Hepatitis C Virus (HCV) Proteins Induce NADPH Oxidase 4 Expression in a Transforming Growth Factor β-Dependent Manner: a New Contributor to HCV-Induced Oxidative Stress (original) (raw)
Abstract
Viral hepatitis-induced oxidative stress accompanied by increased levels of transforming growth factor β (TGF-β) and hepatic fibrosis are hallmarks of hepatitis C virus (HCV) infection. The mechanisms of redox regulation in the pathogenesis of HCV-induced liver disease are not clearly understood. The results of our current studies suggest that reactive oxygen species (ROS) derived from Nox4, a member of the NADPH oxidase (Nox) family, could play a role in HCV-induced liver disease. We found that the expression of HCV (genotype 1a) cDNA constructs (full-length and subgenomic), core protein alone, viral RNA, or replicating HCV (JFH-AM2) induced Nox4 mRNA expression and ROS generation in human hepatocyte cell lines (Huh-7, Huh-7.5, HepG2, and CHL). Conversely, hepatocytes expressing Nox4 short hairpin RNA (shRNA) or an inactive dominant negative form of Nox4 showed decreased ROS production when cells were transfected with HCV. The promoters of both human and murine Nox4 were used to demonstrate transcriptional regulation of Nox4 mRNA by HCV, and a luciferase reporter tied to an ∼2-kb promoter region of Nox4 identified HCV-responsive regulatory regions modulating the expression of Nox4. Furthermore, the human Nox4 promoter was responsive to TGF-β1, and the HCV core-dependent induction of Nox4 was blocked by antibody against TGF-β or the expression of dominant negative TGF-β receptor type II. These findings identified HCV as a regulator of Nox4 gene expression and subsequent ROS production through an autocrine TGF-β-dependent mechanism. Collectively, these data provide evidence that HCV-induced Nox4 contributes to ROS production and may be related to HCV-induced liver disease.
Hepatitis C virus (HCV) is the leading cause of viral hepatitis, which can progress to hepatic steatosis, cirrhosis, and hepatocellular carcinoma (43). Recent observations suggest that reactive oxygen species (ROS) play an important role in the development and progression of inflammatory liver disease mediated by HCV (11, 29). HCV is a 9.6-kb positive-strand RNA virus consisting of 10 genes that encode four structural and six nonstructural proteins. The virus primarily infects and replicates in hepatocytes, utilizing both viral and host proteins. Some HCV proteins regulate host cell gene expression involved in inflammation, apoptosis, fibrosis, and mitogenesis (17). Of the 10 viral proteins, the expression of core, NS3, or NS5a protein has been associated with increased oxidative stress (7, 21, 50, 65). While most hepatitis viruses are associated with increased oxidative stress, HCV induces higher production of ROS than other hepatitis viruses (19). This suggests that ROS-generating enzymes, such as NADPH oxidases (Noxes), are involved in the progression of inflammatory liver disease.
Members of the Nox family generate superoxide by transporting electrons across biological membranes to molecular oxygen. Originally described as the catalytic core of the phagocytic oxidase, Nox2, or gp91phox, is the prototype for six additional nonphagocytic Nox family members (Nox1, -3, -4, and -5 and Duox1 and -2) (3, 23). All Nox enxymes share conserved structural features, including six transmembrane segments that contain highly conserved heme-binding histidines and flavin adenine dinucleotide (FAD) and NADPH binding sequences within their C-terminal cytoplasmic domains. Tissue expression patterns and activation mechanisms vary among the Noxes. Noxes Nox1 to -3 require additional cytosolic regulators for maximum activation and ROS generation, whereas Nox4 exhibits constitutive activity independent of these factors (41).
Nox4 is a 578-amino-acid protein with 39% sequence identity relative to Nox2 (gp91phox) (22). Although originally discovered in the kidney, Nox4 mRNA is detected in several other human and murine tissues, including bone, vascular tissue, and lung (3, 22, 23). Nox4 is primarily localized in perinuclear/endoplasmic reticulum (ER) regions but is also detected at the plasma membrane, at focal adhesions, and within the nucleus (3). In normal liver tissue, Nox4 mRNA is detected at low levels compared with the amount in the kidney (22, 60). Although Nox4 is a constitutively active ROS-generating enzyme, increased expression of mRNA, protein, and ROS has been detected in response to inflammatory stimuli. Recent work suggests that Nox4-derived ROS are involved in transforming growth factor β (TGF-β)-induced fibrosis, ER stress, human immunodeficiency virus type 1-activated cell signaling, beta interferon-regulated transcription, and Toll-like receptor 4-mediated pathways (10, 14, 51-53, 72). However, little is known about the function of Nox4 in the liver under inflammatory conditions. ROS and oxidative stress have been considered critical during the progression and pathogenesis of inflammatory liver diseases, including viral hepatitis (11, 15, 58).
A better understanding of ROS-activated signaling pathways involved in liver disease may provide new strategies to prevent or treat viral hepatitis-induced inflammation and fibrosis. Here, we investigated the potential role of Noxes in HCV-induced oxidative stress and found that of the Nox family enzymes, Nox4 may have a role in HCV-mediated liver disease. We demonstrate that HCV cDNA or viral RNA expression increases Nox4 mRNA and protein levels and subsequent Nox4-dependent superoxide levels. We also observed increased human and murine Nox4 promoter activities in HCV-expressing cells. When we expressed subgenomic structural and nonstructural HCV proteins, increased Nox4 expression and superoxide production were also observed. Furthermore, the structural HCV core protein induced high-level expression of Nox4 and Nox4-dependent ROS generation; this effect was diminished with neutralizing antibodies against TGF-β or dominant negative TGF-β receptor, indicating a novel mechanism where regulation of hepatic Nox4 by HCV core is mediated in an autocrine TGF-β-dependent manner.
MATERIALS AND METHODS
Cell culture.
The human hepatocellular carcinoma cell lines HepG2 and Chang Liver cells (CHL) and the murine hepatocyte cell line Hepa 1-6 were all obtained from the American Type Culture Collection (Rockville, MD). All cell lines were maintained in low-glucose Dulbecco's minimal essential medium (DMEM) (Invitrogen, Carlsbad, CA) and supplemented with 10% heat-inactivated fetal bovine serum (HyClone/Thermo Scientific, Logan, UT) and 100 mg/ml penicillin/streptomycin (Invitrogen). All cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C. All cells were grown on BioCoat collagen I-coated tissue culture plates (BD Biosciences, San Jose, CA).
Transient and stable transfections.
Human cell lines HepG2, Huh-7, and CHL and murine cell line Hepa 1-6 were transfected with Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's protocol. In brief, cells were seeded in numbers dependent upon the cell line (HepG2, 3 × 105; Huh-7, 1.5 × 104; CHL, 2 × 105; and Hepa 1-6, 1.5 × 104) in BioCoat collagen I-coated six-well plates (BD Biosciences) 24 h before transfection. The transfection mixture (serum-free DMEM, FuGENE 6, and plasmid DNA) was incubated at room temperature for 20 min and then added dropwise to cell culture wells. Experiments designed for Nox4 suppression used Nox4-specific Mission short hairpin RNA (shRNA) plasmids (Sigma, St. Louis, MO) stably transfected into HepG2 cells as suggested by the manufacturer's protocol. Briefly, 1.5 × 106 HepG2 cells were seeded into collagen I-coated 10-cm tissue culture dishes 24 h before transfection. Ten micrograms of plasmid DNA was transfected with 20 μl of FuGENE 6 reagent. Forty-eight hours later, the medium was changed to DMEM 10% fetal bovine serum supplemented with 800 μg/ml G418 (Invitrogen). Approximately 2 weeks later, individual clones were selected and analyzed for Nox4 protein expression by Western blot analysis.
Plasmids and primers.
The HCV (genotype 1a) plasmid pCV-H77 (GenBank accession number AF011751) was kindly provided by Suzanne Emerson (NIH/NIAID) (74). The full-length HCV 1a open reading frame (ORF) (nucleotides 342 to 9374) was amplified with Hi-Fi Pfu DNA polymerase (Invitrogen) using the following primers: forward primer 5′-GCACCATGAGCACGAATCCTAAACCTC-3′ and reverse primer 5′-CTATCATCGGTTGGGGAGGAGGTAGATGCC-3′. The PCR conditions were as follows: 94°C for 2 min, denaturation at 94°C for 20 s, annealing at 62°C for 2 min, extension at 68°C for 10 min, and a final extension at 68°C for 10 min. Thirty-five cycles were performed. The amplified PCR product (9,033 bp) was cloned into the pcDNA3.3 TOPO TA mammalian expression vector (Invitrogen) according to the manufacturer's instructions. For HCV structural and nonstructural protein constructs, the following primers were used: the same forward primer as for the full-length ORF, structural protein reverse primer 5′-CTACTATGCGTATGCCCGCTGAGGCAAC-3′, nonstructural protein forward primer 5′-CACCCTGGACACGGAGGTGGCCGCGTC-3′, and the same reverse primer as for the full-length ORF. HCV core and HCV NS3 were cloned into pcDNA3.3 using the following primers: core forward primer 5′-CACCATGAGCACGAATCCTAAACCTC-3′, core reverse primer 5′-CTACTAGGCTGAAGCGGGCACAGTCAGGC-3′, NS3 forward primer 5′-CACCATGGCGCCCATCACGGCGTACGC-3′, and NS3 reverse primer 5′-TCATCACGTGACGACCTCCAGGTCGG-3′. Nox4-ΔCT was generated using full-length human Nox4 (GenBank accession number NM_016931) cDNA subcloned into pcDNA3.3 as the template. Truncation of Nox4 was generated by PCR with the following primers: forward primer 5′-CACCATGGCTGTGTCCTGGAGGA-3′ and reverse primer 5′-AATGATGGTGACTGGCTTATTGCTCCG-3′. The truncated Nox4 PCR product was subsequently cloned into the pcDNA3.1 V5-His directional expression vector. Dominant negative TGF-β receptor type II (TGF-βRII) was generated using a human TGF-βRII cDNA IMAGE clone (GenBank accession number BC040499). Truncated TGF-βRII was subcloned into pcDNA3.3 using the following primers: forward primer 5′-CACCATGGGTCGGGGGCTGCTCAGGG-3′ and reverse primer 5′-CTACTATGAACTCAGCTTTGCTGCCGGTTAACGCG-3′.
Plasmid constructs were verified by automated DNA sequencing with vector-specific primers (Macrogen, Rockville, MD). The HCV ORF was verified by overlapping forward and reverse primers. Table 1 lists additional primers used in this study. The PCR thermocycler conditions were as follows: 94°C for 2 min, denaturation at 94°C for 30 s, annealing at 58°C for 30 s, extension at 72°C for 30 s, and a final extension at 72°C for 5 min. Thirty cycles were performed.
TABLE 1.
Primers used in this study
| Gene product | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Nox1 | AGATGAACAAGCGTGGCTTC | AGATTGAGGGGCAATTAACAAA |
| Nox2 | ACTTGCAGGCTTTGTATGTGAA | ACAGTTTTAAGAATTCCCCGTAGA |
| Nox3 | GACTTCTGGCCGCACTTTC | CAGGGTTGAGGTAGCTCTCG |
| Nox4 | TGGCTCTCCATGAATGTCCT | CTTAGACACAATCCCCCAACA |
| Nox5 | GCTTATCCGAGGTCTCAATCC | AAGAACCCACCAATTCCAGA |
| Duoxl | CACCTCCTGGAGACCTTTTTC | GGCCTGGTTGATGTCCAG |
| Duox2 | TACCCAGGAGGTGAATGAGC | TCCTCCAACTCCGAAATCTG |
| p22phox | ATGGGGCAGATCGAGTGGGCCATG | CTCGTCGGTCACCGGGATGGGGT |
| Actin | CTACAATGAGCTGCGTGTGG | AAGGAAGGCTGGAAGAGTGC |
| GAPDH | ATCTTCCAGGAGCGAGATCC | ACCACTGACACGTTGGCAGT |
| Core | CACCATGAGCACGAATCCTAAACCTC | CTACTAGGCTGAAGCGGGCACAGTC |
| NS3 | CACCGCCATGGCGCCCATCACGGCG | TCATCACGTGACGACCTCCAGGTCG |
JFH-AM2 HCV infection.
Huh 7.5 cells (a gift from Charles Rice) (4) were infected with a mutant (AM2) of HCV strain JFH1 that had acquired cell culture adaptive mutations during serial passage in Huh 7.5 cells (55). Huh 7.5 cells at 50 to 60% confluence in six-well culture plates were incubated with 1 ml of cell culture medium containing approximately 4 × 105 focus-forming units or were mock infected with medium alone. After 5 h of incubation at 37°C in 5% CO2, the liquid was replaced with 3 ml DMEM containing 10% fetal bovine serum, and plates were incubated for an additional 5 days. Cells were confluent by day 2. An aliquot of the day 2 harvest was plated on an eight-well chamber slide and stained on day 5 postinfection with chimpanzee anti-HCV plasma for immunofluorescence microscopy. More than 80% of the cells were positive for HCV antigen.
ROS detection.
Hepatocytes (2.5 × 104) were collected by trypsinization and washed twice with Hank's balanced salt solution buffer (Invitrogen) by centrifugation. Cells were treated for 10 min at 37°C with or without 10 μM diphenylene iodonium chloride (DPI) (Sigma). Kinetic ROS detection measurements were performed by chemiluminescence in 96-well plates at 37°C over a 45-min or 1-h time course using a Luminoskan luminometer (Thermo, Waltham, MA). Extracellular superoxide production was measured as superoxide dismutase-inhibitable chemiluminescence detected using the Diogenes reagent (National Diagnostics, Atlanta, GA). Intracellular hydrogen peroxide production was measured by CM-H2DCF-DA [5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester] oxidation. HepG2 cells (1 × 106/ml) were incubated for 15 min at 37°C in the presence of 1 μg/ml CM-H2DCF-DA (Invitrogen, Carlsbad, CA). Cells were then incubated for an additional 10 min in the presence or absence of 10 μM DPI. CM-H2DCF-DA oxidation was quantified on an LSRII flow cytometer using FACSDiva software (BD, Franklin Lakes, NJ). Data were analyzed in FlowJo (TreeStar, Inc., Ashland, OR).
RNA isolation and RT-PCR.
Total RNA was extracted from cells with Trizol (Invitrogen). One to 2 μg of RNA was used for Thermoscript reverse transcriptase PCR (RT-PCR). Both procedures were conducted according to the manufacturer's protocol (Invitrogen).
In vitro transcription.
In vitro-transcribed full-length HCV-1a or HCVΔNS5b RNA from pCV-H77 was transfected into HepG2 cells with TransMessenger mRNA transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's protocol. Briefly, pCV-H77 was linearized by digestion with XbaI, followed by gel purification and phenol-chloroform extraction. HCVΔNS5b was generated by digestion of plasmid pCV-H77 with XbaI and HindIII. Digestion of the HCV-1a ORF with HindIII at nucleotide 7861 disrupts translation of the NS5b RNA-dependent RNA polymerase protein. One microgram of linear DNA was used for in vitro transcription (4 h) using a MegaScript T7 RNA polymerase kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's protocol. In vitro-transcribed RNA was transfected as described in “Transient and stable transfections.”
Quantitative real-time RT-PCR.
HepG2 Nox4 mRNA was quantified by real-time RT-PCR using an ABI Prism 7500 RT-PCR system (Applied Biosystems). Total cellular RNA was isolated, and 1 μg was used for reverse transcription into cDNA with a ThermoScript RT-PCR kit (Invitrogen). Nox4 mRNA was quantified with SYBR green PCR mix (Invitrogen) with the following human Nox4-specific primers: forward primer 5′-GCTGACGTTGCATGTTTCAG-3′ and reverse primer 5′-CGGGAGGGTGGGTATCTAA-3′. The TGF-β1-specific primers used were forward primer 5′-GCAGCACGTGGAGCTGTA-3′ and reverse primer 5′-CAGCCGGTTGCTGAGGTA-3′. Primers for real-time PCR were designed using Roche Universal ProbeLibrary software. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific forward primer 5′-AGCCACATCGCTCAGACAC-3′ and reverse primer 5′-GCCCAATACGACCAAATCC-3′ were used for an internal reference control. PCR was performed under the following conditions: 10 min at 95°C, followed by 40 cycles of 94°C for 15 s, 50°C for 30 s, and 72°C for 30 s. Relative transcript levels were calculated by using the cycle threshold method as specified by the manufacturer (Applied Biosystems). Results are displayed as the factor of change.
Antibodies and Western blot analysis.
Cells were washed twice with 1× phosphate-buffered saline and scraped in radio immunoprecipitation assay lysis buffer (Sigma). Whole-cell protein extracts were quantified by Bradford assay reagent (Pierce, Rockford, IL). Thirty to 50 μg of whole-cell extracts were resolved electrophoretically on 4-to-12% Bis-Tris NuPAGE gels (Invitrogen) and subsequently transferred to polyvinylidene difluoride membranes (Invitrogen). Membranes were stripped using ReBlot stripping solution (Chemicon/Millipore, Temecula, CA) and reprobed. Western blotting was performed with the following commercially available antibodies: anti-rabbit polyclonal Nox4 antibody (Novus Biologicals, Littleton, CO), rabbit polyclonal antiactin antibody (Sigma), rabbit polyclonal anti-GAPDH (Trevigen, Gaithersburg, MD), mouse monoclonal anti-V5 (Invitrogen), goat polyclonal anti-E2 (BioDesign/Meridian Life Science, Saco, ME), goat polyclonal anti-HCV core (Pierce/ThermoScientific, Rockford, IL), mouse monoclonal anti-NS5a (Pierce/ThermoScientific), goat polyclonal anti-TGF-βRII clone (T-20), and mouse monoclonal anti-TGF-β1 (Santa Cruz) and pan-TGF-β neutralizing monoclonal antibodies (R & D Systems).
Luciferase assay and cloning of Nox4 promoters.
Bacterial artificial chromosome clone RP11-643G5 (Invitrogen) was used as the template to amplify the human Nox4 promoter fragments. The mouse Nox4 promoter was generated from murine genomic DNA. The PCR fragments were generated with _Pfu_-Turbo DNA polymerase (Stratagene, La Jolla, CA). The forward primers amplify fragments from the indicated position to the putative transcription start site downstream. For human Nox4 promoter constructs, all forward primers contain an Mlu1 restriction site (underlined) and the reverse primer contains an XhoI restriction site. The common reverse primer used was 5′-GGCTCGAGGCTGCCCAGACGCCCAGCGC-3′. Forward primers were as follows: (−1356) 5′-GGACGCGTCTGCCAATGAACCTCCTGCCCCC-3′, (−1848) 5′-GGACGCGTGGCTCACCGCAACCTCTGCCTC-3′, and (−741) 5′-CCCACGCGTGTTGCAGTGTCGAATAGGTACCTGGGTCAACTCCACACAC-3′. The forward mouse Nox4 promoter primers contained a BglII restriction site (underlined), and the reverse primer contains an XhoI restriction site. The reverse primer used was 5′-CCATGGGAGTGCTGCGCCCTGCTC-3′. The forward primers were as follows: (−1291) 5′-GAAGATCTGTGGTGGTCAACTAGGACTC-3′, and (−1707) 5′-GAAGATCTCTTTGCAGGCTCAGGCTC-3′. After restriction digestion, promoter fragments were subcloned into the pGL3 Basic reporter plasmid (Promega, Madison, WI) upstream of the luciferase reporter, using KpnI/MluI or NcoI/BglII cloning sites. The luciferase assay was conducted according to the manufacturer's protocol (Promega).
Statistical analysis.
Data are represented as the means ± standard deviations of the results of at least three independent experiments or as the results ± standard errors of the means of an experiment that was representative of at least two other similar experiments. Student's t test was used to calculate significance values; a P value of <0.05 was considered significant.
RESULTS
When hepatocytes are infected with HCV, cells respond by generating a substantial amount of ROS (33, 34). HCV is a ∼9.6-kb positive-strand RNA virus translated into an ∼3,000-amino-acid polypeptide containing 10 viral proteins (4 structural and 6 nonstructural). Once translated, the viral polyprotein is processed and cleaved by host and viral proteases, resulting in the viral protein machinery necessary for HCV replication (Fig. 1A) (44). First, we determined the amount of Nox-dependent ROS generation (DPI-sensitive responses) when hepatocytes are expressing the HCV viral proteins. HCV genotype 1a polypeptide was expressed and properly processed, as indicated by host-dependent cleavage of the core and E2 nonstructural viral proteins (Fig. 1B). Human hepatocellular carcinoma cell lines expressing HCV proteins generated a significant amount of Nox-dependent superoxide 48 h posttransfection (Fig. 1C). We observed increases of approximately two- to fivefold in superoxide production in HCV tranfectants compared to the levels in vector-only transfectants in three human hepatocyte cell lines (Huh-7, ∼fivefold; HepG2, ∼twofold; and CHL, ∼fivefold). While the chemical inhibitor DPI is selective for Nox and other flavoenzymes, the vector-transfected control cells also displayed DPI-sensitive superoxide release. This effect is likely a result of basal ROS production from some Nox enzyme activity. Intracellular H2O2 detected by CM-H2DCF-DA was unaffected by DPI or HCV (Fig. 1D). Together, these results indicate that the expression of HCV proteins results in the production of superoxide released at the plasma membrane.
FIG. 1.

ROS generation by HCV-transfected human hepatocytes. (A) Schematic of the HCV genome. HCV is a positive-strand RNA [(+) RNA] virus whose genes are translated by host cell translational machinery upon infection. Both host and viral proteases cleave the viral polyprotein into 10 individual viral proteins with specific functions. UTR, untranslated region; IRES, internal ribosome entry site. (B) Western blot detection of HCV core and E2 proteins in HepG2 cells transfected with pcDNA3.3 plasmid containing the full-length HCV genome (genotype 1a) (1 μg). Whole-cell lysates were collected 48 h posttransfection, and 30-μg amounts of protein were probed with anti-core, anti-E2, and anti-actin. (C) Human hepatoma cell lines (Huh-7, HepG2, and CHL) were transfected with empty vector or plasmid containing the full-length HCV genome (genotype 1a) (1 μg) for 48 h. Cell lines were assayed for superoxide generation with Diogenes luminescence (45 min) in the presence (+) or absence (−) of Nox inhibitor DPI (10 μM) (n = 5, in triplicate). RLU, relative light units. (D) HepG2 cells were transfected as described for panel C for 48 h. Single-cell intracellular H2O2 was measured by flow cytometry after exposure to CM-H2DCF-DA (DCF; 2 μM) for 15 min and with (+) or without (−) DPI for 10 min (n = 3, in triplicate; *, P < 0.05). MFI, mean fluorescence intensity.
To investigate the potential role of Nox family enzymes in HCV-induced ROS, we asked whether HCV modulates the expression of any of the seven Nox enzymes. When HepG2 cells were transfected with HCV cDNA, we observed a substantial increase in endogenous Nox4 mRNA expression compared with the level in the vector control (Fig. 2A). While other Nox enzymes (Nox1, Duox1, and Duox2) displayed a basal level of mRNA expression, there was no significant change in the level of mRNA in HCV-expressing cells. To further validate these observations, quantification of Nox4 mRNA by quantitative real-time PCR displayed an approximate fivefold increase (Fig. 2B). The increases in Nox4 mRNA expression were observed over a range of concentrations of transfected HCV cDNA (0.25 to 1.0 μg) (Fig. 2C). Constitutive Nox4 activity is reported to be dependent on the transmembrane protein p22phox (41, 69); however, we observed no significant change in p22phox mRNA in the presence of HCV.
FIG. 2.
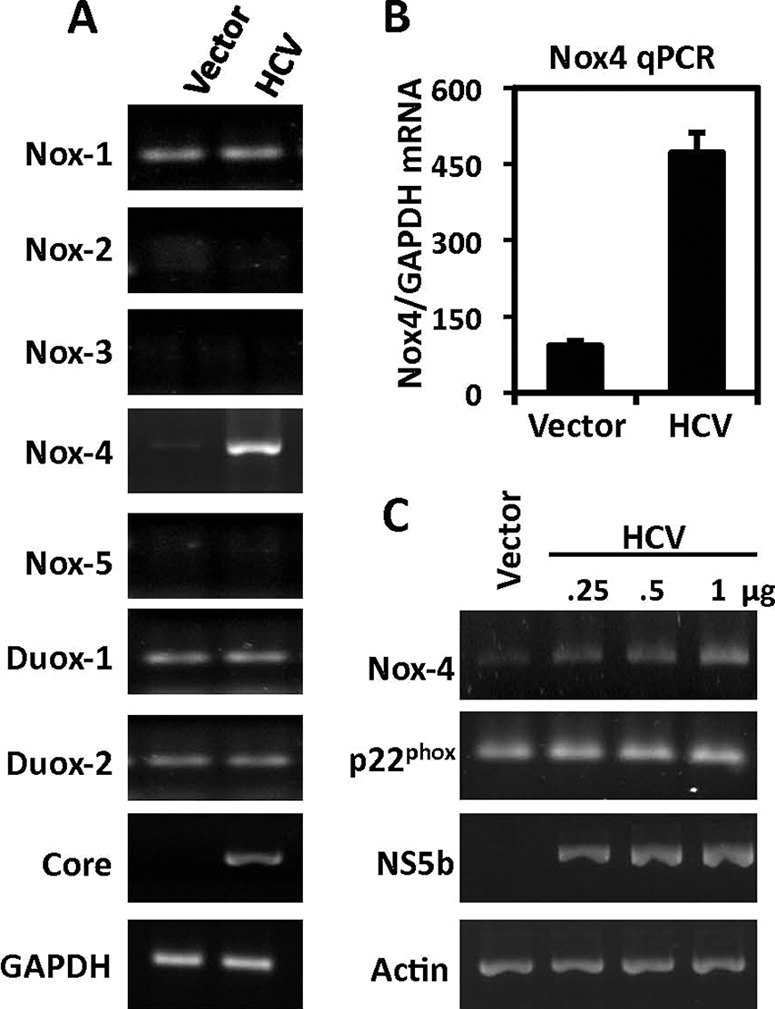
Effect of HCV on the expression of Nox4. (A) Nox-specific PCR amplification of reverse-transcribed total RNA (1 μg) from HepG2 cells transfected 48 h previously with either pcDNA3.1 empty vector plasmid (1 μg) or full-length HCV-1a cDNA plasmid. Results shown are representative of four independent experiments. (B) Quantitative real-time RT-PCR (qPCR) analysis of Nox4 from total RNA extracted from HepG2 cells. RNAs were collected from cells transfected for 48 h with either empty vector plasmid (1 μg) or HCV plasmid (1 μg), followed by reverse transcription. Equal amounts of cDNA were subjected to quantitative RT-PCR using human Nox4-specific primers. GAPDH-specific primers were used as internal control (n = 2, in quadruplicate). (C) HepG2 cells were transfected as described for panel A with increasing amounts of HCV plasmid (0.25, 0.5, and 1.0 μg). p22phox-specific primers were used to determine mRNA expression. Primers specific for the HCV NS5b gene were used to confirm the presence of HCV RNA. Results shown are representative of three independent experiments.
HepG2 cells transfected with in vitro-transcribed full-length HCV RNA also displayed a significant increase in DPI-sensitive superoxide production compared to the level in cells transfected with HCV lacking the gene encoding NS5b, an RNA-dependent RNA polymerase needed for viral replication (Fig. 3A). We observed increased Nox4 mRNA in human hepatocyte cell lines transfected with both HCV cDNA and RNA (Fig. 2 and 3A). To test whether the results could be reproduced in a replicating-HCV cell culture model, Huh7.5 cells were infected with the JFH-AM2 HCV clone. We observed small but significant changes in superoxide generation (Fig. 3B), along with modest Nox4 mRNA changes determined by quantitative PCR (<10%; not shown), in the JFH-AM2-replicating model 2 days postinfection compared with the level of superoxide generation in mock-infected cells. These responses were less robust than those observed in the HCV-transfected Huh-7 and HepG2 models and may reflect the low HCV titers generated in cell culture relative to those observed in HCV-infected patients. Furthermore, adaptations in the viral clone or the host Huh-7.5 cell line (RIG mutations and diminished interferon responsiveness) that render them more permissive to HCV replication (4, 63) may affect Nox4-dependent innate immune responses to HCV.
FIG. 3.

Effect of HCV RNA or infectious virus on superoxide generation and Nox4 mRNA. (A) Left, HepG2 cells were transfected with in vitro-transcribed full-length HCV-1a or in vitro-transcribed HCVΔNS5b (ΔHCV) RNA as mentioned in Materials and Methods. After 48 h, cells were assayed for superoxide generation for 1 h in the presence or absence of DPI (10 μM) by Diogenes chemiluminescence (n = 3, in triplicate). Right, quantitative real-time RT-PCR (qPCR) analysis of Nox4 from total RNA extracted from cells used for experiments whose results are shown at the left using human Nox4-specific primers. Actin-specific primers were used as internal control (n = 3, in triplicate). RLU, relative light units. (B) Huh7.5 cells were infected with cell culture of replicating JFH-AM2 HCV (viral titer 1 × 105 viral copies per milliliter) or were mock infected. Cells were collected at days 2 and 5 postinfection and assayed for superoxide generation. Fold Δ, fold change; w/o, without.
The Nox inhibitor DPI is a general inhibitor of flavoenzymes, including other mitochondrial sources of ROS, and is not specific to Nox family enzymes only. To address this concern, we generated HepG2 cells stably expressing Nox4-specific shRNA. By clonal selection, we obtained a cell line with significantly decreased expression of endogenous Nox4 compared with the level in cells stably expressing a nontargeting shRNA control (Fig. 4A). We observed a decrease in the basal level of superoxide as a result of shRNA knockdown of endogenous Nox4. HCV-transfected HepG2 cells expressing Nox4-specific shRNA displayed a significant reduction (more than twofold) in superoxide generation compared with the level in shRNA control cells transfected with HCV (Fig. 4B). To further validate Nox4 as a contributor to HCV-induced superoxide, we generated a dominant negative Nox4 cDNA construct (Nox4-ΔCT). Deletion of the C-terminal FAD and NADPH binding sites results in a truncated protein that is incapable of transporting electrons and has dominant negative effects on endogenous Nox4 activity (39). HepG2 cells cotransfected with HCV cDNA and Nox4-ΔCT displayed a significant reduction in superoxide generation compared with the level in vector-transfected cells (Fig. 4C and D). These data provide independent confirmation that Nox4 is a key enzyme contributing to HCV-induced cellular ROS production.
FIG. 4.

Dominant negative Nox4 (Nox4-ΔCT) or knockdown of Nox4 decreases HCV-induced superoxide production. (A) HepG2 cells were stably transfected with plasmids encoding shRNAs specific to Nox4 (shRNA Nox4 #88 or shRNA Nox4 #89) or control nontargeting shRNA plasmid and analyzed by Western blotting. The same blot was sequentially probed with anti-Nox4 antibody and anti-GAPDH antibody. Densitometry values given below blots indicate the level of Nox4 protein expression relative to the level of GAPDH protein. (B) Nontargeting control or Nox4 (clone #89) shRNA stable cell lines described for panel A were transfected with HCV and assayed for superoxide generation for 45 min in the presence (+) or absence (−) of DPI (10 μM) (n = 4, in triplicate). (C) Deletion of the FAD and NADPH domains of Nox4 results in a truncated inactive enzyme that has dominant negative effects. (D) HepG2 cells were transfected with vector alone, HCV cDNA, or V5-tagged Nox4-ΔCT or with HCV and V5-tagged Nox4-ΔCT. Forty-eight hours posttransfection, cells were collected and assayed for superoxide generation in the presence or absence (w/o, without) of DPI (n = 3; *, P < 0.05). RLU, relative light units; IB, immunoblot; +, present; −, absent.
To investigate whether HCV protein expression can affect the Nox4 promoter, we generated human and murine Nox4 promoter fragments upstream of the putative transcription start site and subcloned them into a luciferase reporter plasmid (pGL3) (Fig. 5A). HepG2 cells cotransfected with HCV cDNA and the human Nox4 promoter/reporter plasmid pGL3 −1848 displayed a level of luciferase activity approximately threefold higher than the level in control cells cotransfected with green fluorescent protein (GFP) and the Nox4 reporter plasmid (Fig. 5B). This suggests that the expression of HCV proteins has positive regulatory effects on the Nox4 promoter. We then deleted the Nox4 promoter (pGL3 −741 and pGL3 −1356) to delineate HCV-responsive regulatory regions. Similar results were observed when ∼600 bases were deleted (pGL3 −1356). However, deletion of an additional 615 bases revealed an HCV-specific critical regulatory region between −1356 and −741 (Fig. 5C). Two other human hepatocyte cell lines (Huh-7 and CHL) cotransfected with HCV cDNA and the Nox4 promoter/reporter plasmid (pGL3 −1848) also showed increases in luciferase reporter activity, of 4.5-fold and 3.5-fold, respectively, compared with the levels in GFP controls (Fig. 5D). These data correlate with the increased Nox4 mRNA in HCV-transfected cells (Fig. 2). Interestingly, we found similar results in murine hepatocytes cotransfected with HCV and murine Nox4 promoter fragments corresponding to those of the human Nox4 promoter studies (Fig. 5E). Murine cells transfected with HCV also produced significant increases in DPI-sensitive superoxide, at levels similar to the levels of ROS generated by murine Nox4-cDNA-transfected cells (Fig. 5F). These observations suggest that HCV regulates both human and murine Nox4 at the transcriptional level; however, it has not been determined whether this effect is direct or indirect.
FIG. 5.

HCV induces both human and murine Nox4 promoters. (A) 5′-End deletion constructs of the human (top) and murine (bottom) Nox4 promoters were cloned into pGL3 luciferase reporter plasmids. Positive regulatory regions of the Nox4 promoter are indicated in dark gray. ChipMapper, a transcription factor binding site-screening algorithm, was used to analyze 2 kb of promoter sequence upstream of the Nox4 transcription start sites. (B) HepG2 cells were cotransfected with a reporter plasmid (1 μg) containing the luciferase gene under the regulation of the Nox4 promoter (pGL3 −1848) and GFP plasmid (1 μg) or increasing amounts of HCV plasmid (0.25, 0.5, and 1.0 μg). Whole-cell lysates were collected, and luciferase activity was determined by luminescence 48 h later (n = 3, in triplicate). +, present; −, absent. (C) HepG2 cell were cotransfected as described for panel A with serially deleted Nox4 promoter constructs (1 μg) tied to luciferase. Results shown are representative of two independent experiments performed in triplicate. (D) Human Huh-7 or CHL hepatoma cells were cotransfected with Nox4 promoter/reporter plasmid (pGL3 −1848) and GFP or HCV plasmid (1 μg). Whole-cell lysates were collected and analyzed for luciferase activity as described for panel A (n = 4, in triplicate). (E) Hepa 1-6 murine hepatocyte cells were cotransfected with a reporter plasmid containing the luciferase gene under the regulation of the Nox4 murine promoter (pGL3 −1291 or pGL3 −1707) and GFP or HCV plasmid. Luciferase activity was determined 48 h posttransfection (n = 3, in triplicate). (F) Hepa 1-6 cells were transfected with empty vector (1 μg), full-length HCV (1 μg), or murine Nox4 (1 μg) for 48 h. Cells were assayed with Diogenes luminescence for superoxide generation (45 min) in the presence (+) or absence (−) of Nox inhibitor DPI (10 μM) (n = 3, in triplicate; *, P < 0.05). RLU, relative light units.
Previous studies demonstrated that structural or nonstructural HCV constructs can be pro- or antiapoptotic, thereby having different effects on cell fate (24). Next, we examined whether HCV structural and nonstructural genes have an effect on Nox4 levels (Fig. 6A). The expression of either structural or nonstructural HCV cDNA in HepG2 cells resulted in increases in Nox4 protein expression compared with the levels in cells transfected with vector alone (Fig. 6B). Interestingly, expression of the structural genes caused a greater increase in Nox4 protein levels than expression of the full-length HCV or nonstructural specific protein expression. Consistently, the increase in Nox4 expression correlated with an increase in DPI-sensitive superoxide production in cells cotransfected with vector control and full-length HCV or structural or nonstructural genes, whereas in cells cotransfected with Nox4-ΔCT and the HCV constructs, HCV-dependent superoxide generation was significantly blunted (Fig. 6C).
FIG. 6.

HCV structural and nonstructural genes regulate Nox4 expression and Nox4-derived ROS generation. (A) Schematic map of the full-length (FL) HCV genome displaying the structural and nonstructural genomic regions. The structural (Str) genes include core, E1, E2, and p7 viral genes. The nonstructural (NStr) genes include NS2, NS3, NS4a, NS4b, NS5a, and NS5b. HCV deletion constructs (Str and NStr) were generated by PCR and cloned into pcDNA3.3 as described in Materials and Methods. (B) HepG2 cells were transfected with empty-vector (EV), HCV FL, HCV Str, or HCV NStr plasmid (1 μg). After 48 h, whole-cell lysates were collected and 30 μg amounts of protein were analyzed by Western blotting, being probed sequentially with anticore, anti-E2, anti-NS5a, anti-Nox4, and anti-GAPDH. Lower panel displays densitometry values of Nox4 protein relative to GAPDH protein expression. Results are indicated as the factor of change compared to the level in cells transfected with vector alone. IB, immunoblot. (C) HepG2 cells were cotransfected with vector (V) and HCV FL, Str, or NStr constructs or cotransfected with Nox4-ΔCT and HCV FL, Str, or NStr constructs. Cells were assayed by Diogenes luminescence for superoxide generation for 45 min in the presence or absence of DPI (10 μM) (n = 3, in triplicate). (D) HepG2 cells stably expressing Nox4 shRNA (clone #89) were transfected as described for panel B. Cells were assayed with Diogenes reagent for superoxide generation for 45 min in the presence or absence of DPI (10 μM) (n = 3, in triplicate; *, P < 0.05). RLU, relative light units; w/o, without.
To confirm the specificity of Nox4-dependent ROS generation under these conditions, we transfected the HCV subgenomic constructs into HepG2 cells stably expressing Nox4-targeting shRNA. Targeted shRNA prevented Nox4 induction caused by full-length HCV, as well as by structural and nonstructural HCV constructs. These results paralleled our results shown in Fig. 4, indicating that full-length HCV, as well as structural and nonstructural HCV constructs, increases ROS production in response to HCV expression in a Nox4-dependent manner (Fig. 6D). Taken together, these data suggest that specific structural and nonstructural HCV genes play a role in modulating Nox4 expression and subsequent ROS generation.
Previous studies by Okuda et al. demonstrated that the expression of core protein in Huh7 cells induced DPI-sensitive ROS production. These authors attribute these effects to mitochondrion-derived ROS, although DPI is a general flavoprotein inhibitor that also affects all Nox enzymes (50). To further clarify the source of ROS in response to HCV core, we transfected HepG2 cells with plasmid encoding the core viral protein. Analysis of the level of Nox4 in cells transfected with HCV core compared with that in cells transfected with vector alone revealed a significant increase in Nox4 mRNA (Fig. 7A, left). In hepatocytes transfected with HCV NS3, a known inducer of Nox2-derived ROS in monocytes (7, 65), there were no effects on Nox4 expression. In addition, Western blotting analysis detected increased Nox4 levels in cells transfected with the core protein (Fig. 7A, right). Upregulation of Nox4-derived superoxide by core transfection was blunted in Nox4 shRNA-expressing cells (Fig. 7B). Increases in superoxide production were not as closely correlated with Nox4 mRNA changes with larger amounts of transfected core protein cDNA; this likely reflects the effects of transfection reagents or limitations of core protein production at high concentrations (Fig. 7C and D). To further validate HCV core-mediated regulation of Nox4, we examined the effects of core on the Nox4 promoter and observed core-specific effects on Nox4 promoter activity. Cotransfection of NS3 and Nox4 promoter plasmids had no effect on reporter activity (Fig. 7E).
FIG. 7.

HCV core protein induces Nox4 expression and ROS release in human hepatocytes. (A) Left, HepG2 cells were transfected for 48 h with pcDNA3.3 expression plasmid encoding the HCV core protein or NS3 protein (1 μg). Nox4- and actin-specific primers were used to determine mRNA expression by RT-PCR. Right, HepG2 cells were transfected with either empty-vector plasmid or HCV core plasmid (1 μg) for 48 h. Whole-cell lysates were then collected, and 30-μg amounts of protein were analyzed by Western blotting. The blot was sequentially probed with anti-Nox4 and anti-core. IB, immunoblot. (B) HepG2 cells stably expressing Nox4 shRNA (clone #89) were transfected as described for panel A, right. Cells were assayed for superoxide generation with Diogenes reagent for 45 min in the presence or absence of DPI (10 μM) (n = 3, in triplicate). (C) HepG2 cells were transfected for 48 h with empty vector or increasing amounts of pcDNA3.3 core plasmid (0.25, 0.5, 1.0, or 2.0 μg). Total RNA was collected and reverse transcribed to determine the level of Nox4 mRNA expression by quantitative PCR (qPCR) (n = 3, in triplicate). (D) HepG2 cells were transfected for 48 h with empty vector or increasing amounts of pcDNA3.3 core as described for panel D. After 48 h, the cells were collected and analyzed for extracellular superoxide production in the presence or absence of DPI (10 μM). (E) HepG2 cells were cotransfected with the Nox4 promoter-luciferase reporter plasmid pGL3 −1848 and GFP, core, or NS3 plasmids (1.0 μg). Whole-cell lysates were collected, and luciferase activity was determined by luminescence 48 h later (n = 3, in triplicate; *, P < 0.05). RLU, relative light units; w/o, without.
Although the core protein does not bind directly to DNA, previous reports indicate that core-induced changes in gene expression include diverse signaling pathways and downstream transcription factors (1, 26). The profibrotic cytokine TGF-β1 is a plasma serum marker in patients with chronic HCV infection (40, 61). Studies by Taniguchi et al. demonstrated that the expression of core protein in HepG2 cells upregulates TGF-β1 transcription (64). As mentioned previously, TGF-β1 is an inducer of Nox4 and has been implicated in profibrotic processes (5, 14, 62). To test whether TGF-β1 can regulate Nox4 at the level of transcription in human hepatocytes, we utilized our human Nox4 promoter plasmid and found that treatment with TGF-β1 significantly increases reporter activity in HepG2 cells (Fig. 8A). Induction of Nox4 in human hepatocytes by TGF-β1 was further confirmed when cells were treated with increasing amounts of TGF-β1. We found that TGF-β-induced Nox4 mRNA expression peaked in a dose-specific manner (Fig. 8B). Consistently, HepG2 cells treated with TGF-β1 generated a significant amount of DPI-sensitive superoxide in a dose-dependent fashion (Fig. 8C). Given that Nox4 is regulated by the HCV core protein, as well as TGF-β1, we hypothesized that core-induced TGF-β1 can act in an autocrine fashion to modulate hepatic Nox4 expression. To investigate this hypothesis, HepG2 cells were transfected with empty vector or core plasmid and then treated with TGF-β-neutralizing monoclonal antibody or not treated. The exogenous TGF-β antibody would bind TGF-β released from the cells, thereby inhibiting its ability to activate the TGF-β receptor. Our results show that the core protein increases the levels of Nox4 and TGF-β1, as predicted, and interestingly, treatment of cells with TGF-β antibody prevented the HCV core-mediated induction of Nox4 at both the mRNA and protein level (Fig. 8D). Wieser et al. previously described an inactive TGF-β receptor type II lacking signaling domains in the cytoplasmic region that inhibits downstream TGF-β signaling, since extracellular TGF-β ligands can effectively bind the TGF-βRI/TGF-βRII homodimer receptor complex (71). Cotransfection of HepG2 cells with core and a dominant negative form of TGF-βRII or treatment of core-expressing cells with TGF-β-neutralizing antibodies resulted in a reduction of superoxide (Fig. 8E). We were further convinced of this mechanism when Nox4 mRNA expression was decreased in cells cotransfected with core and dominant negative TGF-βRII (Fig. 8F, left). In agreement with previous reports (59, 64), we found that the expression of HCV core upregulated TGF-β mRNA. This effect was still seen in the presence of dominant negative TGF-βRII, indicating that HCV core modulation of TGF-β mRNA is upstream of TGF-β signaling (Fig. 8F, right). Collectively, these data support a new mechanism in which Nox4 expression is induced by HCV core through upregulation of TGF-β1 in an autocrine-dependent signaling pathway.
FIG. 8.

Regulation of Nox4 by HCV core protein involves TGF-β1 autocrine signals. (A) Exogenous TGF-β1 induces the Nox4 promoter. HepG2 cells were transfected with the Nox4 promoter reporter plasmid (pGL3 −1848) with or without GFP plasmid (1.0 μg). Twenty-four hours later, transfected cells were treated with or without TGF-β1 (2 ng/ml) for 24 h. Whole-cell lysates were collected, and luciferase activity was determined by luminescence (n = 4, in triplicate). (B) Nox4 mRNA induction by TGF-β1-treated HepG2 cells. Cells were treated with or without (UT, untreated) increasing concentrations of TGF-β1 (0.5 to 2.0 ng/ml) in full-serum culture medium for 48 h. Total RNA was collected and reverse transcribed. Nox4 and GAPDH expression levels were determined by PCR amplification of total cDNA with gene-specific primers. (C) HepG2 cells were treated with TGF-β1 for 48 h as described for panel B. Cells were assayed with Diogenes reagent for superoxide generation for 45 min in the presence (+) or absence (−) of DPI (10 μM) (n = 3, in triplicate). (D) Left, HepG2 cells were transfected with either empty-vector plasmid or HCV core plasmid (1 μg). Twenty-four hours later, core-transfected cells were treated or not treated (−) with TGF-β monoclonal antibody (MAb; 2 μg/ml) for 24 h. Nox4-, core-, TGF-β1-, and GAPDH-specific PCR products were amplified from reverse-transcribed total RNA (1 μg) from cells subjected to different treatments. Right, HepG2 cells were transfected and treated as described for the left panel. Thirty-microgram amounts of total cell lysate were analyzed for protein expression by Western blotting. The immunoblot (IB) was sequentially probed with antibodies to Nox4, core, TGF-β1, and actin. Densitometry values given below blots indicate Nox4 or TGF-β protein levels relative to GAPDH protein expression. (E) HepG2 cells were transfected with empty-vector plasmid (1 μg), core plasmid (1 μg), or dominant negative TGF-βRII (ΔTβRII; 1 μg). Twenty-four hours later, cells were treated without or with TGF-β monoclonal antibody (TβMAb; 2 μg/ml) for 24 h. Cells were then assayed for superoxide production as described for panel C (n = 3). w/o DPI, without DPI. (F) HepG2 cells were transfected with empty-vector or core plasmid or cotransfected with core plasmid and dominant negative TGF-βRII (ΔTβRII) (1.0 μg each). Forty-eight hours following transfection, total RNA was collected and reverse transcribed to determine Nox4 mRNA expression by quantitative PCR (qPCR) (n = 3, in triplicate; *, P < 0.05). RLU, relative light units; Fold Δ, fold change.
DISCUSSION
Increased oxidative stress is a hallmark of HCV infection in chronic hepatitis, leading eventually to steatosis, cirrhosis, and in many cases, hepatocellular carcinoma (11, 34, 49, 58). Here, we are the first to report modulation of the Nox family member Nox4 by HCV in human and murine hepatocytes, suggesting a role for Nox4-dependent oxidative stress in HCV-induced liver disease. This conclusion is based on the following observations: (i) transfected HCV cDNA or RNA upregulates Nox4 mRNA expression in different human hepatocellular carcinoma cell lines (Fig. 2 and 3); (ii) the effect of dominant negative Nox4 expression or shRNA-mediated Nox4 suppression indicates that a significant amount of HCV-induced ROS is Nox4 derived (Fig. 4 and 6); (iii) the expression of HCV cDNA has positive regulatory effects on human and murine Nox4 promoter regions (Fig. 5); (iv) structural and nonstructural elements of HCV regulate Nox4 mRNA, protein, and ROS generation (Fig. 6 and 7); and (v) Nox4 expression and ROS production are regulated by HCV core protein in a TGF-β1-dependent autocrine-signaling manner (Fig. 8). Our results suggest that Nox4, as a regulator of sustained oxidative injury through a TGF-β-dependent mechanism, is a prime candidate in the progression of HCV inflammatory liver disease, as summarized in the schema in Fig. 9.
FIG. 9.

Nox4-induced oxidative stress in response to HCV is regulated by TGF-β. HCV induces Nox4 expression and Nox4-dependent ROS generation. Expression of the HCV structural core protein upregulates endogenous TGF-β. Thus, TGF-β induces the expression of hepatic Nox4 and superoxide generation in an autocrine fashion. Inhibiting TGF-β signaling with neutralizing TGF-β-specific antibodies or an inactive form of TGF-βRII (TGF-βRII-DN, dominant negative TGF-βRII) abrogates HCV core-mediated induction of Nox4 and subsequent ROS production. MAb, monoclonal antibody.
TGF-β is a profibrogenic cytokine involved in the progression of inflammatory liver disease. Previous clinical studies revealed that serum samples collected from chronic-HCV patients contained elevated levels of serum TGF-β (20, 31, 68). Previously, Nox4 was described as a player in TGF-β-mediated cell differentiation of fibroblasts into a myofibroblast phenotype (14). This report described a Nox4-dependent induction of smooth muscle actin and fibronectin expression, which are indicative of a profibrotic phenotype. Fibroblast differentiation into myofibroblasts is a process heavily dependent on TGF-β signaling and ROS production (54, 73). Recent studies demonstrated that hepatocytes could also transdifferentiate into myofibroblast-like cells (30, 75). When primary or immortalized murine hepatocytes were treated with TGF-β, the cells displayed increased expression of the fibrotic gene products pro-collagen I, connective tissue growth factor, and fibronectin. The significance of TGF-β in fibrogenesis was further supported by in vivo studies with transgenic mice expressing the active form of TGF-β (30, 67). These mice developed liver fibrosis even in the absence of hepatitis virus infection or alcohol treatment. Taken together, these studies provided intriguing evidence of hepatocyte plasticity in the progression of liver disease. Given the increased amounts of circulating TGF-β in HCV patients and the effects of TGF-β on Nox4 described above, we hypothesized that Nox4 contributes to oxidative stress in patients with chronic HCV infection.
Cytokine-induced oxidative stress in hepatocytes is often generated as a direct result of HCV protein expression. Previous reports indicated that ROS induced by the structural core protein has an antiapoptotic and proliferative effect on hepatocytes (48, 56, 57). Hepatocytes expressing the core protein alone were reported to induce TGF-β and connective tissue growth factor mRNA (59). More recently, the core protein was shown to be involved in shifting TGF-β responses from tumor suppression to epithelial-to-mesenchymal transition in human and murine hepatocytes (2). In support of these findings, HCV core transgenic mice showed a significant increase in hepatic oxidative stress, fibrotic lesions on the liver, and steatosis, and many developed hepatocellular carcinoma (45-47). Together, these data provide a strong correlation between HCV core, TGF-β, and oxidative stress in HCV pathogenesis.
Previous reports also suggested that core-mediated ROS was derived from depolarization of the mitochondrial electron transport chain, thereby increasing hepatocyte oxidative stress (35). While these studies suggested mitochondrial ROS as the primary source of oxidative stress, it was undetermined if any of the Nox enzymes contributed to core-mediated ROS. Our studies reported here indicate that the core protein is a regulator of Nox-dependent oxidative stress in human hepatocytes. We found that core-regulated ROS decreases in the presence of DPI, a general flavoenzyme inhibitor that can affect Nox sources of ROS, as well as mitochondrial enzymes. We demonstrated that the expression of HCV core significantly increased Nox4 mRNA, protein, and ROS and appeared to have an effect on Nox4 promoter activity. All of these HCV core effects on Nox4 involve TGF-β, since inhibition of TGF-β signaling with neutralizing antibodies diminished core-mediated Nox4 expression, ROS production, and Nox4 promoter activity. In support of our TGF-β-dependent Nox4 mechanism, Ismail et al. recently reported hypoxia-induced upregulation of insulin-like growth factor binding protein 3 and Nox4 in human pulmonary artery smooth muscle cells through a TGF-β-dependent autocrine mechanism (28). They demonstrated that TGF-β-dependent Nox4 expression was involved in hypoxia-mediated cell proliferation. Here, we demonstrate that TGF-β-dependent autocrine regulation of Nox4 is a new contributor to HCV-induced oxidative stress.
Our studies also indicate that Nox4-derived ROS in hepatocytes are produced as superoxide released at the plasma membrane. Nox4 has been detected in several cellular compartments, such as the nucleus; perinuclear organelles, including the ER; the plasma membrane; and focal adhesions (3). Unlike the other Nox family enzymes, Nox4 does not require additional cytosolic regulators for activation, which may explain how Nox4 functions as a constitutively active enzyme regulated primarily at the transcriptional level. In several transfected-cell models, Nox4 is detected in perinuclear regions and H2O2 is the principal source of ROS detected outside of cells (9, 41). Further investigation into Nox4's function and localization in hepatocytes may reveal additional Nox4 partners influencing its localization at the plasma membrane.
Not only has the core protein been identified as an oxidative stress regulator, but other reports showed that HCV nonstructural proteins induce ROS production. NS5a, a viral phosphoprotein, led to dysregulation of cellular Ca2+ concentrations, resulting in an ER stress response and increased mitochondrion-derived ROS. At the transcriptional level, core and NS5a were associated with redox-dependent NF-κB transactivation, mitogen-activated protein kinase activation, and TGF-β-induced gene regulation (12, 25, 27, 38). Whether NS5a or other nonstructural proteins (NS4a, NS4b, and NS5b) affect the Nox family oxidases remains unknown. However, previous reports showed that the NS3 protein activated Nox2 in phagocytes and led to apoptosis of T cells, natural killer cells, and killer T cells (7, 65). In our studies, we did not observe changes in Nox4 expression or activity in response to NS3 in hepatocyte cell lines (Fig. 7).
HCV proteins induced changes in host gene expression (6, 13, 16, 42, 70). Our studies demonstrated that full-length HCV or expression of the core protein upregulated Nox4 mRNA and promoter activity. Interestingly, sequence analysis of the Nox4 promoter revealed putative binding sites for many of the important transcription factors in signaling pathways activated by HCV infection (SMAD3/4, NF-κB, AP-1, Myc, Chop-cEBP, IRF-1, IRF-2, and PPARγ) (Fig. 5A) (8, 18, 32, 36, 37, 66). Further investigation of the Nox4 promoter is necessary to establish the precise mechanisms of transcriptional regulation of Nox4 by HCV. Our studies demonstrate that the Nox4 promoter is responsive to TGF-β as a consequence of HCV core expression. Together, these findings may reflect a more general role of Nox4 in response to fibrotic insults.
Acknowledgments
The U.S. National Institutes of Health Intramural Research Program, National Institutes of Allergy and Infectious Diseases, supported this work.
Footnotes
▿
Published ahead of print on 7 October 2009.
REFERENCES
- 1.Basu, A., K. Meyer, K. K. Lai, K. Saito, A. M. Di Bisceglie, L. E. Grosso, R. B. Ray, and R. Ray. 2006. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 349**:**347-358. [DOI] [PubMed] [Google Scholar]
- 2.Battaglia, S., N. Benzoubir, S. Nobilet, P. Charneau, D. Samuel, A. L. Zignego, A. Atfi, C. Brechot, and M. F. Bourgeade. 2009. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS ONE 4**:**e4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bedard, K., and K. H. Krause. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87**:**245-313. [DOI] [PubMed] [Google Scholar]
- 4.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76**:**13001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Block, K., A. Eid, K. K. Griendling, D. Y. Lee, Y. Wittrant, and Y. Gorin. 2008. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 283**:**24061-24076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budhu, A., Y. Chen, J. W. Kim, M. Forgues, K. Valerie, C. C. Harris, and X. W. Wang. 2007. Induction of a unique gene expression profile in primary human hepatocytes by hepatitis C virus core, NS3 and NS5A proteins. Carcinogenesis 28**:**1552-1560. [DOI] [PubMed] [Google Scholar]
- 7.Bureau, C., J. Bernad, N. Chaouche, C. Orfila, M. Beraud, C. Gonindard, L. Alric, J. P. Vinel, and B. Pipy. 2001. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J. Biol. Chem. 276**:**23077-23083. [DOI] [PubMed] [Google Scholar]
- 8.Chan, S. W., and P. A. Egan. 2005. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19**:**1510-1512. [DOI] [PubMed] [Google Scholar]
- 9.Chen, K., M. T. Kirber, H. Xiao, Y. Yang, and J. F. Keaney, Jr. 2008. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181**:**1129-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang, E., O. Dang, K. Anderson, A. Matsuzawa, H. Ichijo, and M. David. 2006. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J. Immunol. 176**:**5720-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi, J., and J. H. Ou. 2006. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 290**:**G847-G851. [DOI] [PubMed] [Google Scholar]
- 12.Choi, S. H., and S. B. Hwang. 2006. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J. Biol. Chem. 281**:**7468-7478. [DOI] [PubMed] [Google Scholar]
- 13.Ciccaglione, A. R., C. Marcantonio, E. Tritarelli, P. Tataseo, A. Ferraris, R. Bruni, B. Dallapiccola, G. Gerosolimo, A. Costantino, and M. Rapicetta. 2008. Microarray analysis identifies a common set of cellular genes modulated by different HCV replicon clones. BMC Genomics 9**:**309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cucoranu, I., R. Clempus, A. Dikalova, P. J. Phelan, S. Ariyan, S. Dikalov, and D. Sorescu. 2005. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97**:**900-907. [DOI] [PubMed] [Google Scholar]
- 15.De Minicis, S., and D. A. Brenner. 2007. NOX in liver fibrosis. Arch. Biochem. Biophys. 462**:**266-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou, J., P. Liu, and X. Zhang. 2005. Cellular response to gene expression profiles of different hepatitis C virus core proteins in the Huh-7 cell line with microarray analysis. J. Nanosci. Nanotechnol. 5**:**1230-1235. [DOI] [PubMed] [Google Scholar]
- 17.Dubuisson, J. 2007. Hepatitis C virus proteins. World J. Gastroenterol. 13**:**2406-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farinati, F., R. Cardin, M. Bortolami, M. Guido, and M. Rugge. 2006. Oxidative damage, pro-inflammatory cytokines, TGF-alpha and c-myc in chronic HCV-related hepatitis and cirrhosis. World J. Gastroenterol. 12**:**2065-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farinati, F., R. Cardin, N. De Maria, G. Della Libera, C. Marafin, E. Lecis, P. Burra, A. Floreani, A. Cecchetto, and R. Naccarato. 1995. Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J. Hepatol. 22**:**449-456. [DOI] [PubMed] [Google Scholar]
- 20.Gabriel, A., A. Ziolkowski, P. Radlowski, K. Tomaszek, and A. Dziambor. 2008. Hepatocyte steatosis in HCV patients promotes fibrosis by enhancing TGF-beta liver expression. Hepatol. Res. 38**:**141-146. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Mediavilla, M. V., S. Sanchez-Campos, P. Gonzalez-Perez, M. Gomez-Gonzalo, P. L. Majano, M. Lopez-Cabrera, G. Clemente, C. Garcia-Monzon, and J. Gonzalez-Gallego. 2005. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 43**:**606-613. [DOI] [PubMed] [Google Scholar]
- 22.Geiszt, M., J. B. Kopp, P. Varnai, and T. L. Leto. 2000. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 97**:**8010-8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geiszt, M., and T. L. Leto. 2004. The Nox family of NAD(P)H oxidases: host defense and beyond. J. Biol. Chem. 279**:**51715-51718. [DOI] [PubMed] [Google Scholar]
- 24.Giannini, C., and C. Brechot. 2003. Hepatitis C virus biology. Cell Death Differ. 10(Suppl. 1)**:**S27-S38. [DOI] [PubMed] [Google Scholar]
- 25.Gong, G., G. Waris, R. Tanveer, and A. Siddiqui. 2001. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 98**:**9599-9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassan, M., D. Selimovic, H. Ghozlan, and O. Abdel-kader. 2009. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 49**:**1469-1482. [DOI] [PubMed] [Google Scholar]
- 27.He, Y., S. L. Tan, S. U. Tareen, S. Vijaysri, J. O. Langland, B. L. Jacobs, and M. G. Katze. 2001. Regulation of mRNA translation and cellular signaling by hepatitis C virus nonstructural protein NS5A. J. Virol. 75**:**5090-5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail, S., A. Sturrock, P. Wu, B. Cahill, K. Norman, T. Huecksteadt, K. Sanders, T. Kennedy, and J. Hoidal. 2009. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 296**:**L489-L499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain, S. K., P. W. Pemberton, A. Smith, R. F. McMahon, P. C. Burrows, A. Aboutwerat, and T. W. Warnes. 2002. Oxidative stress in chronic hepatitis C: not just a feature of late stage disease. J. Hepatol. 36**:**805-811. [DOI] [PubMed] [Google Scholar]
- 30.Kaimori, A., J. Potter, J. Y. Kaimori, C. Wang, E. Mezey, and A. Koteish. 2007. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 282**:**22089-22101. [DOI] [PubMed] [Google Scholar]
- 31.Kanzler, S., M. Baumann, P. Schirmacher, V. Dries, E. Bayer, G. Gerken, H. P. Dienes, and A. W. Lohse. 2001. Prediction of progressive liver fibrosis in hepatitis C infection by serum and tissue levels of transforming growth factor-beta. J. Viral Hepat. 8**:**430-437. [DOI] [PubMed] [Google Scholar]
- 32.Kim, K. H., S. P. Hong, K. Kim, M. J. Park, K. J. Kim, and J. Cheong. 2007. HCV core protein induces hepatic lipid accumulation by activating SREBP1 and PPARgamma. Biochem. Biophys. Res. Commun. 355**:**883-888. [DOI] [PubMed] [Google Scholar]
- 33.Koike, K. 2006. Oxidative stress and apoptosis in hepatitis C: the core issue. J. Gastroenterol. 41**:**292-294. [DOI] [PubMed] [Google Scholar]
- 34.Koike, K., and H. Miyoshi. 2006. Oxidative stress and hepatitis C viral infection. Hepatol. Res. 34**:**65-73. [DOI] [PubMed] [Google Scholar]
- 35.Korenaga, M., T. Wang, Y. Li, L. A. Showalter, T. Chan, J. Sun, and S. A. Weinman. 2005. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 280**:**37481-37488. [DOI] [PubMed] [Google Scholar]
- 36.Larrea, E., A. Alberdi, Y. Castelruiz, P. Boya, M. P. Civeira, and J. Prieto. 2001. Expression of interferon-alpha subtypes in peripheral mononuclear cells from patients with chronic hepatitis C: a role for interferon-alpha5. J. Viral Hepat. 8**:**103-110. [DOI] [PubMed] [Google Scholar]
- 37.Liao, Q. J., L. B. Ye, K. A. Timani, Y. L. She, X. J. Yang, L. Ye, and Z. H. Wu. 2005. Hepatitis C virus non-structural 5A protein can enhance full-length core protein-induced nuclear factor-kappaB activation. World J. Gastroenterol. 11**:**6433-6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macdonald, A., K. Crowder, A. Street, C. McCormick, K. Saksela, and M. Harris. 2003. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J. Biol. Chem. 278**:**17775-17784. [DOI] [PubMed] [Google Scholar]
- 39.Mahadev, K., H. Motoshima, X. Wu, J. M. Ruddy, R. S. Arnold, G. Cheng, J. D. Lambeth, and B. J. Goldstein. 2004. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 24**:**1844-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marek, B., D. Kajdaniuk, U. Mazurek, E. Janczewska-Kazek, B. Kos-Kudla, B. Strzalka, A. Fila, D. Niedziolka, M. Beniowski, Z. Ostrowska, H. Borgiel-Marek, J. Kajdaniuk, L. Sieminska, M. Nowak, T. Wilczok, D. Pakula, and P. Filipczyk. 2005. TGF-beta1 mRNA expression in liver biopsy specimens and TGF-beta1 serum levels in patients with chronic hepatitis C before and after antiviral therapy. J. Clin. Pharm. Ther. 30**:**271-277. [DOI] [PubMed] [Google Scholar]
- 41.Martyn, K. D., L. M. Frederick, K. von Loehneysen, M. C. Dinauer, and U. G. Knaus. 2006. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 18**:**69-82. [DOI] [PubMed] [Google Scholar]
- 42.Mas, V. R., D. G. Maluf, K. J. Archer, K. Yanek, X. Kong, L. Kulik, C. E. Freise, K. M. Olthoff, R. M. Ghobrial, P. McIver, and R. Fisher. 2009. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol. Med. 15**:**85-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mengshol, J. A., L. Golden-Mason, and H. R. Rosen. 2007. Mechanisms of disease: HCV-induced liver injury. Nat. Clin. Pract. Gastroenterol. Hepatol. 4**:**622-634. [DOI] [PubMed] [Google Scholar]
- 44.Moradpour, D., F. Penin, and C. M. Rice. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5**:**453-463. [DOI] [PubMed] [Google Scholar]
- 45.Moriya, K., H. Fujie, Y. Shintani, H. Yotsuyanagi, T. Tsutsumi, K. Ishibashi, Y. Matsuura, S. Kimura, T. Miyamura, and K. Koike. 1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4**:**1065-1067. [DOI] [PubMed] [Google Scholar]
- 46.Moriya, K., K. Nakagawa, T. Santa, Y. Shintani, H. Fujie, H. Miyoshi, T. Tsutsumi, T. Miyazawa, K. Ishibashi, T. Horie, K. Imai, T. Todoroki, S. Kimura, and K. Koike. 2001. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61**:**4365-4370. [PubMed] [Google Scholar]
- 47.Moriya, K., H. Yotsuyanagi, Y. Shintani, H. Fujie, K. Ishibashi, Y. Matsuura, T. Miyamura, and K. Koike. 1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78(Pt. 7)**:**1527-1531. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen, H., S. Sankaran, and S. Dandekar. 2006. Hepatitis C virus core protein induces expression of genes regulating immune evasion and anti-apoptosis in hepatocytes. Virology 354**:**58-68. [DOI] [PubMed] [Google Scholar]
- 49.Nishikawa, T., T. Nakajima, T. Katagishi, Y. Okada, M. Jo, K. Kagawa, T. Okanoue, Y. Itoh, and T. Yoshikawa. 2009. Oxidative stress may enhance the malignant potential of human hepatocellular carcinoma by telomerase activation. Liver Int. 29**:**846-856. [DOI] [PubMed] [Google Scholar]
- 50.Okuda, M., K. Li, M. R. Beard, L. A. Showalter, F. Scholle, S. M. Lemon, and S. A. Weinman. 2002. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122**:**366-375. [DOI] [PubMed] [Google Scholar]
- 51.Park, H. S., H. Y. Jung, E. Y. Park, J. Kim, W. J. Lee, and Y. S. Bae. 2004. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 173**:**3589-3593. [DOI] [PubMed] [Google Scholar]
- 52.Patel, D. N., S. R. Bailey, J. K. Gresham, D. B. Schuchman, J. H. Shelhamer, B. J. Goldstein, B. M. Foxwell, M. B. Stemerman, J. K. Maranchie, A. J. Valente, S. Mummidi, and B. Chandrasekar. 2006. TLR4-NOX4-AP-1 signaling mediates lipopolysaccharide-induced CXCR6 expression in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 347**:**1113-1120. [DOI] [PubMed] [Google Scholar]
- 53.Pedruzzi, E., C. Guichard, V. Ollivier, F. Driss, M. Fay, C. Prunet, J. C. Marie, C. Pouzet, M. Samadi, C. Elbim, Y. O'Dowd, M. Bens, A. Vandewalle, M. A. Gougerot-Pocidalo, G. Lizard, and E. Ogier-Denis. 2004. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 24**:**10703-10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poli, G. 2000. Pathogenesis of liver fibrosis: role of oxidative stress. Mol. Aspects Med. 21**:**49-98. [DOI] [PubMed] [Google Scholar]
- 55.Russell, R. S., J. C. Meunier, S. Takikawa, K. Faulk, R. E. Engle, J. Bukh, R. H. Purcell, and S. U. Emerson. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. USA 105**:**4370-4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saito, K., K. Meyer, R. Warner, A. Basu, R. B. Ray, and R. Ray. 2006. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 80**:**4372-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato, Y., J. Kato, R. Takimoto, K. Takada, Y. Kawano, K. Miyanishi, M. Kobune, T. Takayama, T. Matunaga, and Y. Niitsu. 2006. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 55**:**1801-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seronello, S., M. Y. Sheikh, and J. Choi. 2007. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic. Biol. Med. 43**:**869-882. [DOI] [PubMed] [Google Scholar]
- 59.Shin, J. Y., W. Hur, J. S. Wang, J. W. Jang, C. W. Kim, S. H. Bae, S. K. Jang, S. H. Yang, Y. C. Sung, O. J. Kwon, and S. K. Yoon. 2005. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-beta1. Exp. Mol. Med. 37**:**138-145. [DOI] [PubMed] [Google Scholar]
- 60.Shiose, A., J. Kuroda, K. Tsuruya, M. Hirai, H. Hirakata, S. Naito, M. Hattori, Y. Sakaki, and H. Sumimoto. 2001. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem. 276**:**1417-1423. [DOI] [PubMed] [Google Scholar]
- 61.Shirai, Y., S. Kawata, S. Tamura, N. Ito, H. Tsushima, K. Takaishi, S. Kiso, and Y. Matsuzawa. 1994. Plasma transforming growth factor-beta 1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer 73**:**2275-2279. [DOI] [PubMed] [Google Scholar]
- 62.Sturrock, A., T. P. Huecksteadt, K. Norman, K. Sanders, T. M. Murphy, P. Chitano, K. Wilson, J. R. Hoidal, and T. P. Kennedy. 2007. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 292**:**L1543-L1555. [DOI] [PubMed] [Google Scholar]
- 63.Sumpter, R., Jr., Y. M. Loo, E. Foy, K. Li, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79**:**2689-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taniguchi, H., N. Kato, M. Otsuka, T. Goto, H. Yoshida, Y. Shiratori, and M. Omata. 2004. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J. Med. Virol. 72**:**52-59. [DOI] [PubMed] [Google Scholar]
- 65.Thoren, F., A. Romero, M. Lindh, C. Dahlgren, and K. Hellstrand. 2004. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J. Leukoc. Biol. 76**:**1180-1186. [DOI] [PubMed] [Google Scholar]
- 66.Tsutsumi, T., T. Suzuki, K. Moriya, H. Yotsuyanagi, Y. Shintani, H. Fujie, Y. Matsuura, S. Kimura, K. Koike, and T. Miyamura. 2002. Alteration of intrahepatic cytokine expression and AP-1 activation in transgenic mice expressing hepatitis C virus core protein. Virology 304**:**415-424. [DOI] [PubMed] [Google Scholar]
- 67.Ueberham, E., R. Low, U. Ueberham, K. Schonig, H. Bujard, and R. Gebhardt. 2003. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology 37**:**1067-1078. [DOI] [PubMed] [Google Scholar]
- 68.Verma, V., A. Chakravarti, and P. Kar. 2008. Cytokine levels of TGF-beta, IL-10, and sTNFalphaRII in type C chronic liver disease. Dig. Dis. Sci. 53**:**2233-2237. [DOI] [PubMed] [Google Scholar]
- 69.von Lohneysen, K., D. Noack, A. J. Jesaitis, M. C. Dinauer, and U. G. Knaus. 2008. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J. Biol. Chem. 283**:**35273-35282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walters, K. A., A. J. Syder, S. L. Lederer, D. L. Diamond, B. Paeper, C. M. Rice, and M. G. Katze. 2009. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 5**:**e1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wieser, R., L. Attisano, J. L. Wrana, and J. Massague. 1993. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol. Cell. Biol. 13**:**7239-7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu, R. F., Z. Ma, D. P. Myers, and L. S. Terada. 2007. HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. J. Biol. Chem. 282**:**37412-37419. [DOI] [PubMed] [Google Scholar]
- 73.Xu, J., S. Lamouille, and R. Derynck. 2009. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19**:**156-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA 94**:**8738-8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeisberg, M., C. Yang, M. Martino, M. B. Duncan, F. Rieder, H. Tanjore, and R. Kalluri. 2007. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 282**:**23337-23347. [DOI] [PubMed] [Google Scholar]