edgeR: a Bioconductor package for differential expression analysis of digital gene expression data (original) (raw)
Abstract
Summary: It is expected that emerging digital gene expression (DGE) technologies will overtake microarray technologies in the near future for many functional genomics applications. One of the fundamental data analysis tasks, especially for gene expression studies, involves determining whether there is evidence that counts for a transcript or exon are significantly different across experimental conditions. edgeR is a Bioconductor software package for examining differential expression of replicated count data. An overdispersed Poisson model is used to account for both biological and technical variability. Empirical Bayes methods are used to moderate the degree of overdispersion across transcripts, improving the reliability of inference. The methodology can be used even with the most minimal levels of replication, provided at least one phenotype or experimental condition is replicated. The software may have other applications beyond sequencing data, such as proteome peptide count data.
Availability: The package is freely available under the LGPL licence from the Bioconductor web site (http://bioconductor.org).
Contact: mrobinson@wehi.edu.au
1 INTRODUCTION
Modern molecular biology data present major challenges for the statistical methods that are used to detect differential expression, such as the requirement of multiple testing procedures and increasingly, empirical Bayes or similar methods that share information across all observations to improve inference. For microarrays, the abundance of a particular transcript is measured as a fluorescence intensity, effectively a continuous response, whereas for digital gene expression (DGE) data the abundance is observed as a count. Therefore, procedures that are successful for microarray data are not directly applicable to DGE data.
This note describes the software package edgeR (empirical analysis of DGE in R), which forms part of the Bioconductor project (Gentleman et al., 2004). edgeR is designed for the analysis of replicated count-based expression data and is an implementation of methology developed by Robinson and Smyth (2007, 2008). Although initially developed for serial analysis of gene expression (SAGE), the methods and software should be equally applicable to emerging technologies such as RNA-seq (Li et al., 2008; Marioni et al., 2008) giving rise to digital expression data. edgeR may also be useful in other experiments that generate counts, such as ChIP-seq, in proteomics experiments where spectral counts are used to summarize the peptide abundance (Wong et al., 2008), or in barcoding experiments where several species are counted (Andersson et al., 2008). The software is designed for finding changes between two or more groups when at least one of the groups has replicated measurements.
2 MODEL
Bioinformatics researchers have learned many things from the analysis of microarray data. For instance, power to detect differential expression can be improved and false discoveries reduced by sharing information across all probes. One such procedure is limma (Smyth, 2004), where an empirical Bayes model is used to moderate the probe-wise variances. The moderated variances replace the probe-wise variances in the _t_- and _F_-statistic calculations. In a closely analogous but mathematically more complex procedure, edgeR models count data using an overdispersed Poisson model, and uses an empirical Bayes procedure to moderate the degree of overdispersion across genes.
We assume the data can be summarized into a table of counts, with rows corresponding to genes (or tags or exons or transcripts) and columns to samples. For RNA-seq experiments, these may be counts at the exon, transcript or gene-level. We model the data as negative binomial (NB) distributed,
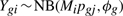 |
(1) |
|---|
for gene g and sample i. Here, M i is the library size (total number of reads), ϕ_g_ is the dispersion and p gj is the relative abundance of gene g in experimental group j to which sample i belongs. We use the NB parameterization where the mean is μ_gi_=M i p gj and variance is μ_gi_ (1+μ_gi_ϕ_g_). For differential expression analysis, the parameters of interest are p gj.
The NB distribution reduces to Poisson when ϕ_g_=0. In some DGE applications, technical variation can be treated as Poisson. In general, ϕ_g_ represents the coefficient of variation of biological variation between the samples. In this way, our model is able to separate biological from technical variation.
edgeR estimates the genewise dispersions by conditional maximum likelihood, conditioning on the total count for that gene (Smyth and Verbyla, 1996). An empirical Bayes procedure is used to shrink the dispersions towards a consensus value, effectively borrowing information between genes (Robinson and Smyth, 2007). Finally, differential expression is assessed for each gene using an exact test analogous to Fisher's exact test, but adapted for overdispersed data (Robinson and Smyth, 2008).
3 FEATURES
The required inputs for edgeR are the table of counts and two vectors annotating the samples: the vector of the library sizes (i.e. total number of reads) and a factor specifying the experimental group or condition for each sample.
For users of limma, the edgeR package has a number of analogous functions. Once the data have been processed and the dispersion estimates are moderated, the topTags function can be used to tabulate the top differentially expressed genes (or tags or exons, etc.). Also, MA (log ratio versus abundance) plots can be created using the plotSmear function, allowing the same visualizations for DGE data as used for microarray data analysis (Fig. 1).
Fig. 1.

DGE data can be visualized as ‘MA’ plots (log ratio versus abundance), just as with microarray data where each dot represents a gene. This plot shows RNA-seq gene expression for DHT-stimulated versus Control LNCaP cells, as described in Li et al. (2008). The smear of points on the left side signifies that genes were observed in only one group of replicate samples and the points marked ‘×’ denote the top 500 differentially expressed genes.
A number of features have been added to the edgeR package since the initial publications. The initial methodology worked only for a two-group comparison. The extension to estimating and moderating the dispersion for multiple groups is straightforward and has been implemented recently. At present, testing for differential expression is supported only for pairwise comparisons; the user must specify which two groups to compare. We are currently investigating tests for more general cases.
Many of the early RNA-seq datasets involve sequence reads from technical replicates (e.g. same source of RNA) as opposed to biological replicates (e.g. RNA from different individuals). Technical replicates will generally have lower variability than biological replicates and in our experience, the dispersion parameter (and the moderation procedure in edgeR) may not be necessary. For experiments with technical replicates, the data may be fitted well by the Poisson distribution, as demonstrated in Marioni et al. (2008). Since the Poisson distribution is a special case of the NB distribution (ϕ=0), edgeR can perform a Poisson-based analysis. The pairwise exact testing procedure will still be useful for these datasets.
4 DISCUSSION
We have developed a Bioconductor package edgeR that addresses one of the fundamental downstream data analysis tasks for count-based expression data: determining differential expression. The package and methods are general, and can work on other sources of count data, such as barcoding experiments and peptide counts. To the authors' knowledge, edgeR is the only software for SAGE or DGE data at this time which can account for biological variability when there are only one or two replicate samples.
Funding: National Health and Medical Research Council Program (Grant 406657 to G.K.S.); NHMRC, Independent Research Institutes Infrastructure Support Scheme (Grant 361646); Victorian State Government OIS grant (awarded to the WEHI); a Melbourne International Research Scholarship (to M.D.R.); Belz, Harris and IBS Honours scholarships (to D.J.M.).
Conflict of Interest: none declared.
REFERENCES
- Andersson AF, et al. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model. Proc. Natl Acad. Sci. USA. 2008;105:20179–20184. doi: 10.1073/pnas.0807121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Small sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;1 doi: 10.2202/1544-6115.1027. Art 3. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Verbyla AP. A conditional approach to residual maximum likelihood estimation in generalized linear models. J. R. Stat. Soc. B. 1996;58:565–572. [Google Scholar]
- Wong JWH, et al. Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments. Brief. Bioinform. 2008;9:156–165. doi: 10.1093/bib/bbm046. [DOI] [PubMed] [Google Scholar]