial cells, in which PAR-3 also acts through TIAM1 on the Rac GTPase independently of binding to either aPKC or PAR-6 (Chen and Macara, 2005). What then is the function of PAR-6 and aPKC during spine morphogenesis? We have found that PAR-6 regulates dendritic spine biogenesis and maintenance rather than spine maturation. PAR-6 acts through aPKC, which is linked to RhoA GTPase activity through the p190 RhoGAP. Taken together, these results reveal an important role for PAR-6 and aPKC in dendritic spines morphogenesis that is independent of PAR-3 and which, unexpectedly, involves the Rho GTPase.
Results
PAR-6 is essential for spine morphogenesis
We earlier demonstrated that the polarity protein PAR-3 is essential for normal spine morphogenesis, and that a splice variant of PAR-3 lacking the aPKC binding site (PAR-3c) can functionally replace the wild type protein in this process (Zhang and Macara, 2006). Thus, PAR-3 acts independently of aPKC binding and phosphorylation to regulate spine maturation. However, aPKC can also interact with PAR-3 indirectly, through the association of PAR-6 with the first PDZ domain in PAR-3. To determine whether binding of PAR-6 is necessary for the function of PAR-3 in spines, we knocked down endogenous PAR-3 in hippocampal neurons and co-expressed a PAR-3 fragment (PAR-3c-C) that does not bind PAR-6. As shown previously (Zhang and Macara, 2006), depletion of PAR-3 caused the formation of multiple filopodia-like protrusions and a dramatic decrease in the number of normal, mushroom-shaped spines. Co-expression of PAR-3c-C efficiently rescued the spine formation defects in PAR-3 knockdown neurons, suggesting that binding of PAR-6 is not necessary for the function of PAR-3 in spines (supplementary data, Fig. S1). We were interested, therefore, in whether PAR-6 functions in spine morphogenesis, and whether this pathway is distinct from that involving PAR-3.
Initially, we examined the subcellular distribution of endogenous PAR-6C in DIV 14 cultured hippocampal neurons. As we have reported previously (Joberty et al., 2000), PAR-6C is the predominant isoform in neurons. Immunofluorescence of endogenous PAR-6C revealed a punctate staining pattern along the dendrites (Fig. 1A). These puncta colocalized with PSD-95, a marker for excitatory synapses, showing that PAR-6 is synaptic. To validate the specificity of staining by the anti-PAR-6C antibody, we silenced endogenous PAR-6C in hippocampal neurons using RNAi. ShRNAs were introduced using the pSUPER vector, and their efficiency was tested in Rat2 fibroblasts by immunoblotting (Fig. 1B). ShRNA #3 and #8 both knocked down the expression of endogenous PAR-6C effectively and were used in subsequent studies (Fig. 1B and supplementary data, Fig. S2). Both shRNAs substantially reduced the punctate immunostaining observed in dendrites (Supplementary data, Fig. S2B, and data not shown), confirming that the puncta represent authentic, endogenous PAR-6C.
Figure 1. PAR-6C is necessary for spine maintenance.
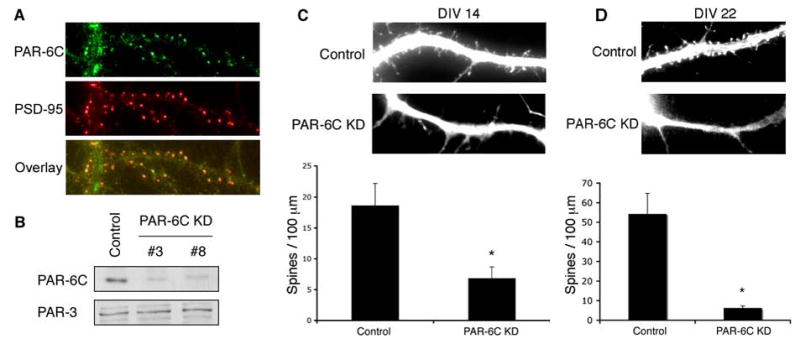
A. PAR-6C localizes to excitatory synapses. Hippocampal neurons were fixed and co-immunostained with PAR-6C and PSD-95 antibodies.
B. Efficiencies of PAR-6C shRNA constructs. Rat2 fibroblasts were electroporated with pSUPER-luciferase (Control), pSUPER-PAR-6C shRNA#3 or pSUPER-shRNA#8 (PAR-6C KD). 48 hours after transfection, cells were lysed and analyzed by SDS-PAGE and western blotting.
C. Effects of PAR-6C knockdown on spine formation. Hippocampal neurons were transfected with either pSUPER-luciferase (Control) or pSUPER-PAR-6C shRNA#3 (PAR-6C KD) at DIV 6–7 and imaged at DIV 14. Yellow fluorescent protein (YFP) was co-expressed as a marker for transfected neurons and to visualize cell morphology. Knockdown of PAR-6C significantly reduced the number of spines. *p<0.001 by Student’s t-test.
D. PAR-6C is necessary for spine maintenance. Hippocampal neurons were transfected with either pSUPER-luciferase (Control) or pSUPER-PAR-6C shRNA#3 (PAR-6C KD) at DIV 17 and imaged at DIV 22. Yellow fluorescent protein (YFP) was co-expressed as a marker for transfected neurons and to visualize cell morphology. There was a significant reduction in the number of dendritic spines even though PAR-6C was knocked down after the neurons have formed numerous spines, suggesting that PAR-6C is necessary for spine maintenance. *p<0.001 by Student’s t-test.
To isolate the effects on spine formation from axonal specification and neurite outgrowth, we transfected neurons with the pSUPER constructs at DIV6–7 and imaged them at DIV14. In each case, soluble YFP was co-expressed as a marker for transfected neurons and also to visualize cell morphology. As shown in Fig. 1C, the spine density along dendrites of neurons transfected with shRNA#3 was significantly lower than that of control neurons expressing an shRNA targeting luciferase. ShRNA #8 gave a similar phenotype (data not shown). Note that filopodia were not induced by loss of PAR-6, as occurs when PAR-3 expression is silenced (Zhang and Macara, 2006). These results demonstrate that PAR-6C is required for spine morphogenesis, and that its function is distinct from that of PAR-3, which regulates spine maturation but not spine density.
To determine whether PAR-6 is required for the formation or maintenance of spines, we knocked down PAR-6 at DIV17, when neurons have formed numerous spines. Neurons were then imaged at DIV22 to examine the effects of PAR-6 depletion. As shown in Fig. 1D, silencing of PAR-6 at this stage still caused a dramatic decrease in spine density. This result indicates that PAR-6 is required for the maintenance of dendritic spines.
If PAR-6 controls spine density, one might predict that elevated PAR-6C expression would trigger the formation of supernumerary spines. Indeed, the ectopic expression of myc-tagged PAR-6C produced a higher density of spines along the dendrites, and these spines were longer as compared with the control. A slight increase in spine width was also observed (Fig. 2A). We conclude that PAR-6C promotes spine formation, and that the effect is dose-dependent.
Figure 2. Overexpression of PAR-6C promotes spine morphogenesis.

A. Effects of PAR-6C overexpression on spine formation. Hippocampal neurons were transfected with either an empty myc vector, or myc-tagged PAR-6C. YFP was co-expressed to visualize the transfected cells. Overexpression of PAR-6C increases the density and length of spines. *p<0.001 by Student’s t-test.
B. Effects of PAR-6C on the number of functional synapses as visualized by FM1-43 dye uptake.
C. Quantification of the results shown in B. *p<0.001 by Student’s t-test.
Are the supernumerary spines triggered by PAR-6C overexpression functional? To address this question, we performed FM1-43 dye uptake to evaluate the number of functional synapses in neurons either overexpressing or depleted of PAR-6C. FM1-43 is an amphipathic styryl dye that reversibly stains membranes. Therefore, when neurons are exposed to the FM dye during stimulation and are subsequently washed, only the endocytosed membranes retain the dye. As a result, only functional synapses that have undergone synaptic vesicle recycling will show FM1-43 staining. Neurons overexpressing PAR-6C showed increased number of FM1-43 puncta, whereas PAR-6C knockdown neurons exhibited a significant decrease in the number of FM1-43 puncta, as compared to the control cells (Fig. 2B and C). These data suggest that PAR-6C promotes the formation of functional spines.
We next tested myc-PAR-6B, which is an isoform that is more widely expressed than PAR-6C, and has been extensively studied in epithelial cells. Over-expression of this isoform produced a phenotype indistinguishable from that caused by PAR-6C, indicating that the effect is not isoform-specific (Fig. 3B and C). To identify the domains within PAR-6 that are responsible for promoting spine density, we expressed various PAR-6B mutants and analyzed their effects on spine formation (Fig. 3A–C and supplementary data, Fig. S3). The PAR-6BβPro mutant lacks an essential proline residue (Pro 136) in its semi-CRIB domain, and cannot effectively bind Cdc42; however, this mutant was still able to promote spine formation in a manner similar to its wild type counterpart, indicating that the association with Cdc42-GTP is unnecessary. In contrast, when the N-terminal PB1 domain was deleted, which binds aPKC, the resulting mutant was no longer able to promote spine formation. Finally, a dominant-negative effect was seen with PAR-6BmutPDZ, which has four point mutations (K167A, P168A, L169A, G170A) that abolish the ligand-binding capacity of the PDZ domain.
Figure 3. Effects of PAR-6 mutants on spine morphogenesis.

A. Schematic diagram of PAR-6 mutants used in this study.
B. Effects of overexpressing different PAR-6 mutants on spine formation. Hippocampal neurons were transfected with the indicated constructs. YFP was co-expressed to visualize the transfected cells.
C. Quantification of spine density in neurons expressing different PAR-6 mutants. *p<0.001 by Student’s t-test.
D. The aPKC binding domain and the PDZ domain are necessary for rescuing the PAR-6C knockdown defect. Hippocampal neurons were transfected with pSUPER-luciferase (Control), pSUPER-PAR-6C shRNA#3, or shRNA #3 together with the indicated PAR-6 constructs. YFP was co-expressed to visualize the transfected cells.
E. Quantification of the rescue results shown in D. *p<0.001 by Student’s t-test.
To verify that the aPKC binding and the PDZ domains are required, we performed rescue experiments using the various PAR-6 mutants expressed in neurons depleted of endogenous PAR-6C (Fig. 3D and E). Importantly, the shRNA #3 sequence targets rat PAR-6C, and cannot recognize human PAR-6. First, we found that both wild type PAR-6C and PAR-6B efficiently rescued spine biogenesis, confirming that the defects in spine formation are specifically caused by loss of PAR-6, and are not caused by off-target effects of the shRNA. Second, neither the PB1 domain mutant nor the PDZ domain mutant was able to rescue the knockdown defect, showing that these domains are both required for PAR-6 to promote spine formation. These data are consistent with a widely accepted role for PAR-6 as a targeting subunit of aPKC, in which the PDZ domain recruits substrates for phosphorylation (or, conversely, recruits aPKC to the substrates). The PB1 mutant cannot form a complex with aPKC. Therefore, it will bind substrates but not recruit them to the kinase. Conversely, the PDZ mutant will not bind substrates but will sequester aPKC and disrupt its ability to phosphorylate appropriate substrates. Taken together, these data argue that spine density is controlled by the phosphorylation of an aPKC substrate that is recruited by PAR-6.
Effects of PKCβ mutants on spine morphogenesis
To test this hypothesis, we examined the effects of PKCβ, one of the two mammalian atypical PKC isoforms, on spine morphogenesis (Fig. 4A and B). Ectopic expression of a constitutively active PKCβ mutant (PKCβT410E) significantly increased the overall density of spines, similar to what was seen with PAR-6 overexpression. By contrast, a kinase-dead mutant of PKCβ (PKCβT410A) reduced the number of mature spines, but also resulted in the appearance of some filopodia-like extensions. We speculate that this effect might arise from an aPKC-driven mislocalization of PAR-3. In any case, these results support the idea that the kinase activity of PKCβ is necessary for spine morphogenesis.
Figure 4. PKCβ functions downstream of PAR-6 in spine morphogenesis.

A. Effects of different PKCβ mutants on spine morphogenesis. Hippocampal neurons were transfected with YFP and the indicated PKCβ mutants. A kinase-active PKCβ (PKCβ T410E) promotes spine formation, whereas a kinase-inactive mutant (PKCβ T410A) inhibits spine formation.
B. Quantification of the results in A. *p<0.001 by Student’s t-test.
C. Hippocampal neurons were transfected with YFP and the indicated constructs. PKCβT410E cannot rescue the spine formation defects in PAR-6C knockdown neurons or the defects in neurons expressing PAR-6BmutPDZ, indicating that targeting information provided by PAR-6 is necessary for the function of aPKC.
D. Quantification of the results in C. *p<0.001 by Student’s t-test.
E. Localization of PKCβT410E depends on the presence of PAR-6. The localization of PKCβT410E was examined in control neurons and neurons depleted of PAR-6. PKCβT410E localizes to spines in control neurons but fails to accumulate in spines in neurons depleted of PAR-6.
F. PKCβ functions downstream of PAR-6. Hippocampal neurons were co-transfected with PAR-6B and either PKCβT410E or PKCβT410A. Co-expression of PAR-6 and PKCβT410E does not increase spine number to a level higher than each construct singly expressed. PKCβT410A efficiently inhibited spine formation induced by PAR-6B.
G. Quantification of the results shown in F. *p<0.001 by Student’s t-test.
We then examined whether aPKC kinase activity alone is sufficient for spine formation in the absence of the targeting information provided by PAR-6. To address this issue, we knocked down endogenous PAR-6C and tried to rescue spine morphogenesis by expressing the constitutively active PKCβT410E. However, this mutant failed to reverse the spine defect (Fig. 4C and D) even though its expression in wild type neurons can promote spine formation. This observation suggests that PAR-6 is needed to provide targeting information for aPKC. As a further test, we asked if expression of PKCβT410E would reverse the defect caused by the PAR-6BmutPDZ dominant-negative mutant. Again, PKCβT410E failed to rescue normal spine morphogenesis (Fig. 4C and D). We conclude, therefore, that PAR-6 is necessary either to recruit a target protein for phosphorylation by aPKC, or conversely to recruit aPKC to a target.
If PAR-6 is necessary to provide the targeting information for aPKC, one might expect aPKC to be mislocalized in PAR-6 knockdown neurons. To test this prediction, we examined the localization of PKCβT410E in either control neurons or neurons depleted of PAR-6. As shown in Fig. 4E, PKCβT410E localized to spines in control neurons but failed to accumulate in spines in neurons depleted of PAR-6. This result argues that PAR-6 is necessary for the proper targeting of aPKC.
Finally, to verify that aPKC functions downstream of PAR-6, we co-expressed PAR-6 with either PKCβT410E or PKCβT410A. Co-expression of PAR-6 with PKCβT410E did not increase the number of spines (36+/−12 spines/100μm) beyond that induced by each construct expressed singly (36+/−8 spines/100μm for PAR-6; and 38 +/−5 for PKCβT410E; see Figs. 3C, and 4B & C), suggesting that the two proteins function on the same pathway. Moreover, PKCβT410A efficiently inhibited the spine formation induced by PAR-6 overexpression (Fig. 4F and G), further showing that aPKC functions downstream of PAR-6.
RhoA functions downstream of PAR-6/aPKC
What are the downstream effectors of the PAR-6/aPKC complex in spine morphogenesis? We initially focused on Smurf1, an E3 ubiquitin ligase that has been reported to associate with, and be phosphorylated by, the PAR-6/aPKC complex (Wang et al., 2003). Smurf1 targets RhoA for local degradation. Therefore, the activation of Smurf1 by aPKC should reduce RhoA in the dendrites, while a decrease in aPKC activity, produced for example by knockdown of PAR-6, should elevate RhoA. We were unable to detect any differences, however, either in the total level or dendritic concentration of RhoA, when PAR-6 was silenced or over-expressed in hippocampal neurons. It is unlikely, therefore, that Smurf1 is involved in the control of dendritic spine density.
Nonetheless, RhoA activity is a pivotal regulator of spine density, because the expression of a constitutively active RhoA mutant (RhoAV14) blocked spine morphogenesis (Fig. 5A), phenocopying the PAR-6 knockdown. Expression of RhoAV14 at DIV17 also caused a dramatic loss of spines (data not shown), similar to what was observed with PAR-6C knockdown. On the other hand, a dominant-negative mutant of RhoA (N19) increased spine density along the dendrites (Fig. 5A), as we had observed when PAR-6 expression was increased. Similar effects of RhoA on spine density have been reported previously (Govek et al., 2005; Tashiro et al., 2000).
Figure 5. RhoA functions downstream of PAR-6 in spine morphogenesis.

A. Effects of RhoA mutants on spine morphogenesis. Hippocampal neurons were transfected with YFP and the indicated RhoA constructs. Constitutively active RhoA (RhoA V14) inhibits spine formation, whereas dominant negative RhoA (RhoA N19) promotes spine formation. *p<0.001 by Student’s t-test.
B. Dominant negative RhoA rescues the spine formation defects in PAR-6C knockdown neurons. Hippocampal neurons were transfected with YFP and the indicated constructs. RhoA N19 reverses the spine formation defects in PAR-6C shRNA#3 expressing neurons. *p<0.001 by Student’s t-test.
C. Treatment of PAR-6C knockdown neurons with a ROCK inhibitor H-1152 (10 μM) reverses the spine formation defects in these neurons. *p<0.001 by Student’s t-test.
D. Constitutively active RhoA inhibits spine formation induced by PAR-6 overexpression. Hippocampal neurons were transfected with YFP and the indicated constructs. RhoA V14 inhibited spine formation induced by PAR-6B. *p<0.001 by Student’s t-test.
E. PAR-6 affects RhoA activity. Hippocampal neurons were transfected with an empty vector, myc-tagged PAR-6C or pSUPER-PAR-6C shRNA#3. A pRaichu-RhoA FRET biosensor was co-expressed to examine RhoA activity in these neurons. Representative FRET images are shown.
F. Quantification of mean FRET values shown in D. *p<0.001 by Student’s t-test.
A key effector of RhoA is Rho Kinase (ROCK). To see if ROCK is downstream of RhoA in regulating spine morphogenesis, we treated neurons with H-1152, a specific inhibitor of ROCK. H-1152 treatment caused an increase in spine density in control neurons, similar to what was observed with RhoAN19 expression (supplementary data, Fig. S4A). H-1152 also efficiently rescued the spine formation defects in RhoAV14 expressing neurons (supplementary data, Fig. S4B). Taken together, our data demonstrate that the RhoA-ROCK pathway negatively regulates spine morphogenesis.
To test whether the RhoA pathway is linked to PAR-6 function, we asked if the expression of a dominant-negative RhoA mutant could restore spine morphogenesis in cells depleted of PAR-6. Indeed, as shown in Fig. 5B, RhoAN19 efficiently reversed the spine defect caused by PAR-6 silencing. To confirm that the Rho pathway acts in concert with PAR-6, we treated PAR-6 depleted neurons with H-1152. Treatment with H-1152 completely rescued the PAR-6 knockdown defect (Fig. 5C). Finally, the constitutively active RhoA, RhoAV14, was able to inhibit the spine formation induced by PAR-6 (Fig. 5D). We conclude, therefore, that the Rho signaling pathway is inversely coupled to PAR-6/aPKC, such that PAR-6 suppresses RhoA function. However, these data do not distinguish whether RhoA acts downstream of PAR-6 or instead acts in a parallel but independent pathway that converges on spine formation.
To address this issue, we examined endogenous RhoA activity levels using a Raichu FRET (fluorescence resonance energy transfer) biosensor (Yoshizaki et al., 2003). The Raichu RhoA probe is a fusion protein consisting of YFP plus the RhoA-binding domain (RBD) of PKN, RhoA, and CFP. When the RhoA binds GTP, the neighboring RBD undergoes an intramolecular association with RhoA that triggers a conformational switch. This switch brings the CFP FRET donor closer to the YFP acceptor, resulting in higher FRET efficiency. To determine whether changes in PAR-6 levels affect RhoA activity, we expressed the Raichu probe in neurons that either over-expressed or were depleted of PAR-6. Over-expression of PAR-6 caused a reproducible decrease in FRET efficiency as compared with the controls, whereas knockdown of PAR-6 resulted in a significant increase in FRET efficiency (Fig. 5E and F). We conclude that PAR-6 negatively regulates RhoA activity.
PAR-6 regulates Rho activity through p190 RhoGAP
RhoA, like other small GTPases, is regulated by guanine nucleotide exchange factors (GEFs) that promote GTP loading, and by GTPase activating proteins (GAPs) that return them to the inactive state. The GEFs are frequently auto-inhibited, and can be activated by signaling inputs that release the catalytic domain and relieve the inhibition (Rossman et al., 2005). In contrast, no common mechanism has yet emerged for the regulation of Rho GAPs and some are activated while others are inhibited by upstream signals. Since PAR-6 suppresses Rho-GTP levels, we considered it unlikely that it would inhibit a constitutively active GEF, and we focused instead on the GAPs. Although there are about 70 Rho family GAP genes in the mammalian genome, many are not Rho-specific, or are not expressed in neurons. However, p190A RhoGAP is expressed at high levels in the brain, is present in spines and has been implicated in fear memory formation (Lamprecht et al., 2002; Settleman, 2003). To investigate a possible link between the PAR-6/aPKC complex and p190, we first performed endogenous co-immunoprecipitations from hippocampal lysate. A small but reproducible amount of p190 was detected in association with PKCβ (Fig. 6A).
Figure 6. p190 RhoGAP functions downstream of PAR-6/aPKC in spine morphogenesis.

A. Co-immunoprecipitation of endogenous PKCβ and p190 RhoGAP from hippocampal neurons. Hippocampal neurons (DIV8) were lysed and immunoprecipitated with either a GFP antibody or a PKCβ antibody. Immunoprecipitates were washed and analyzed with SDS-PAGE and western blot. IP: immunoprecipitation. WB: western blot.
B. Efficiency of the p190 RhoGAP shRNA construct. Hippocampal neurons were transfected with YFP and pSUPER-p190 shRNA. At DIV14 cells were fixed and immunostained for p190 RhoGAP. The staining intensity was dramatically reduced in pSUPER-p190 shRNA expressing neurons as compared with nearby untransfected neurons. IF: immunofluorescence.
C. Knockdown of p190 RhoGAP reduces spine density. Hippocampal neurons were transfected with either pSUPER-luciferase or pSUPER-p190 shRNA. YFP was co-expressed to visualize the transfected cells. *p<0.001 by Student’s t-test.
D. The GAP activity of p190 RhoGAP is necessary for spine formation. Hippocampal neurons were transfected with an empty vector (Control), wild type p190 RhoGAP, or a GAP-deficient mutant of p190 RhoGAP (p190 RhoGAP R1283A). YFP was co-expressed to visualize the transfected cells. *p<0.001 by Student’s t-test.
E. P190 RhoGAP functions downstream of PAR-6. Hippocampal neurons were co-transfected with PAR-6B and either wild type or GAP-deficient p190 RhoGAP. The GAP-deficient p190 RhoGAP inhibited the spine formation induced by PAR-6B overexpression. *p<0.001 by Student’s t-test.
F. PAR-6 regulates RhoA activity through p190 RhoGAP. Hippocampal neurons were transfected with either an empty vector plus pSUPER-p190 shRNA, or myc-PAR-6C plus pSUPER-p190 shRNA. pRaichu-RhoA was co-expressed to visualize RhoA activity. Representative FRET images are shown.
G. Quantification of mean FRET values shown in F. *p<0.001 by Student’s t-test.
Next, we silenced endogenous p190A expression by RNAi. A second isoform, p190B, is not expressed in the hippocampus. As illustrated in Fig. 6B, the shRNA construct efficiently knocked down p190 expression in the transfected neuron as compared with a neighboring untransfected neuron. Strikingly, depletion of p190 caused a substantial reduction in spine density (Fig. 6C). To determine whether the GAP activity of p190 is necessary for spine formation, neurons were transfected with a p190 mutant, which has a single amino acid mutation (R1283A) that abolishes its GAP activity (Tatsis et al., 1998). Expression of this mutant significantly reduced spine density (Fig. 6D), showing that GAP activity is necessary for spine formation. The effects of p190 knockdown and p190R1283A can be effectively reversed by treating the neurons with H-1152 (supplementary data, Fig. S4C and D), confirming that, as expected, p190 controls the Rho-ROCK pathway.
To address whether p190 is coupled to PAR-6/aPKC function, we co-expressed PAR-6 with either wild type p190 or the GAP-deficient p190 mutant. The mutant significantly inhibited the increase in spine density caused by PAR-6 (Fig. 6E), consistent with the idea that p190 RhoGAP might mediate the action of PAR-6 in promoting spine formation. Next, we asked whether PAR-6 regulates RhoA activity through the p190RhoGAP. FRET efficiency of the Raichu probe was examined in neurons depleted of p190. Importantly, silencing of p190 caused a significant increase in FRET efficiency, indicating an increase in RhoA activity (Fig. 6F and G). Thus, p190 is a pivotal RhoGAP required for maintenance of low RhoGTP in hippocampal neurons. We then examined the FRET efficiency in neurons co-expressing myc-PAR-6C plus the p190 shRNA. If PAR-6 decreases Rho activity through p190, knockdown of the RhoGAP should prevent this decrease, and we would predict that the FRET efficiency in these neurons would be similar to that in neurons expressing p190 shRNA alone. On the other hand, if PAR-6 acts on Rho through a distinct pathway, independent of p190, then depletion of p190 would not abolish the decrease, and the neurons would show lower FRET as compared with p190 shRNA expressing neurons. As demonstrated in Fig. 6F and G, neurons co-expressing PAR-6 and p190 shRNA showed similar FRET efficiency to that of neurons expressing p190 shRNA alone. We conclude, therefore, that PAR-6 regulates RhoA activity through p190RhoGAP.
Discussion
Previously, we had shown that the polarity protein PAR-3 is essential for spine maturation, and operates through the small GTPase Rac, which is a key regulator of the actin cytoskeleton (Zhang and Macara, 2006). The same signaling pathway accounts for PAR-3 function in the assembly of epithelial tight junctions (Chen and Macara, 2005). Surprisingly, however, although PAR-3 is often coupled in a signaling complex with PAR-6 and aPKC, neither protein needs to bind PAR-3 in order to drive tight junction formation. Moreover, we found that a PAR-3 splice variant that cannot bind aPKC, as well as a mutant of PAR-3 that lacks the PAR-6 binding domain, PDZ1 (Fig. S1), both can efficiently rescue spine maturation in neurons depleted of endogenous PAR-3. Taken together, these observations suggest that PAR-6 and aPKC might perform functions in dendritic spine morphogenesis that are distinct from those of PAR-3. We now show that these polarity proteins are required for spine biogenesis and maintenance. In addition, we have established an unexpected link between the PAR-6/aPKC complex and the RhoA/ROCK signaling pathway, which is another central regulator of actin dynamics. Thus, the PAR-3/6 polarity proteins signal through two different Rho GTPases that ultimately target the actin cytoskeleton to regulate spine morphogenesis.
PAR-6 is an adaptor protein that is believed to function as a scaffold, bringing aPKC and its substrate together to facilitate downstream signaling. In this study, we found that both the N-terminal PB1 domain of PAR-6, which binds aPKC, and the PDZ domain, which binds substrates, are each required for normal spine morphogenesis. What upstream inputs regulate PAR-6 in neurons remains unclear. In other systems, such as epithelial cysts and neuronal progenitors, PAR-6 is recruited to the cortex by CDC42-GTP, which associates with the CRIB domain of PAR-6. However, we found that a mutant of PAR-6, which is defective in CDC42-binding, can drive normal spine morphogenesis. Moreover, we see little effect on spine density when a dominant-negative mutant of CDC42 is expressed (data not shown). Therefore, the requirement for PAR-6 in spine morphogenesis seems to be independent of CDC42.
Unexpectedly, activation of the PAR-6/aPKC signaling pathway facilitates spine formation through RhoA. A dominant negative Rho mutant overcame the suppression of spine formation caused by silencing of PAR-6. Previously, PAR-6 has been shown to regulate local RhoA protein levels through association with an E3 ubiquitin ligase, Smurf1 (Wang et al., 2003). However, we did not detect any changes in RhoA protein levels when PAR-6 levels were manipulated in the neurons. Instead, RhoA activity was altered by PAR-6, as visualized using a FRET probe that senses the balance of GAP and GEF activities in the cell. This effect was mediated by the p190A RhoGAP, which is expressed at high levels in the brain (Brouns et al., 2000). It remains unclear exactly how PAR-6/aPKC regulates p190 RhoGAP. One possibility is PKCβ may directly phosphorylate p190 on serine residues and activate it. Alternatively, PKCβ could regulate one of the upstream activators of p190. We have examined a potential role for the Src family of tyrosine kinases, which are major upstream activators of p190 (Brouns et al., 2001; Moon and Zheng, 2003), in PAR-6/aPKC induced spine morphogenesis. Treatment of neurons with a Src family inhibitor PP2 had no effect on PAR-6 induced spine formation (data not shown), however, suggesting that Src is not involved in this pathway.
A distinct RhoGAP called oligophrenin-1 has also been implicated in spine morphogenesis, and is mutated in some cases of X-linked mental retardation. Notably, however, oligophrenin-1 controls spine length rather than biogenesis (Govek et al., 2005). Other studies have implicated a variety of Rho family regulators in spine morphogenesis, including Kalirin, a GEF that activates multiple GTPases in this family (but not RhoA or RhoB) (Penzes et al., 2001). Depletion of Kalirin also reduces spine density, but through an unknown mechanism (Ma et al., 2003). Additionally, Tiam1/2 and βPIX, which are Rac GEFs, are necessary for normal spine maturation (Zhang and Macara, 2006; Zhang et al., 2003). Together, these data point to an exquisite spatial and temporal control over multiple small GTPases, most of which impact actin dynamics.
The PAR-3/6 polarity complex is involved in cell polarity establishment in many contexts from worms to humans; however, the mechanisms by which this complex functions have not been fully elucidated. While in some cases the proteins appear to act together, or function in the same process (Nakaya et al., 2000; Ooshio et al., 2007; Watts et al., 1996), in others they do not colocalize and may function independently (Harris and Peifer, 2005). Dendritic spine morphogenesis provides an ideal model in which to address this dichotomy. A striking conclusion is that both PAR-3 and PAR-6/aPKC modulate the actin cytoskeleton through Rho family GTPases, but that they operate on distinct steps in spine morphogenesis, and through different GTPases. While PAR-3 spatially sequesters Tiam1 and restricts Rac activation, PAR-6 functions through p190RhoGAP to suppress Rho activation. Moreover, the two GTPases have opposing effects: while Rac activity is essential for spine maturation, Rho activity suppresses spine biogenesis. Elucidation of the upstream regulators of the PAR-3/6 complex, possibly including neurotransmitter receptors and other cell surface receptors, will shed more light on the role of PAR proteins in spine morphogenesis.
Experimental Procedures
Plasmids
PAR-6B and PAR-6C constructs have been described previously (Gao et al., 2002; Joberty et al., 2000). Par-6C and p190RhoGAP shRNA constructs were generated by inserting the following annealed oligonucleotides into the pSUPER vector. PAR-6C shRNA#3: 5′-gatccccCCAGCGTAATAATGTGGTAttcaagagaTACCACATTATTACGCTGGtttttggaaa-3′ (forward), 5′-agcttttccaaaaaCCAGCGTAATAATGTGGTAtctcttgaaTACCACATTATTACGCTGGggg (reverse). PAR-6C shRNA #8: 5′-gatccccATGAGATCCTCGAGGTCAAttcaagagaTTGACCTCGAGGATCTCATtttttggaaa-3′ (forward), 5′-agcttttccaaaaaATGAGATCCTCGAGGTCAAtctcttgaaTTGACCTCGAGGATCTCATggg-3′ (reverse). P190A shRNA: 5′-gatccccGACTCTGCACAGCTTAATAttcaagagaTATTAAGCTGTGCAGAGTCtttttggaaa-3′ (forward), 5′-agcttttccaaaaaGACTCTGCACAGCTTAATAtctcttgaaTATTAAGCTGTGCAGAGTCggg-3′ (reverse). The PAR-3 shRNA#3 construct has been described previously (Zhang and Macara, 2006). PKCβ constructs were generously provided by Margaret Chou (University of Pennsylvania, Philadelphia, PA). The p190A RhoGAP constructs have been described previously (Tatsis et al., 1998). The Raichu RhoA biosensor was a gift from Dr. Michiyuki Matsuda (Kyoto University, Kyoto, Japan).
Reagents and antibodies
The primary antibodies used in this study include: goat polyclonal anti-PAR-6C (T-20 from Santa Cruz, 1:100 for immunostaining), rabbit polyclonal anti-PAR-6C (H-90 from Santa Cruz, 1:100 for immunostaining and 1:1000 for western blotting), rabbit polyclonal PAR-3 antibody (Zymed, 1:2000), monoclonal anti-p190A RhoGAP (BD Biosciences, 1:200). Secondary antibodies used include: FITC conjugated donkey anti-goat IgG, Texas Red-conjugated sheep anti-mouse IgG, and Alexa594 conjugated goat anti-mouse IgG. The ROCK inhibitor H-1152 has been described previously (Chen and Macara, 2005).
Neuronal culture and transfection
Hippocampal cultures were prepared and transfected with calcium phosphate precipitation as previously described (Zhang et al., 2003). For transfections involving more than one plasmid, the expression of each construct was confirmed by immunofluorescence and co-expression was found to occur in >90% of the neurons.
Immunofluorescence Microscopy and Image Quantification
Hippocampal neurons were fixed in 4% paraformaldehyde with 4% sucrose in phosphate buffered saline (PBS) at room temperature for 15 minutes, and then permeabilized in 0.2% Triton X-100 in PBS for 5 minutes. Alternatively, they were fixed and permeabilized simultaneously in 100% methanol for 20 minutes at −20°C. After blocking with 10% BSA in PBS for 1 hour at room temperature, the neurons were incubated with primary antibodies diluted in 3% BSA in PBS overnight at 4°C. Neurons were then washed and incubated with secondary antibodies for 1 hour at room temperature. Coverslips were then washed and mounted using Gel/Mount (Biomeda Corp., Foster City, CA). FM dye uptake experiments were performed as previously described (Zhang and Macara, 2006). Epifluorescence images were collected using an inverted microscope (Nikon TE-200)) with a 60x water-immersion lens (Plan Achromatic, NA 1.2) coupled to a CCD camera (Hamamatsu Orca), controlled by Openlab 5.0.1 software (Improvision, Boston, MA). Spine density and spine morphology were quantified as previously described (Zhang and Macara, 2006). For spine density, 80–100 primary and secondary dendrites from 15–20 neurons were quantified for each experimental condition. For spine length and width, over 300 spines from 15–20 neurons were quantified for each experimental condition. Two-tailed, two-sample unequal variance Student’s t-test was used to calculate the p-values.
FRET Microscopy
Ratio imaging of the FRET biosensor was performed using excitation filters S436/10X for CFP and S500/20X for TFP and emission filters S470/30m for CFP and S535/30m for YFP (Chroma Technology Corp.). Cells were illuminated through both the ND4 and ND8 filters to reduce photobleaching. After background subtraction, FRET images were created using the Openlab 5.0.1 FRET module.
Immunoprecipitation and Western Blotting
Immunoprecipitations and western blotting were done as described previously (Zhang and Macara, 2006).
Supplementary Material
Acknowledgments
We would like to thank Dr. Margaret Chou (University of Pennsylvania, Philadelphia, PA) and Dr. Michiyuki Matsuda (Kyoto University, Kyoto, Japan) for generously providing reagents. This work was supported by grant GM 070902 from the National Institutes of Health, and in part by the Alzheimer’s and Related Diseases Research Award Fund from the Virginia Center on Aging. H.Z. was supported in part by the John Douglas French Alzheimer’s Foundation postdoctoral fellowship.
References
- Brouns MR, Matheson SF, Hu KQ, Delalle I, Caviness VS, Silver J, Bronson RT, Settleman J. The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development. 2000;127:4891–4903. doi: 10.1242/dev.127.22.4891. [DOI] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- Chen X, Macara IG. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Gao L, Joberty G, Macara IG. Assembly of epithelial tight junctions is negatively regulated by Par6. Curr Biol. 2002;12:221–225. doi: 10.1016/s0960-9822(01)00663-7. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. 2003;40:243–264. doi: 10.1016/s0896-6273(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Harris TJ, Peifer M. The positioning and segregation of apical cues during epithelial polarity establishment in Drosophila. J Cell Biol. 2005;170:813–823. doi: 10.1083/jcb.200505127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, LeDoux JE. Fear memory formation involves p190 RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron. 2002;36:727–738. doi: 10.1016/s0896-6273(02)01047-4. [DOI] [PubMed] [Google Scholar]
- Ma XM, Huang J, Wang Y, Eipper BA, Mains RE. Kalirin, a multifunctional Rho guanine nucleotide exchange factor, is necessary for maintenance of hippocampal pyramidal neuron dendrites and dendritic spines. J Neurosci. 2003;23:10593–10603. doi: 10.1523/JNEUROSCI.23-33-10593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macara IG. Parsing the polarity code. Nat Rev Mol Cell Biol. 2004;5:220–231. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- Matus A. Growth of dendritic spines: a continuing story. Curr Opin Neurobiol. 2005;15:67–72. doi: 10.1016/j.conb.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Fukui A, Izumi Y, Akimoto K, Asashima M, Ohno S. Meiotic maturation induces animal-vegetal asymmetric distribution of aPKC and ASIP/PAR-3 in Xenopus oocytes. Development. 2000;127:5021–5031. doi: 10.1242/dev.127.23.5021. [DOI] [PubMed] [Google Scholar]
- Ooshio T, Fujita N, Yamada A, Sato T, Kitagawa Y, Okamoto R, Nakata S, Miki A, Irie K, Takai Y. Cooperative roles of Par-3 and afadin in the formation of adherens and tight junctions. J Cell Sci. 2007;120:2352–2365. doi: 10.1242/jcs.03470. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Sattler R, Zhang X, Huganir RL, Kambampati V, Mains RE, Eipper BA. The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron. 2001;29:229–242. doi: 10.1016/s0896-6273(01)00193-3. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Settleman J. A memory GAP. Trends Neurosci. 2003;26:285–287. doi: 10.1016/S0166-2236(03)00103-6. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The Postsynaptic Architecture of Excitatory Synapses: A More Quantitative View. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Tatsis N, Lannigan DA, Macara IG. The function of the p190 Rho GTPase-activating protein is controlled by its N-terminal GTP binding domain. J Biol Chem. 1998;273:34631–34638. doi: 10.1074/jbc.273.51.34631. [DOI] [PubMed] [Google Scholar]
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- Watts JL, Etemad-Moghadam B, Guo S, Boyd L, Draper BW, Mello CC, Priess JR, Kemphues KJ. par-6, a gene involved in the establishment of asymmetry in early C. elegans embryos, mediates the asymmetric localization of PAR-3. Development. 1996;122:3133–3140. doi: 10.1242/dev.122.10.3133. [DOI] [PubMed] [Google Scholar]
- Yamada S, Nelson WJ. Synapses: Sites of Cell Recognition, Adhesion, and Functional Specification. Annu Rev Biochem. 2007;76:267–294. doi: 10.1146/annurev.biochem.75.103004.142811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223–232. doi: 10.1083/jcb.200212049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol. 2003;161:131–142. doi: 10.1083/jcb.200211002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.