Novel antagonists for proteinase-activated receptor 2: inhibition of cellular and vascular responses in vitro and in vivo (original) (raw)
Abstract
Background and purpose:
Proteinase-activated receptor 2 (PAR2) is a G-protein coupled receptor associated with many pathophysiological functions. To date, the development of PAR2 antagonists has been limited. Here, we identify a number of novel peptide-mimetic PAR2 antagonists and demonstrate inhibitory effects on PAR2-mediated intracellular signalling pathways and vascular responses.
Experimental approach:
The peptide-mimetic compound library based on the structures of PAR2 agonist peptides were screened for inhibition of PAR2-induced calcium mobilisation in human keratinocytes. Representative compounds were further evaluated by radioligand binding and inhibition of NFκB transcriptional activity and IL-8 production. The vascular effects of the antagonists were assessed using in vitro and in vivo models.
Key results:
Two compounds, K-12940 and K-14585, significantly reduced SLIGKV-induced Ca2+ mobilisation in primary human keratinocytes. Both K-12940 and K-14585 exhibited competitive inhibition for the binding of a high-affinity radiolabelled PAR2-ligand, [3H]-2-furoyl-LIGRL-NH2, to human PAR2 with Ki values of 1.94 and 0.627 µM respectively. NFκB reporter activity and IL-8 production were also significantly reduced. Furthermore, relaxation of rat-isolated aorta induced by SLIGRL-NH2 was inhibited competitively by K-14585. K-14585 also significantly lowered plasma extravasation in the dorsal skin of guinea pigs and reduced salivation in mice.
Conclusions and implications:
K-12940 and K-14585 antagonized PAR2 competitively, resulting in inhibition of PAR2-mediated signalling and physiological responses both in vitro and in vivo. These peptide-mimetic PAR2 antagonists could be useful in evaluating PAR2-mediated biological events and might lead to a new generation of therapeutically useful antagonists.
Keywords: Proteinase-activated receptor 2 (PAR2), antagonist, Ca2+ mobilization, keratinocytes, radioligand-binding
Introduction
Proteinase-activated receptor 2 (PAR2), a member of the family of G-protein-coupled receptors [nomenclature follows Alexander et al. (2008)], is activated by proteases such as trypsin, tryptase and coagulation factors VIIa and Xa (Nystedt et al., 1994; Molino et al., 1997; Camerer et al., 2000) cleaving the receptor to reveal an N-terminal ‘tethered ligand’, SLIGKV and SLIGRL for human and mouse/rat PAR2, respectively, which subsequently interacts with the activation domain of the receptor, initiating intracellular signalling pathways and functional responses (Macfarlane et al., 2001; Hollenberg, 2005; Steinhoff et al., 2005).
A number of studies have demonstrated a wide range of important physiological roles for PAR2. Although PAR2 plays protective roles in several biological systems, such as the lung (Cocks and Moffatt, 2000; Lan et al., 2000; 2001;) and gastrointestinal tract (Kawabata et al., 2001; Kawao et al., 2002), there is much evidence that activation of PAR2 is pro-inflammatory. For instance, the PAR2 activating peptide elicits inflammatory responses in the rat hind paw (Kawabata et al., 1998; Vergnolle et al., 1998), mouse knee joint (Ferrell et al., 2003) and mouse colon (Cenac et al., 2002). A critical role of PAR2 has also been demonstrated in skin (Kawagoe et al., 2002) and neurogenic inflammation (Ricciardolo et al., 2000; Steinhoff et al., 2000). Therefore, antagonists for PAR2 would be appropriate for the treatment of these inflammatory conditions. Currently, however, there is only limited published information available for the development of PAR2 antagonists.
Recently, several approaches have been demonstrated to inhibit PAR2-mediated responses utilizing pepducins (Kaneider et al., 2007), siRNA and monoclonal antibodies (Kelso et al., 2006). A low molecular weight compound which mimics the peptide structure of truncated PAR2 agonist peptide inhibited PAR2-induced joint inflammation in vivo (Kelso et al., 2006), although its molecular mechanisms of receptor blockade were not clear. So far, there are no reports of competitive antagonists for this receptor.
We have previously identified a series of modified PAR2 agonist peptides, substituted with 2-furoyl on the N-terminal serine residue of native PAR2-activating peptides, which were shown to be potent and metabolically stable both in vivo and in vitro (Ferrell et al., 2003; Kawabata et al., 2004). Utilizing the radiolabelled potent PAR2-activating peptide, [3H]2-furoyl-LIGRL-NH2, we have established a PAR2 receptor binding system, which enabled us to characterize binding profile of agonists/antagonists to human PAR2 (Kanke et al., 2005). By analysing the structure-activity relationship (SAR) of PAR2 agonists, we found the affinity of the ligand and the receptor activation potency were regulated relatively independently by several critical sites of the peptide structures. Therefore, we thought it could be possible to identify PAR2 antagonists, possessing high affinity to the receptor binding site but not activating the receptor, through screening of peptide-mimetic compound libraries. As a result of extensive screening, we have identified a series of peptide-mimetic compounds that inhibit Ca2+ responses in human keratinocytes. Two representative compounds, K-12940 and K-14585 (Figure 1), were competitive inhibitors of the binding of [3H]-2-furoyl-LIGRL-NH2, to human PAR2. Both compounds were also found to be able to inhibit a variety of previously characterized intracellular responses including activation of NFκB and the production of IL-8. Furthermore, the inhibitory effects of K-14585 on PAR2-mediated in vitro and in vivo tissue responses were demonstrated including relaxation of the rat aorta, increased vascular permeability and saliva production. These results have identified novel peptide mimetic antagonists for PAR2 which could be therapeutically useful for the treatment of PAR2-related inflammatory diseases.
Figure 1.
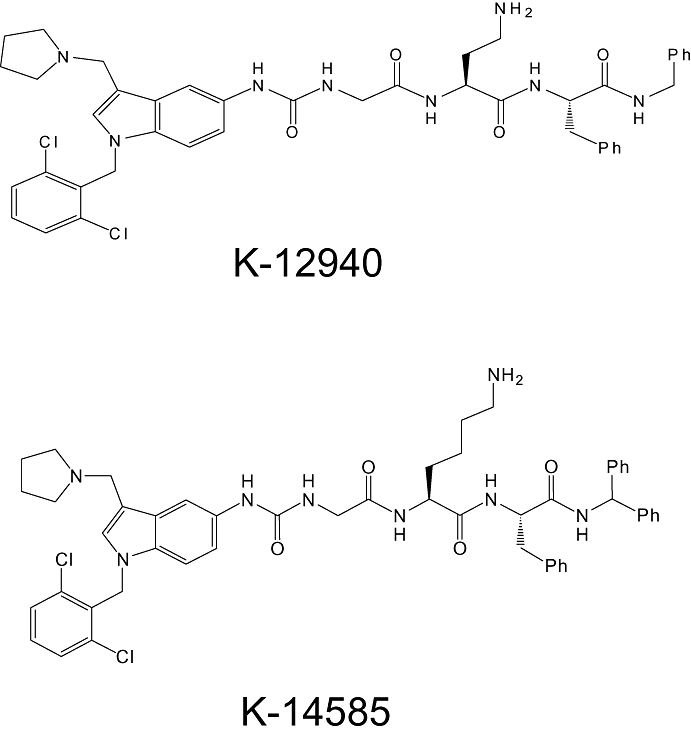
Chemical structures of peptide-mimetic proteinase-activated receptor 2 antagonists K-12940 and K-14585.
Methods
Cell culture
Normal human epidermal keratinocytes (NHEK) which strongly expressed PAR2 (Santulli et al., 1995; Scott et al., 2004) were obtained from Cambrex (Walkersville, MD, USA) and cultured in the complete keratinocyte growth medium. The human skin epithelial cell line NCTC2544 expressing human PAR2, designated as NCTC2544-PAR2 cells, was described previously (Kanke et al., 2001). Control NCTC2544 cells (WT-NCTC2544 cells) were grown in Medium 199 with Earle's salts (Sigma-Aldrich, MO, USA) containing 10% (v·v−1) fetal calf serum (FCS), sodium bicarbonate (50 mM), L-glutamine (2 mM), penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1). NCTC2544-PAR2 cells were cultured in complete medium containing geneticin (400 µg·mL−1) to maintain selection pressure. Human embryonic kidney 293 cells (HEK293 cells) were maintained in 10% minimal essential medium (MEM with Earle's salts, L-glutamine supplemented with 10% (v·v−1) FCS, 1% penicillin, 1% streptomycin, 1% non-essential amino acids). All cells were grown at 37°C in an incubator with saturated humidity and 5% CO2 and NCTC2544 cells were passaged using Versene [0.53 mM ethylenediaminetetraacetic acid (EDTA) in phosphate buffered saline (PBS)] to avoid trypsin exposure. NHEK were passaged with trypsin, and passages at 4–8 were used for the experiments. HEK293 cells were passaged using 1xSSC (sodium citrate, pH 7.4).
Transient transfection of PAR1, PAR2 and PAR4 in HEK293 cells
HEK293 cells were grown to 70–80% confluence on 12 well plates and transiently transfected with 1 µg/well Wild type PAR1, PAR2 or PAR4 plasmid DNA using the Lipofectamine™ 2000 transfection system according to manufacturer's guidelines. After a 4 h incubation period with the DNA mixture in OptiMEM (Invitrogen Ltd, Paisley, UK) with no serum or antibiotics, the OptiMEM was replaced with complete medium for another 20 h. Measurement of [3H]inositol phosphate accumulation stimulated by PAR-specific activating peptide was measured in these cells pre-labelled overnight with [3H]2-myoinositol in serum free medium as outlined previously (Plevin et al., 1994). During this time, cells were rendered quiescent by serum deprivation. Test compounds were added 30 min prior to agonist stimulation with various PAR-specific activating peptides for 1 h.
Measurement of Ca2_+_ mobilization
PAR2-mediated intracellular calcium mobilization in normal human keratinocytes was measured with minor modifications of a previously described method (Kawabata et al., 2004) using a 96-well scanning fluorimeter, FlexStation (Molecular Devices, Wokingham, UK). Human normal keratinocytes (30 000 cells per well) were seeded in black-wall clear-bottom 96-well plates (Corning B.V. Life Sciences, Koolhovenlaan, the Netherlands) 24 h prior to the assay and grown to reach confluence. Cells were washed once with keratinocyte basic medium (KBM) and replaced with 80 mL of KBM. Subsequently, 80 µL of calcium assay dye solution (FLEXstation Calcium 3 Assay kit, Molecular Devices) dissolved in Hank's balanced salt solution (HBSS)(pH 7.4) containing 2.5 mM probenecid was added and incubated for 60 min at 37°C. Test compounds were added 30 min prior to the agonist stimulation. Then cells were stimulated with various concentrations of agonists prepared in HBSS and the fluorescence change measured at 25°C (excitation 485 nm and emission 525 nm). Antagonists were added 30 min prior to the agonist stimulation. Inhibitory effects of antagonists on calcium responses (max–min) were expressed as percentage of the reference calcium response induced by either PAR2 agonist peptide SLIGKV-OH (30 µM) or histamine (30 µM) alone. The half-maximal effective concentration values (IC50) were estimated from the concentration-response curve.
NFκB-luciferase reporter gene assay
PAR2 agonist-activated transcriptional activation of NFκB was determined utilizing NFκB-luciferase reporter gene system as described previously (Macfarlane et al., 2005) The plasmid vector containing repeated consensus sequence of NFκB binding cis-elements, ×4 NFκB, connected to the firefly luciferase gene was stably transfected into the NCTC2544-PAR2 cells and a positive clone was used for the assay. In the assay, the cells were detached from flasks using Versene and seeded into white 96-well plates (96F Nunclon Delta, Thermo Scientific Nunc, Leicestershire, UK) with 30 000 cells per well. Cells were grown until sub-confluent (18–24 h) at 37°C. Then, cells were washed with serum free medium once and rendered quiescent with serum free medium for 18 h. Test compounds dissolved in dimethyl sulfoxide (DMSO) were diluted in serum free medium at appropriate concentrations and was added to the cells to preincubate for 15–30 min. Subsequently, cells are stimulated with agonists such as SLIGKV, trypsin or phorbol myristate acetate (PMA) for 6 h at 37°C. Stimulation was terminated by adding 100 µL per well of luciferase assay reagent (Bright-Glo, Promega UK Ltd., Hampshire, UK) and luminescence was measured with 96-well luminometer (MicroLUMAT LB96P, EG&G Berthold UK, Herts, UK).
ELISA for interleukin (IL)-8
NCTC2544-PAR2 cells seeded in 24-well plates were stimulated with PBS (Control), SLIGKV (100 µM) or tumour necrosis factorα (TNFα;10 ng·mL−1) in the absence or presence of antagonists (5 µg·mL−1) for 24 h. IL-8 levels in the culture medium were determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions TECHNE (Bibby-Scientific Ltd, Staffordshire, UK). Data are expressed as the mean and SEM of three experiments.
PAR2 ligand-binding assay
PAR2 ligand-binding assay was performed using [3H]2-furoyl-LIGRL-NH2 as described previously (Kanke et al., 2005). NCTC2544-PAR2 cell suspension (0.2 mL, 3.0 × 105 cells) were incubated at 25°C along with [3H]2-furoyl-LIGRL-NH2 (9.2 nM, 1 µCi·mL−1) in either absence or presence of agonists and antagonists. The cell-bound radioligand was separated by centrifugation and the radioactivity was measured by scintillation counting (Tri-Carb 2700TR, PerkinElmer, MA, USA). The amount of specific binding was calculated by subtraction from the total amount of the radioligand bound in the absence of competing ligands, non-specific binding in the presence of an excess (100 µM) of unlabeled 2-furoyl-LIGRL-NH2. Competition studies with various PAR2 agonists and antagonists were performed and the displacement curve for each agonist/antagonist was constructed by measuring the percentage of the specific [3H]2-furoyl-LIGRL-NH2 binding (% specific binding) in the presence of each peptide concentration, relative to the maximum specific binding in the absence of unlabelled 2-furoyl-LIGRL-NH2.
Experimental animals
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Relaxation of rat-isolated thoracic aorta
Male Wistar rats (8–9 weeks, 300–350 g; Japan SLC. Inc., Kotocho, Nishi-Ku, Japan) were killed by decapitation under urethane (1.5 g·kg−1, i.p.) anaesthesia, and the thoracic aorta was removed. Endothelium-intact, rings of thoracic aorta (4 mm long) were placed in 2 mL organ bath filled with gassed Krebs Hensleit solution (composition in mM: NaCl, 118; KCl, 4.7; CaCl2, 2.5; MgCl2, 1.2; NaHCO3, 25; KH2PO4, 1.2; glucose, 10) maintained at 37°C and bubbled with 95% O2, 5% CO2. The segment was allowed to equilibrate for 1 h under a resting tension of 10 mN, and isometric tension was recorded through a force-displacement transducer (UL-10GR, Minebea Co., Ltd., Meguro-Ku, Tokyo, Japan). SLIGRL-NH2 (1-100 µM) was cumulatively applied in the absence or presence of K-14585 (10 µM) to aortic rings precontracted with phenylephrine (1 µM) and relaxation of the rings was measured. Data are expressed as percent of relaxation calculated as percent of the maximum relaxation induced by papaverine (100 µM).
Cutaneous microvascular permeability in guinea pigs
Male Hartley guinea pigs (450–550 g, Japan SLC. Inc., Japan) were used for the experiments. Under urethane (1.5 g·kg−1, i.p.) anaesthesia, Evans blue dye (50 mg·kg−1) was injected intravenously, and immediately after, either vehicle (10% DMSO), SLIGRL-NH2 (300 nM) or a mixture of SLIGRL-NH2 (300 nM) and K-14585 (300 µg) was injected intradermally. We used eight sites per guinea pig, approximately 20 mm apart, on dorsal skin shaved a day before the experiments. Intradermal injections were made in a volume of 100 µL per site. Thirty minutes after the intradermal injections, the animal was killed and the increase of vascular permeability was assessed by measuring the amount of extravasated dye in the injection sites. Dorsal skin was removed from the animal and the sites of injection (approximately 15 mm in diameter) were punched out and dissolved in 1.4 mL of 1N KOH at 37°C overnight. The dye was extracted with 18.6 mL of a mixture of 0.6N H3PO4 and acetone (5:13, v: v−1) per site and quantified using a spectrophotometer at 620 nm with standard curve of Evans blue in the same solvent at concentrations of 0.1–30 µg·mL−1.
Secretion of saliva in anaesthetized mice in vivo
Salivary exocrine secretion was assessed in male ddY mice (8–10 weeks old; 20–25 g) under urethane anaesthesia (i.p., 1.5 g·kg−1), as described elsewhere (Takeda and Krause, 1989; Kawabata et al., 2000). Small pre-weighed pads of cottonwool were placed in the mouth and replaced with new pads every 5 min. The difference in weight of the pads before and after being placed in the mouth was taken to represent the amount of saliva secreted in each period. Salivation was monitored for 15 min after i.p. challenge with agonists. Amastatin (Peptide Institute, Minoh, Japan), an inhibitor of aminopeptidase, a degradation enzyme for peptides, was administered i.p. immediately before agonists. Antagonists were given i.p. 5 min before the agonist challenge.
Data analysis
For calcium mobilization studies, the peak fluorescence change was plotted versus the concentration of agonists and the concentration-response curve fitted using a four-parameter logistic equation to determine the EC50 value. Radioligand binding data were analysed by nonlinear regression analysis using GraphPad Prism 4 (GraphPad Software, CA, USA). The displacement curves by agonists/antagonists were assumed to fit to a one-site model to determine IC50 values. The inhibitor constant, Ki of each agonist/antagonist was then derived from the IC50 and the Kd of 2-furoyl-LIGRL-NH2.
Data are represented as mean ± SEM. Statistical significance was analyzed by Student's _t_-test for two group data. Significance was set at a P < 0.05 level.
Materials
Preparation of PAR2 antagonists
Peptide mimetic PAR2 antagonists (Figure 1), K-12940, {(N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl)aminocarbonyl}-glycinyl-L-α,γ-diaminobutyryl-L-phenylalaninyl-N-benzylamide and K-14585 {(N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl)aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyle-N-benzhydrylamide were synthesized at Kowa Tokyo New Drug Research Laboratories (Tokyo, Japan). The chemical structures were confirmed by nuclear magnetic resonance and mass spectrometry (MS). The purity of compounds (>95%) was determined by high-performance liquid chromatography (HPLC). The compounds were dissolved in DMSO and aliquots were kept at −20°C until use.
PAR2-activating peptide and other chemicals
The human PAR2-activating peptide, Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH), mouse/rat PAR2-activating peptide, SLIGRL-NH2, Ser-Leu-Ile-Gly-Arg-Leu-amide; and a highly potent PAR2-activating peptide, 2-furoyl-Leu-Ile-Gly-Arg-Leu-amide (2-furoyl-LIGRL-NH2) (Kawabata et al., 2004) were synthesized and purified (>95%) by HPLC, and the structures were confirmed by MS. The radiolabelled PAR2-activating peptide, [3H]2-furoyl-LIGRL-NH2 (radiochemical purity of 99.4%, specific activity of 4.03 TBq·mM), was prepared for us by Amersham Biosciences (Buckinghamshire, UK) as described previously (Kanke et al., 2005). Trypsin from bovine pancreas (9300 unit·mg−1), A23187 and probenecid were obtained from Sigma-Aldrich Co. Histamine dihydrochloride, dibutyl phthalate and dinonyl phthalate were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Results
Inhibitory effects of peptide antagonists on PAR2 agonist peptide-induced Ca2_+_ mobilization in human keratinocytes
Initially, a series of potential PAR2 antagonist compounds including K-14584 and K-12940 (Figure 1) were screened for their ability to inhibit PAR2 mediated Ca2+ mobilization in primary cultures of human keratinocytes, a cell type known to express PAR2 (Santulli et al., 1995). SLIGKV-OH stimulated a rapid increase in intracellular Ca2+ which peaked within 60 s of stimulation (Figure 2). Pre-incubation with K-14585 (Figure 2) resulted in a significant decrease in this induced Ca2+ mobilization, of approximately 60% over a number of experiments (Table 1). Several additional compounds including K-12940 were screened and inhibition assessed (see Table 1). From these compounds, K-14585 and K-12940 were then assessed in a number of other assays and in vitro and in vivo systems to determine if the relative difference in effectiveness in the Ca2+ mobilization assays between the two compounds were reflected in other test systems.
Figure 2.
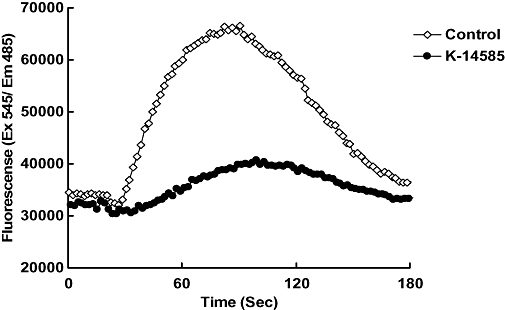
Representative trace for inhibitory effect of K-14585 on SLIGKV-induced Ca2+ mobilization in normal human epidermal keratinocytes. Cells were stimulated with SLIGKV (100 µM), and intracellular Ca2+ mobilization was measured using a fluorescence method as described in the Methods section. K-14585 (10 µM) was incubated with cells 15 min prior to addition of agonist peptide. Fluorescence was measured over 180 s.
Table 1.
Inhibitory effects of PAR2 antagonists on Ca2+ mobilization in human keratinocytes
| Compound | % Inhibition | |
|---|---|---|
| Mean | SEM | |
| K-12940 | 33.6 | 16.8 |
| K-13412 | 15.1 | 10.5 |
| K-14324 | 14.2 | 10.0 |
| K-14458 | 32.0 | 2.3 |
| K-14585 | 58.4 | 13.0 |
| K-14773 | 52.2 | 1.8 |
To examine the direct effect of both K-12940 and K-14585 upon PAR2 receptor binding, we utilized a radioligand binding assay previously established and characterized in the laboratory (Kanke et al., 2005). Initially, using competition curves for the binding of [3H]-2-furoyl-LIGRL-NH2 to plasma membranes from NCTC2544-PAR2 cells we established an order of potency for the agonist peptides to confirm the pharmacological identity of the binding site (Table 2). All PAR2-activating peptides exhibited concentration-dependent displacement of specific [3H]-2-furoyl-LIGRL-NH2 binding to NCTC2544-PAR2 cells. The order of the affinity of the displacing peptides was: 2-furoyl-LIGRL-NH2 > 2-furoyl-LIGKV-NH2 > 2-furoyl-LIGRL-OH > 2-furoyl-LIGKV-OH > SLIGRL-NH2 > SLIGKV-NH2 > SLIGRL-OH > SLIGKV-OH. The calculated Ki value from the displacement curve for each agonist and its relative binding affinity to the original peptide, SLIGKV-OH, are shown in Table 2 and compared with EC50 values for the Ca2+ assay in the same cells as described previously (Kawabata et al., 2004). There was minimal binding competition for [3H]-2-furoyl-LIGRL-NH2 binding by the inactive reverse PAR2 peptide LRGILS-NH2 while the PAR1 selective agonist peptide, TFLLRN-NH2, reduced the [3H]-2-furoyl-LIGRL-NH2 binding only at high concentrations and its estimated Ki value was higher than 1 mM.
Table 2.
Comparison of PAR2 agonist peptides on binding competition and potency on Ca2+ mobilization in NCTC2544PAR2 cells
| Binding assay | Ca2+mobilization | |||||
|---|---|---|---|---|---|---|
| Ki (µM) | Relative to SLIGKV-OH | EC50 (µ M) | Relative to SLIGKV-OH | |||
| Mean | (SEM) | Mean | (SEM) | |||
| SLIGKV-OH | 50.3 | (4.77) | 1 | 0.54 | (0.078) | 1 |
| SLIGRL-OH | 15.5 | (4.76) | 3.25 | 0.20 | (0.070) | 2.70 |
| SLIGKV-NH2 | 9.64 | (1.21) | 5.24 | 0.075 | (0.0072) | 7.20 |
| SLIGRL-NH2 | 2.61 | (0.37) | 19.3 | 0.046 | (0.019) | 11.7 |
| 2-furoyl-LIGKV-OH | 2.57 | (0.67) | 19.6 | 0.067 | (0.011) | 8.06 |
| 2-furoyl-LIGRL-OH | 0.65 | (0.064) | 77.9 | 0.024 | (0.0069) | 22.5 |
| 2-furoyl-LIGKV-NH2 | 0.30 | (0.053) | 166 | 0.0076 | (0.00076) | 71.1 |
| 2-furoyl-LIGRL-NH2 | 0.11 | (0.011) | 449 | 0.0050 | (0.0017) | 108 |
| tc-LIGRLO- NH2 | 14.0 | (2.49) | 3.59 | n.d. | n.d. | n.d. |
| TFLLR- NH2 | 1177 | (952.2) | 0.043 | n.d. | n.d. | n.d. |
Using the radioligand binding assay, the characteristics of displacement by K-12940 and K-14585 were further analysed (Figure 3). Both compounds displaced [3H]-2-furoyl-LIGRL-NH2 binding in the low to mid-micromolar range, giving Ki values of 1.94 and 0.627 µM respectively (Table 3). Hill slopes of close to unity suggested one site binding although insufficient material was available to generate enough data points to assess this fully. Furthermore, the peptide-mimetic antagonists did not reduce non-specific [3H]2-furoyl-LIGRL-NH2 binding to either PAR2 expressing parental cells (data not shown) confirming specific inhibition of PAR2 receptor binding.
Figure 3.
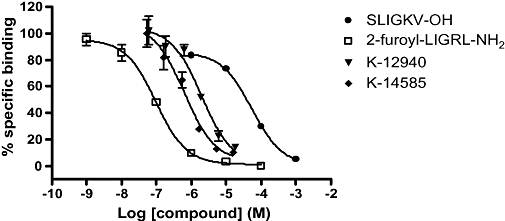
Displacement of [3H]2-furoyl-LIGRL-NH2 binding in NCTC2544-PAR2 cells by K-12940 and K-14585. Cells were incubated with [3H]2-furoyl-LIGRL-NH2 (9.2 nM, 1 µCi·mL−1) in either absence or presence of increasing concentrations of unlabelled agonist/antagonist under conditions as outlined in the Methods section. Binding competition curves for unlabelled compounds are presented as % specific binding. Results are shown as the mean ± SEM (_n_= 3).
Table 3.
Comparative Ki values for displacement of [3H]2-furoyl-LIGRL-NH2 binding to PAR2 and inhibition of luciferase expression
| Binding assay Ki (µM) | Luciferase assay IC50 (µM) | ||||
|---|---|---|---|---|---|
| Mean | (SEM) | Relative to SLIGKV-OH | Mean | (SEM) | |
| SLIGKV-OH | 50.3 | (4.77) | 1 | n.d | n.d |
| 2-furoyl-LIGRL-NH2 | 0.11 | (0.011) | 449 | n.d | n.d |
| K-12940 | 1.94 | (0.47) | 27.9 | 2.87 | 0.48 |
| K-14585 | 0.63 | (0.14) | 86.3 | 1.10 | 0.07 |
We next sought to assess the antagonist effect of K-12940 and K-14585 on a number of cellular markers relevant to functional end points. Using PAR2 expressing NCTC2544 cells, we initially measured [3H]IP production and found significant inhibition of SLIGKV stimulation using the maximum concentration of antagonist (10 µM) (% inhibition, mean ± SEM; K-12940 = 36 ± 1.1% _n_= 4, K-14858 = 62 ± 3.2%, _n_= 4, P > 0.01, compared with SLIGKV alone). We used the same cell line additionally expressing the NFκB reporter luciferase gene, which had been used to study the role of intermediate signalling events including Gq11 and Ca2+– dependent pathways (Macfarlane et al., 2005). We found that while SLIGKV-OH, given alone, caused a 20-fold increase in reporter activity luciferase measured at the 4 h time point, both K-12940 and K-14585 caused a concentration-dependent inhibition of luciferase activity over the low micromolar range which compared well with their effects upon radioligand binding (Figure 4 and Table 3). Maximal inhibition was as much as 80%. Trypsin-stimulated luciferase was also significantly inhibited but less effectively, maximal inhibition was approximately 25% at 10 µM concentration of either compound. Neither agent inhibited luciferase activity stimulated by PMA (% inhibition; K-12940 5.0 ± 2.9, K-14585 = 1.0 ± 1.7 _n_= 3, non-specific (ns) compared with PMA alone), confirming the receptor specificity of the inhibition.
Figure 4.
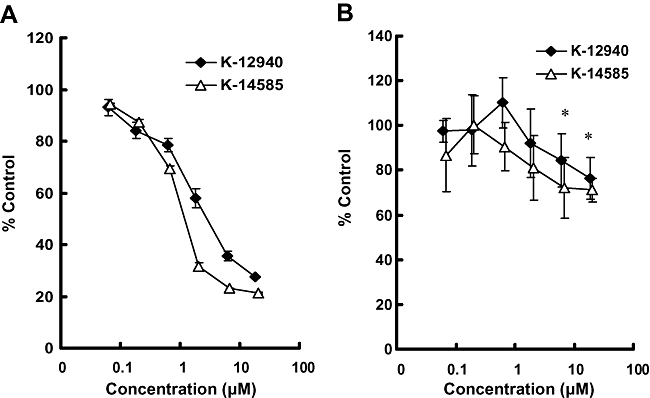
Concentration-dependent inhibitory effects of K-12940 and K-14585 on SLIGKV- or trypsin-mediated NFκB-luciferase activation in NCTC2544 cells. Cells were stimulated with SLIGKV (30 µM) (Panel A) or trypsin (10 nM) (Panel B) in the presence of increasing concentrations of either K-12940 or K-14585. NFκB reporter activity was assayed as outlined in the Methods section. Data represent % of the control activity stimulated with agonist alone (_n_= 6). *P < 0.05, compared with control stimulation either peptide or trypsin alone.
We then examined the effect of K-12940 and K-14585 upon IL-8 production, known to be regulated at least in part by NFκB activation (Yoshida et al., 2007). Alone, SLIGKV-OH stimulated a 40-fold increase in IL-8 production measured at the 24 h time point (Figure 5). Pre-incubation with both compounds causes a substantial and significant inhibition, K-14585 reducing IL-8 production by over 90%. In contrast, both compounds had no significant effect upon Il-8 production stimulated by TNFα.
Figure 5.
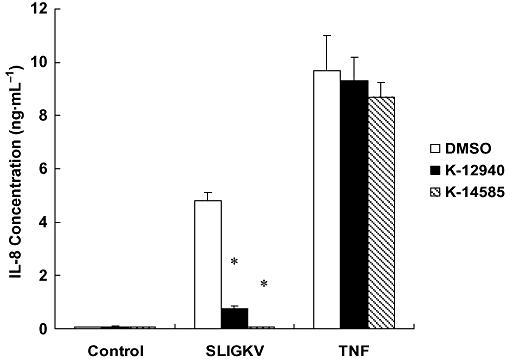
Effect of K-12940 and K-14585 on IL-8 production in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were stimulated with vehicle (Control), SLIGKV (100 µM) or tumuor necrosis factorα (TNFα) (10 ng·mL−1) in the absence (DMSO) or presence of antagonists (5 µM) for 24 h. IL-8 levels in the culture medium were determined by ELISA. Data is expressed as the mean ± SEM of three experiments. *P < 0.05 compared with SLIGKV stimulated control.
Having established the potential for PAR2 antagonists to affect a series of parameters at the cellular level, we sought to investigate possible effects of K-14585, the more potent of the two compounds on in vitro and in vivo vascular responses. However, because tissue systems often contain other PARs such as PAR1 and PAR4, we first of all tested whether K-14585 was able to cross-react with either of these receptors. Human PARs 1, 2 and 4 were transfected into HEK293 cells and stimulated with the relevant PAR activating peptides (Table 4). While K-14585 (10 µM) caused a significant decrease in PAR2 peptide-induced [3H]IP accumulation, the compound displayed no equivalent inhibitory effect upon PAR1- or PAR4-mediated responses, suggesting selectivity of action.
Table 4.
Inhibitory effects of K-14585 on PAR-mediated [3H]-inositol phosphate accumulation in HEK293 cells
| HEK293 | Agonist | Fold stimulation | |||
|---|---|---|---|---|---|
| Control | K-14585 (10 µM) | ||||
| Mean | SEM | Mean | SEM | ||
| PAR1 | TFLLR-NH2 | 4.76 | 1.23 | 5.30 | 1.61 |
| PAR2 | SLIGKV-OH | 5.34 | 1.60 | 2.39* | 0.48 |
| PAR4 | AYPGKF-NH2 | 4.97 | 0.57 | 5.57 | 0.81 |
Having established a lack of cross reactivity of K-14585 on PAR1 and PAR4, we examined the endothelium-dependent relaxation of isolated aorta, a well-recognized effect of PAR2 activation (Figure 6). The rodent-specific activating peptide SLIGRL-NH2 caused a concentration-dependent decrease in contractile tone, with an EC50 value of 1.2 µM. However, in the presence of 10 µM, K-14585, this curve was shifted to the right in a parallel fashion giving an EC50 value of 3.2 µM which was significantly different from the control value (P < 0.001 Bonferroni's test, Figure 6B). Although this displacement was small and precluded an accurate estimation of Kd, an approximate value of 3.86 µM, derived from the Gaddum equation, was close to the Ki values obtained in the ligand binding experiments and was in the same range of effect observed in the cellular assays. In contrast, relaxant responses to histamine were not modified by K-14585, EC50 values for histamine alone, 3.1 µM, and in the presence of K-14858, 4.2 µM, were not significantly different (Figure 6C).
Figure 6.
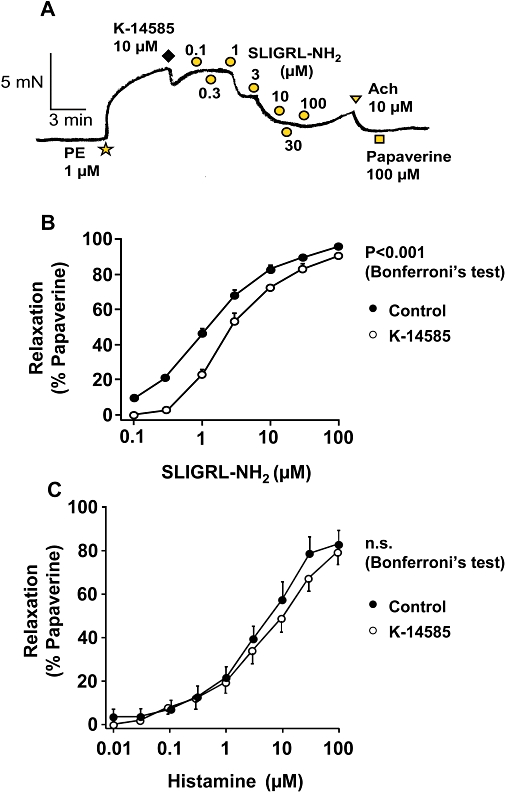
Effect of K-14585 on relaxation of rat aortic rings induced by the proteinase-activated receptor 2 (PAR2) agonist, Ser-Leu-Ile-Gly-Arg-Leu-amide (SLIGRL-NH2). In A, SLIGRL-NH2 was applied cumulatively to the aortic ring preparation pre-contracted with 1 µM phenylephrine (PE). K-14585 (10 µM) was added to the bath 2 min prior to the addition of SLIGRL-NH2. Summary data (in B) are the mean ± SEM of nine control experiments and 16 with K-14585. The response curve was significantly shifted to the right by K-14585; P < 0.001 compared with the control curve (Bonferoni's test). In C, results from experiments using histamine instead of PAR2 activating peptide and data shown are the mean ± SEM of eight experiments. n.s., not significant.
We then examined the effect of K-14585 on two variables in vivo, vascular permeability assessed by exudation of Evans blue dye (Figure 7) and secretion of saliva (Figure 8). The agonist peptide, SLIGRL-NH2, caused a substantial increase in dye exudation, assessed in four different animals (Figure 7A) and this effect was significantly reduced by pre-treatment with K-14585 (P < 0.05). A similar effect was observed when saliva secretion was examined. Two doses of the PAR2 activating peptide were used, 1.5 and 2.5 mmol·kg−1. Both stimulated a marked increase in saliva reaching a peak within 10 min before decreasing again at 15 min (Figure 8A). Pre-treatment with K-14585 reduced saliva secretion in response to the lower dose of agonist at 15 min, but the responses to the higher dose were not significantly affected. Similarly, when the response to the lower dose of SLIGRL-NH2 was expressed as the total saliva collected over 15 min, this total was significantly reduced by K-14585, whereas saliva secretion in response to the higher dose of agonist was unaffected (Figure 8B)
Figure 7.
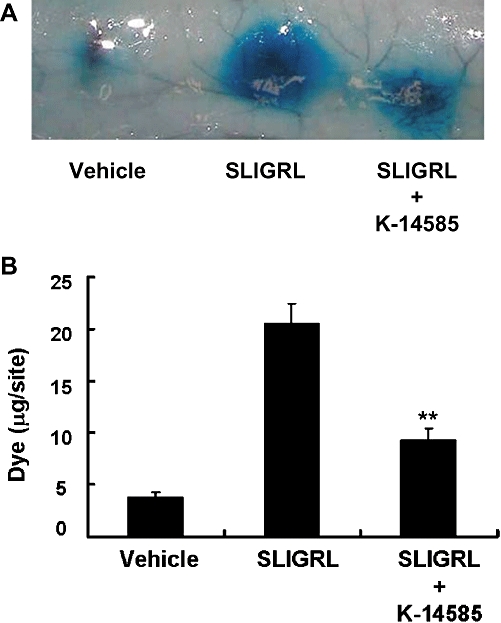
Effect of K-14585 on the plasma exudation caused by the proteinase-activated receptor 2 agonist, Ser-Leu-Ile-Gly-Arg-Leu-amide (SLIGRL-NH2), in guinea pig dorsal skin. Evans blue dye (50 mg·kg−1) was injected intravenously in male Hartley guinea pigs. Immediately after the dye injection, vehicle (10% DMSO), SLIGRL- NH2 (300 nM) or a mixture of SLIGRL-NH2 (300 nM) and K-14585 (300 µg) was injected intradermally on dorsal skin in a volume of 100 µL per site. Dye extravasation (in A) was quantified (in B) spectrophotometrically, as outlined in the Methods section. Each value represents the mean ± SEM from at least four experiments. *P < 0.01 compared with test control (SLIGRL-NH2).
Figure 8.
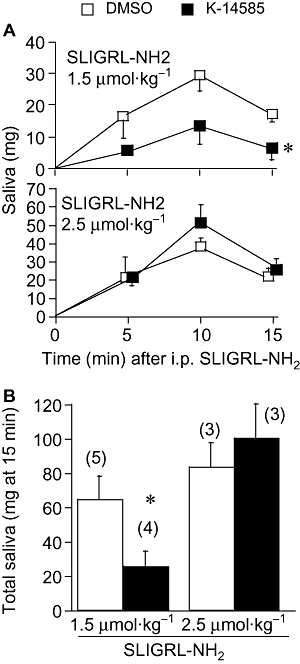
The effect of K-14585 on saliva secretion in anaesthetized mice. K-14585 at 10 µmol·kg−1 dissolved in DMSO was administered i.p. in a volume of 10 µL per 10 g body weight, 5 min before i.p. Ser-Leu-Ile-Gly-Arg-Leu-amide (SLIGRL-NH2) at 1.5 or 2.5 µM, in combination with amastatin at 2.5 µM. Saliva secretion was measured at 5 min intervals for 15 min [basal levels at time 0 were below detection (1 mg)]. Control animals received only the same volume of vehicle (DMSO) before the agonist peptide. Summary results from the whole 15 min collection period are shown in histogram form (in B) and each value represents the mean ± SEM. *P < 0.001 compared with control; numbers of animals are shown in parentheses.
Discussion
In this study, we examined the effects of novel PAR2 antagonist peptides in a series of assays both in vitro and in vivo. Initially, we examined a series of compounds on PAR2 -induced intracellular Ca2+ mobilization and identified K-12490 and K-14585 as moderately effective inhibitors. PAR2 has been shown to mediate Ca2+ mobilization in a large number of cell types, including keratinocytes (Santulli et al., 1995), neurons (Bushell et al., 2006) and endothelial cells. In these cell types, Ca2+ mobilization is predominantly Pertussis toxin-independent (Bushell et al., 2006) and involves Gq11 in the coupling to InsP3 production and subsequent intracellular Ca2+ release, although this has not been investigated directly. Recently, we have shown that the novel Gq11 inhibitor YM254890 is able to inhibit InsP production (Goon Goh et al., 2008) and intracellular Ca2+ mobilization in NCTC2544-PAR2 cells (Bushell and Plevin, unpublished), results consistent with this model. In other experiments, we also demonstrated that both K-12490 and K-14585 inhibited [3H]InsP in the same cell line, suggesting inhibition of a Gq11 coupled PAR2 response.
Nevertheless, in order to establish that these compounds were directly interacting with the receptor, we examined the compounds in a peptide ligand binding assay utilizing a clonal cell line exogenously expressing PAR2 (Kanke et al., 2001). This cell line was initially found to express no endogenous PAR2, no PAR3 or PAR4 and a minor amount of PAR1 (Kawabata et al., 2004; Kanke et al., 2005). Competition curves indicated that these compounds competed with [3H]2-furoyl-LIGRL-NH2 binding to the receptor with affinities close to their inhibitory potency at the level of Ca2+ mobilization. Slopes close to unity suggested that K-12940 and K-14585 are competitive PAR2 antagonists, however, this needs to be examined in more detail. As there is no binding of radiolabelled peptide in cells which do not express the receptor (Kanke et al., 2005), this suggests competitive binding to PAR2; however, it is possible, although unlikely, that displacement of radiolabelled 2-furoyl-LIGRL-NH2 is artifactual and that the K compounds bind directly to the radioligand.
We next assessed the potential of K-14585 to inhibit a number of selected cellular events relevant to the biological actions of PAR2. Activation of this receptor has been shown by ourselves and others to link strongly to the NFκB pathway primarily via Gq11 and Ca2+-dependent mediated events (Kanke et al., 2001; Macfarlane et al., 2005; Goon Goh et al., 2008) which in turn is involved in regulation of IL-8 production (Yoshida et al., 2007). Although both compounds were active against agonist peptide stimulation of NFκB and IL-8 production, we consistently found that trypsin-stimulated responses, including that of NFκB were inhibited to a much lesser extent. The reasons for this are unclear and given the Ki values of K-14585 for binding to PAR2 (0.627 µM) and the maximum concentration used in several assays of 10 µM, one might theoretically expect greater inhibition. It is therefore possible that K-14585 competes more effectively for peptide binding to a site which is distinct from, or at least only partially overlapping, that occupied by the tethered ligand. This possibility has been raised although not conclusively proved for PAR1 activating peptides (Blackhart et al., 2000). In addition, the data tentatively suggest the potential for agonist-directed signalling; peptide and tethered ligand activate a different pattern of signalling pathways such that inhibition of downstream parameters such as NFκB activity by the K compounds reflects inhibition of only the signalling pathways that the peptide can induce and not the tethered ligand. This possibility is currently being examined in our laboratory.
This difference in affinity between the activating serine protease and cognate activating peptide has been observed before, for the other PARs (Macfarlane et al., 2001). Recently, however, one group successfully developed a competitive PAR1 antagonist, RWJ-56110, which can effectively inhibit both agonist peptide-activated and proteolytically activated receptor (Andrade-Gordon et al., 1999; Maryanoff et al., 2003), suggesting a common site of receptor interaction for the PAR1 peptide and tethered ligand in these examples. Therefore, the PAR2 antagonists identified in this study still may represent appropriate leads for the future development of high affinity antagonists.
A limitation of our current investigation is the structure of the compounds studied. There was a need for a peptide structure and a precise requirement for bulky moieties at the C-terminus, which made the compounds more complicated in structure and less soluble in water. This limited the use of these compounds at higher concentrations because of problems of solubility. Nevertheless, the SAR analysis of these compounds highlighted some strict structural requirement for PAR2 antagonism. Firstly, the importance of the N-terminal N-heteroaromatic urea portion; secondly, the basic functionality on the amino side chain; and thirdly, the C-terminal hydrophobic group. For example the indol-5-yl isomer of K-14585 lost all antagonist activity (not shown). Furthermore, there may be additional structural requirements to aid resistance to degradation in tissues; this aspect has not been examined in this study but was a feature of the substituted agonist peptide, 2-furoyl-LIGRL-OH (Kawabata et al., 2004).
Despite the limitations of the antagonists described in this study in terms of potency, any potential actions in vitro and in vivo would be an important aspect of antagonist actions. Endothelium-dependent relaxations of blood vessels is an established feature of the actions of PAR2 mediated by a predominantly nitric oxide (NO)-dependent mechanism (AlAni et al., 1995; Hwa et al., 1996; Sobey and Cocks, 1998). The most potent compound, K-14585, caused a parallel shift in the concentration indicative of competitive antagonism with the estimated Kd value in the low micromolar range consistent with that obtained in other assays. However, the actions of PAR2–mediated effects upon endothelial function are not limited to vasodilatation and we recorded similar inhibitory effects of K-14585 upon vascular permeability. For both these parameters, inhibition was not always complete and sometimes less effective at higher concentrations, suggestive of solubility problems (Figures 6 and 8). Nevertheless, PAR2 has been shown to directly regulate endothelial barrier function as a component of a cellular response which includes leucocyte rolling, adhesion and extravasation in endothelial cells (Vergnolle et al., 1999a) and the inhibition described here is consistent with blocking this action. These effects are manifested in vivo, as intraplantar injection of PAR activating peptide induced significant oedema in the rat hind paw which was followed by a disruption of tissue architecture along with an inflammatory cell infiltrate (Vergnolle et al., 1999b) and PAR2 deficient mice show a delayed onset of leucocyte rolling and adhesion following inflammatory challenge (Lindner et al., 2000). These actions may be via a direct effect on the prostanoids (Kong et al., 1997) and NO (Hwa et al., 1996), although a neurogenic mechanism may also be involved, in turn, via the release of calcitonin gene-related peptide and substance P (Steinhoff et al., 2000). Similar mechanisms of regulation are also observed for PAR2 regulation of salivary gland function (Kawabata et al., 1999; 2000; 2002;), the inhibition of this process by K-14585 suggests a common receptor existing in different tissues, susceptible to competitive antagonism.
In conclusion, this study identifies a moderately potent competitive PAR2 antagonist, K-14585 which was effective against PAR2 activating peptides in cellular assays and in vitro systems and may be utilized to further characterize the effects mediated by PAR2. However, at present, its usefulness in probing PAR2 functions mediated by protease activators of PARs is limited. Nevertheless, while further studies are required to develop potent bio-available, non-peptide, antagonists, the compound described in this study provides an attractive template for such future studies.
Acknowledgments
This work was supported by Kowa Company Ltd., Japan. Margaret R Cunningham is a recipient of an AJ Clark studentship award from The British Pharmacological Society.
Glossary
Abbreviations:
2-furoyl-LIGRL-NH2
2-furoyl-Leu-Ile-Gly-Arg-Leu-amide
HUVEC
human umbilical vein endothelial cells
PAR
proteinase-activated receptor
PMA
phorbol myristate acetate
SAR
structure-activity relationship
SLIGKV-OH
Ser-Leu-Ile-Gly-Lys-Val
SLIGRL-NH2
Ser-Leu-Ile-Gly-Arg-Leu-amide
TNFα
tumour necrosis factorα
Statement of conflict of interest
None.
References
- AlAni B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol. 1995;73:1203–1207. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257–12262. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackhart BD, Ruslim-Litrus L, Lu CC, Alves VL, Teng W, Scarborough RM, et al. Extracellular mutations of protease-activated receptor-1 result in differential activation by thrombin and thrombin receptor agonist peptide. Mol Pharmacol. 2000;58:1178–1187. doi: 10.1124/mol.58.6.1178. [DOI] [PubMed] [Google Scholar]
- Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50:714–725. doi: 10.1016/j.neuropharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Camerer E, Gjernes E, Wiiger M, Pringle S, Prydz H. Binding of Factor VIIa to tissue factor on keratinocytes induces gene expression. J Biol Chem. 2000;275:6580–6585. doi: 10.1074/jbc.275.9.6580. [DOI] [PubMed] [Google Scholar]
- Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks TM, Moffatt JD. Protease-activated receptors: sentries for inflammation? Trends Pharmacol Sci. 2000;21:103–108. doi: 10.1016/s0165-6147(99)01440-6. [DOI] [PubMed] [Google Scholar]
- Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goon Goh F, Sloss CM, Cunningham MR, Nilsson M, Cadalbert L, Plevin R. G-protein-dependent and -independent pathways regulate proteinase-activated receptor-2 mediated p65 NFkappaB serine 536 phosphorylation in human keratinocytes. Cell Signal. 2008;20:1267–1274. doi: 10.1016/j.cellsig.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD. Physiology and pathophysiology of proteinase-activated receptors (PARs): proteinases as hormone-like signal messengers: PARs and more. J Pharmacol Sci. 2005;97:8–13. doi: 10.1254/jphs.fmj04005x2. [DOI] [PubMed] [Google Scholar]
- Hwa JJ, Ghibaudi L, Williams P, Chintala M, Zhang RM, Chatterjee M, et al. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ Res. 1996;78:581–588. doi: 10.1161/01.res.78.4.581. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanke T, Macfarlane SR, Seatter MJ, Davenport E, Paul A, McKenzie RC, et al. Proteinase-activated receptor-2-mediated activation of stress-activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J Biol Chem. 2001;276:31657–31666. doi: 10.1074/jbc.M100377200. [DOI] [PubMed] [Google Scholar]
- Kanke T, Ishiwata H, Kabeya M, Saka M, Doi T, Hattori Y, Kawabata A, Plevin R. Binding of a highly potent protease-activated receptor-2 (PAR2) activating peptide, [3H]2-furoyl-LIGRL-NH2, to human PAR2. Br J Pharmacol. 2005;145:255–263. doi: 10.1038/sj.bjp.0706189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Hollenberg MD, Kuroda R. Effects of trypsin and a selective agonist peptide of proteinase-activated receptor 2 on vascular permeability in rat hindpaw. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:3797. [Google Scholar]
- Kawabata A, Kuroda R, Nishikawa H, Kawai K. Modulation by protease-activated receptors of the rat duodenal motility in vitro: possible mechanisms underlying the evoked contraction and relaxation. Br J Pharmacol. 1999;128:865–872. doi: 10.1038/sj.bjp.0702755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Morimoto N, Nishikawa H, Kuroda R, Oda Y, Kakehi K. Activation of protease-activated receptor-2 (PAR-2) triggers mucin secretion in the rat sublingual gland. Biochem Biophys Res Commun. 2000;270:298–302. doi: 10.1006/bbrc.2000.2404. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Kinoshita M, Nishikawa H, Kuroda R, Nishida M, Araki H, et al. The protease-activated receptor-2 agonist induces gastric mucus secretion and mucosal cytoprotection. J Clin Invest. 2001;107:1443–1450. doi: 10.1172/JCI10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Kuroda R, Nishida M, Nagata N, Sakaguchi Y, Kawao N, et al. Protease-activated receptor-2 (PAR-2) in the pancreas and parotid gland: immunolocalization and involvement of nitric oxide in the evoked amylase secretion. Life Sci. 2002;71:2435–2446. doi: 10.1016/s0024-3205(02)02044-1. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Kanke T, Yonezawa D, Ishiki T, Saka M, Kabeya M, et al. Potent and metabolically stable agonists for protease-activated receptor-2: evaluation of activity in multiple assay systems in vitro and in vivo. J Pharmacol Exp Ther. 2004;309:1098–1107. doi: 10.1124/jpet.103.061010. [DOI] [PubMed] [Google Scholar]
- Kawagoe J, Takizawa T, Matsumoto J, Tamiya M, Meek SE, Smith AJ, et al. Effect of protease-activated receptor-2 deficiency on allergic dermatitis in the mouse ear. Jpn J Pharmacol. 2002;88:77–84. doi: 10.1254/jjp.88.77. [DOI] [PubMed] [Google Scholar]
- Kawao N, Sakaguchi Y, Tagome A, Kuroda R, Nishida S, Irimajiri K, et al. Protease-activated receptor-2 (PAR-2) in the rat gastric mucosa: immunolocalization and facilitation of pepsin/pepsinogen secretion. Br J Pharmacol. 2002;135:1292–1296. doi: 10.1038/sj.bjp.0704562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, et al. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316:1017–1024. doi: 10.1124/jpet.105.093807. [DOI] [PubMed] [Google Scholar]
- Kong W, McConalogue K, Khitin LM, Hollenberg MD, Payan DG, Bohm SK, et al. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc Natl Acad Sci (USA) 1997;94:8884–8889. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan RS, Stewart GA, Henry PJ. Modulation of airway smooth muscle tone by protease activated receptor-1,-2,-3 and-4 in trachea isolated from influenza A virus-infected mice. Br J Pharmacol. 2000;129:63–70. doi: 10.1038/sj.bjp.0703007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan RS, Knight DA, Stewart GA, Henry PJ. Role of PGE(2) in protease-activated receptor-1, -2 and -4 mediated relaxation in the mouse isolated trachea. Br J Pharmacol. 2001;132:93–100. doi: 10.1038/sj.bjp.0703776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner JR, Kahn ML, Coughlin SR, Sambrano GR, Schauble E, Bernstein D, et al. Delayed onset of inflammation in protease-activated receptor-2- deficient mice. J Immunol. 2000;165:6504–6510. doi: 10.4049/jimmunol.165.11.6504. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Macfarlane SR, Sloss CM, Cameron P, Kanke T, McKenzie RC, Plevin R. The role of intracellular Ca2+ in the regulation of proteinase-activated receptor-2 mediated nuclear factor kappa B signalling in keratinocytes. Br J Pharmacol. 2005;145:535–544. doi: 10.1038/sj.bjp.0706204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanoff BE, Zhang HC, Andrade-Gordon P, Derian CK. Discovery of potent peptide-mimetic antagonists for the human thrombin receptor, protease-activated receptor-1 (PAR-1) Curr Med Chem Cardiovasc Hematol Agents. 2003;1:13–36. doi: 10.2174/1568016033356724. [DOI] [PubMed] [Google Scholar]
- Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- Mototsugu K, Kyoko Y, Toru K, Hioryuki I, Junya T. PAR-2 antagonists EP1806141 (A1); 2007/7/11.
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor [see comments. Proc Natl Acad Sci USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plevin R, Kellock NA, Wakelam MJ, Wadsworth R. Regulation by hypoxia of endothelin-1-stimulated phospholipase D activity in sheep pulmonary artery cultured smooth muscle cells. Br J Pharmacol. 1994;112:311–315. doi: 10.1111/j.1476-5381.1994.tb13070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardolo FLM, Steinhoff M, Amadesi S, Guerrini R, Tognetto M, Trevisani M, et al. Presence and bronchomotor activity of protease-activated receptor-2 in guinea pig airways. Am J Respir Crit Care Med. 2000;161:1672–1680. doi: 10.1164/ajrccm.161.5.9907133. [DOI] [PubMed] [Google Scholar]
- Santulli RJ, Derian CK, Darrow AL, Tomko KA, Eckardt AJ, Seiberg M, et al. Evidence for the presence of a protease-activated receptor distinct from the thrombin receptor in human keratinocytes. Proc Natl Acad Sci U S A. 1995;92:9151–9155. doi: 10.1073/pnas.92.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott G, Leopardi S, Printup S, Malhi N, Seiberg M, Lapoint R. Proteinase-activated receptor-2 stimulates prostaglandin production in keratinocytes: analysis of prostaglandin receptors on human melanocytes and effects of PGE2 and PGF2alpha on melanocyte dendricity. J Invest Dermatol. 2004;122:1214–1224. doi: 10.1111/j.0022-202X.2004.22516.x. [DOI] [PubMed] [Google Scholar]
- Sobey CG, Cocks TM. Activation of protease-activated receptor-2 (PAR-2) elicits nitric oxide-dependent dilatation of the basilar artery in vivo. Stroke. 1998;29:1439–1444. doi: 10.1161/01.str.29.7.1439. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Krause JE. Neuropeptide K potently stimulates salivery gland secretion and potentiates substance P-induced salivation. PNAS. 1989;86:392–396. doi: 10.1073/pnas.86.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle N, McKnight W, Befus AD, Hollenberg MD, Wallace JL. Induction of an inflammatory response by activation of protease-activated receptor 2 (PAR-2) Naunyn Schmiedebergs Arch Pharmacol. 1998;358:573. [Google Scholar]
- Vergnolle N, Hollenberg D, Wallace JL, Morley MD. Activation of proteinase-activated receptor-2 (PAR-2) induces leukocyte adhesion. FASEB J. 1999a;13:A668. [Google Scholar]
- Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR(2))-activating peptides in the rat paw. Br J Pharmacol. 1999b;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Katada K, Handa O, Takagi T, Kokura S, Naito Y, et al. Interleukin-8 production via protease-activated receptor 2 in human esophageal epithelial cells. Int J Mol Med. 2007;19:335–340. doi: 10.3892/ijmm.19.2.335. [DOI] [PubMed] [Google Scholar]