RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development (original) (raw)
Abstract
Blood vessel formation is a complex morphological process that is only beginning to be understood at the molecular level. In this study, we demonstrate a novel and critical role for the small GTPase, RhoB, in vascular development. RhoB null mice have retarded vascular development in the retina characterized by altered sprout morphology. Moreover, pharmaceutical means to deplete RhoB in neonatal rats is associated with apoptosis in the sprouting endothelial cells of newly forming vessels. Similarly, acute depletion of RhoB by antisense or dominant-negative strategies in primary endothelial cell culture models led to apoptosis and failures in tube formation. We identified a novel link between RhoB and the Akt survival signaling pathway to explain these changes. Confocal microscopy revealed that RhoB is highly localized to the nuclear margin with a small percentage found inside the nucleus. Similarly, total Akt is throughout the cell but has increased accumulation at the nuclear margin, and active phosphorylated Akt is found primarily inside the nucleoplasm, where it partially colocalizes with the RhoB therein. We show that this colocalization is functionally relevant, because when RhoB was depleted, Akt was excluded from the nucleus and total cellular Akt protein was decreased in a proteosome-dependent manner. Because the function of RhoB in vivo appears to only be rate limiting for endothelial cell sprouting, we propose that RhoB has a novel stage-specific function to regulate endothelial cell survival during vascular development. RhoB may offer a therapeutic target in diseases such as cancer, diabetic retinopathy, and macular degeneration, where the disruption of sprouting angiogenesis would be desirable.
Keywords: RhoB, farnesyl transferase inhibitor, angiogenesis, Akt, retina, yolk sac
At the earliest stages of vascular development, mesodermal precursors differentiate into endothelial cells that subsequently assemble to form the first blood vessels in a process referred to as vasculogenesis. Following this stage, fully formed blood vessels are capable of further expansion via endothelial cell replication. This type of vessel formation from existing vessels is termed angiogenesis (Risau 1997). The goal of physiological angiogenesis is to increase nutrient delivery to local tissues. Angiogenesis can be accomplished by one of two routes, either by enlarging preexisting blood vessels or by forming new blood vessels, termed sprouting angiogenesis. Nonsprouting angiogenesis is a major route of collateral formation when it applies to arterial vessels (arteriogenesis; Schaper and Buschmann 1999). Sprouting angiogenesis, on the other hand, is thought to be the major source of new blood vessels after the earliest stages of embryonic development and in processes such as wound healing, tumor growth, diabetic retinopathy, and macular degeneration. Both vessel enlargement and sprouting angiogenesis require endothelial cell proliferation, but sprouting angiogenesis requires additional cellular processes such as endothelial cell migration, vessel assembly, and tube formation. The molecular controls of these distinct, stage-specific morphological processes have yet to be elucidated.
In this study, we have investigated a role for Rho signaling in angiogenesis. The Rho/Rac/Cdc42 family of small GTPases has been implicated in cell motility and migration in multiple cell lineages including endothelial cells (Aepfelbacher et al. 1997; Mackay and Hall 1998). Although the Rho proteins in this family are highly homologous, evidence is accumulating that each member has distinct functions in the cell, perhaps related in part to unique subcellular localizations. Interest in a role for RhoB during vascular development relates in part to evidence that it is up-regulated in a stage-specific manner during formation of the endocardial cushion. During heart development, RhoB is turned on in endocardial cells undergo an endothelial-mesenchyme transition and migration that is required for cardiac valve formation (Henderson et al. 2000). Recent studies of RhoB suggest that it may have unique roles in vesicle trafficking, Akt control, and cell survival. For example, RhoB has been shown to regulate EGF-R trafficking through the endosomal compartment (Gampel et al. 1999). In cancer cells, RhoB induces growth inhibition and apoptosis and in some cells it can suppress Akt activity (Prendergast and Oliff 2000). In addition to its localization to endosomes and possibly other cellular membranes, RhoB has also been observed at the nucleus, suggesting a possible role in trafficking processes there (Zalcman et al. 1995; Lebowitz and Prendergast 1998a). There are some reports of Akt localization at the nucleus, but virtually nothing is known about subcellular trafficking of Akt (Andjelkovic et al. 1997; Camper-Kirby et al. 2001). Filling this gap in knowledge may be important to understanding how Akt regulates cell survival, because several physiologically important substrates of Akt are located in the nucleus. For example, Akt phosphorylation of forkhead (FOXO) transcription factors such as FKHR/FOXO1 stimulate nuclear exit through a mechanism involving association with the 14-3-3 protein, thereby down-regulating transcription of proapoptotic FKHR/FOXO1 target genes (Brunet et al. 1999, 2001, 2002). We investigated a role for RhoB in endothelial cells, where Akt is a critical survival factor, based on previous reports that suggest that RhoB functions in the control of cell survival and intracellular transport. Our findings suggest that RhoB is an important determinant of Akt stability and trafficking to the nucleus in endothelial cells, and that this function has a stage-specific role in the survival of sprouting endothelial cells that contribute to blood vessel assembly during development.
Results
RhoB null mice exhibit retarded vascularization of the retina consistent with impaired capillary sprouting
RhoB null mice are viable and fertile, although they are noticeably smaller than wild-type and heterozygous littermates (data not shown). Among the many possibilities to explain their small size, one possible explanation is a retardation in angiogenesis, known to limit tissue growth. Many of the complex and redundant stages of angiogenesis can be visualized in the developing mouse retina. This model permits a clear view of blood vessel formation, from endothelial cell sprouting to vessel enlargement, organization, and remodeling. In the rodent, blood vessel formation occurs during the first 3 wk after birth. Vessels grow radially outward from the optic disc in week 1, and then sprout inward to form a second parallel plexus beneath the first layer in week 2. By focusing on these first 2 wk, whole-mount immunofluorescence can be used to visualize the entire vasculature as it progresses through multiple stages of vessel formation and remodeling. The spatial and temporal specificity of vessel formation in this system provides a unique setting for studies of various stage-specific steps in vascular development.
Examination of the RhoB null retina showed that newborn mice had retardations in the outgrowth of the primary vascular plexus (Fig. 1). In postnatal day 4 (P4) mice, the initial vascular plexus extends partway from the central optic disc across the retinal surface and the active sprouting at the edges show nicely extended cellular processes (Fig. 1A). Examination of the early outgrowth in RhoB null mice revealed that the sprouting vessels had not yet formed a capillary plexus and had only just begun to expand from the optic disc (Fig. 1B). A measurement of the distance from the optic disc covered by microvessels revealed that at P4, the vasculature of the RhoB null mice covered between 30% and 40% of the area covered in wild-type animals, depending on the retina and location measured. In addition, null blood vessels had morphological abnormalities in their tips, failing to exhibit the characteristic robust cytoplasmic extensions (Fig. 1C,D). Examination of the neural retina in H&E sections as well as a general examination upon necropsy did not reveal other developmental abnormalities. The neural portions of the retina appeared normal in structure and cellularity (Fig. 1E,F), as did the other parts of the eye such as the lens and cornea; thus, we believe the overall vascular defect is likely to account for the retarded growth of the animal.
Figure 1.
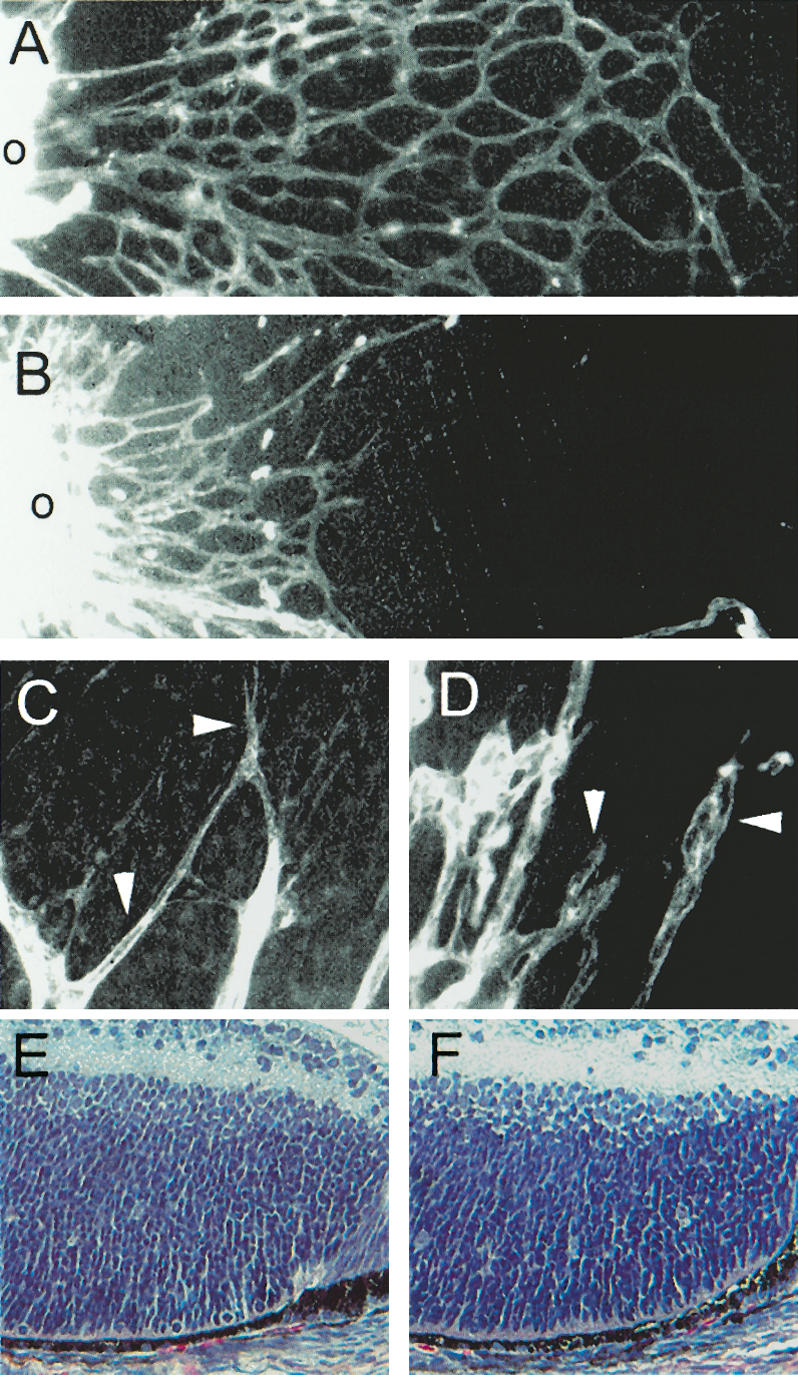
RhoB null mice have retarded vessel outgrowth in the retina and are insensitive to FTI. (A) Vessel outgrowth in P4 129 mice. (B) Vessel outgrowth in P4 RhoB null mice. (C) Vessel tips with cytoplasmic extensions in 129. (D) Vessel tips of RhoB null mice. (E) Neural retina 129. (F) Neural retina RhoB null. Blood vessels were visualized with whole-mount lectin staining. (o) Optic disc.
Definition of a stage-specific cell survival mechanism in sprouting endothelia
As an additional strategy to explore a possible role for RhoB in vascular biology, we exploited a well-characterized pharmacological inhibitor of farnesyl transferase, FTI L-744,832 (Kohl et al. 1995), which rapidly and effectively alters the lipid modification and function of RhoB (Lebowitz et al. 1997). Previous studies have established that RhoB is a crucial target of the effects of this FTI in vitro and in vivo, particularly with regard to apoptosis (Liu et al. 2000; Prendergast 2001). At P2-P4 and P8-P10, representing two independent stages of retinal development, we observed that treatment of neonatal rats for 48 h with FTI dramatically affected the formation of blood vessels in the retina (Fig. 2). Specifically, FTI caused a selective inhibition in vessel sprouting at both stages of vascular development. Retinas from untreated P2-P4 animals displayed a robust phase of vessel assembly very similar to that which occurs in the embyronic day 7.5-8.5 (E7.5-E8.5) yolk sac. This stage contains many proliferating endothelial cells and extensive new vessel formation. The extensive sprouting and vessel formation results in a dense vascular web by P4 (Fig. 2A) with well-defined subcellular processes that indicate active sprouting (Fig. 2C). FTI treatment resulted in a striking decrease in vessel density at P4 (Fig. 2B). Evidence that this event was the result of abortive sprouting was indicated by the presence of blind-ended microvessels (see arrow in Fig. 2B) and collapsed subcellular processes in the cells at the leading edge of the expanding plexus (Fig. 2D). The area covered by capillary in the control P4 retinas was an average of zas1.7-fold higher than the FTI-treated retinas. Vessel density was quantified by measuring the total area covered by blood vessels from digital images using NIH image 1.63 analysis software.
Figure 2.
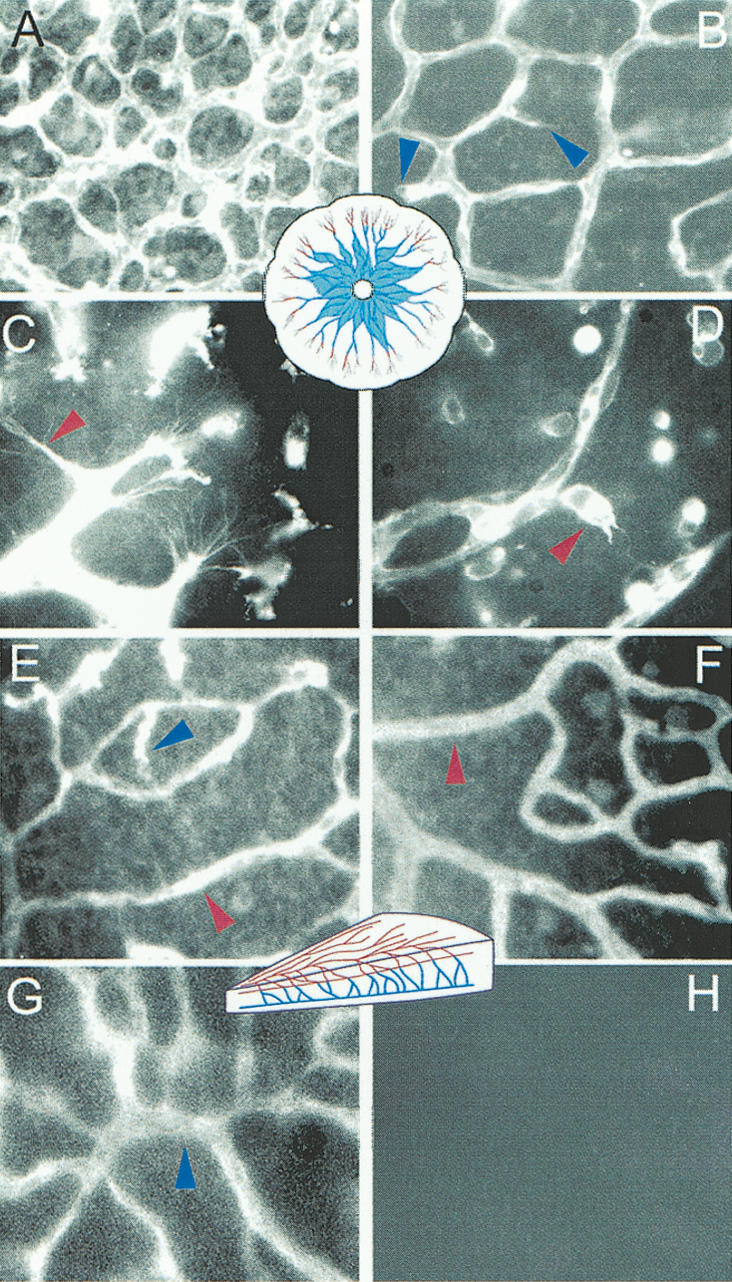
FTI inhibits developmental retinal angiogenesis. Panels A-D show effects of FTI on blood vessels in the P4 retina. (A,C) DMSO treated animals. (B,D) Effects of FTI on the plexus and sprouts. Diagrammatic representations of the retinal vessels at the stage analyzed are shown in the center of the relevant panels and are color-coded to match arrows used to indicate the location of the blood vessels shown in higher magnifications. (A,B) Low-power images of the blood vessels show alterations in vessel density between controls (A) and FTI-treated animals (B). Blue arrows highlight aborted sprouting, which in one case contains erythrocytes that have become lodged in the “dead end” vessel. (C,D) In high-power images of vessel tips we have used red arrows to highlight the dramatic morphological alterations between the normal subcytoplasmic projections that occur during sprouting in control retinas (C) and the misshapen sprouts in the FTI-treated animals (D). E-H illustrate that the formation of the second layer of retinal blood vessels depends on sprouting from the first layer and is entirely eliminated by FTI treatment between P8 and P11. Again, a schematic drawing of this stage color codes the older vessels (red) and the sprouting vessels (blue). The first layer of vessels, which was formed before FTI treatment in control animals (E), and in FTI-treated animals (F) are not dramatically altered. Yet, the second vessel layer that forms during P8-P11 (G) is completely prevented by FTI treatment (H).
To examine the effects of FTI on later processes that required additional sprouting and vessel assembly, we investigated animals that were treated at P8-P10. During this period, a second layer of retinal blood vessels forms by sprouting perpendicularly from the first plexus, inwards from the ganglion cell layer (GCL) toward the inner nuclear layer (INL) before forming a second parallel plexus. In addition, older blood vessels at this stage undergo a trimming and remodeling that is highly dependent on survival signaling from the vascular endothelial growth factor VEGF-A. If VEGF-A signaling is blocked at this stage, a massive regression and death of blood vessels occurs, not only in the areas that are actively sprouting and forming new vessels, but also in the the older vessels that predominate near the optic disk (Alon et al. 1995; Benjamin et al. 1998). In stark contrast to the effects of inhibiting VEGF-A signaling, FTI treatment did not affect the preformed blood vessels but instead only blocked the sprouting processes that form the lower vascular plexus. Figure 2E shows the sprouting process in untreated tissue (note corkscrew descending vessels indicated by blue arrowheads). The descending vessels that were readily observed in untreated tissue could not be seen in retinas from FTI-treated animals (Fig. 2G). Figure 1F shows images of the second plexus taken from untreated animals, visualized by altering the focal plane of the fluorescence microscope. In FTI-treated littermates, there was a complete absence of the sprouting vessels that form the lower plexus (Fig. 2H). We saw no loss of the existing blood vessels in the superficial layer, suggesting that regression was not induced in vessels that were already formed when FTI treatment was initiated. The quite different phenotype of retinas from FTI-treated animals with those deprived of VEGF-A survival signals indicated that FTI targeted a survival signal that was distinct from that regulated by VEGF-A.
TUNEL staining revealed that the FTI-induced block to endothelial cell sprouting and new vessel formation was associated with apoptosis (Fig. 3), even though VEGF-A signaling was not compromised. The highly ordered and reproducible pattern of vessel formation in this model was valuable because it permitted the visualization of sprouting vessels that could be identified spatially as they invaded the INL to form the second vessel plexus. Notably, we did not observe apoptotic cells in larger blood vessels, but only in small vessels in regions where new vessels were sprouting or in isolated endothelial cells. We also did not observe apoptotic endothelial cells in control retinas, not surprisingly because endothelial turnover is very rare. Figure 3A shows a bright-field image of the two layers of the retina where blood vessels form. In P11 retinas from FTI-treated animals, the earliest vessels formed in the GCL were unlabeled whereas the sprouting endothelium in the INL displayed cells that were positive for TUNEL assay. Double labeling of apoptosis and blood vessels is presented in adjacent panels of Figure 3B and C with apoptotic nuclei surrounded by lectin staining for endothelial cells in the matched frame. Proliferation was still occuring in the more mature vessels, as indicated by robust staining of the S phase marker PCNA in the vessel walls of large preexisting blood vessels (Fig. 3D). This result was consistent with other evidence that this FTI has little ifany significant effect on the proliferation and survival of most normal cells (Prendergast and Oliff 2000). The evidence that apoptosis in blood vessels from FTI-treated animals was limited to the youngest and smallest blood vessels supported the conclusion that a stage-specific mechanism of cell survival during sprouting angiogenesis was specifically targeted.
Figure 3.

Apoptosis but not proliferation is blocked in the wild-type developing retinas treated with FTI. (A) Hematoxylin staining of retinal sections during sprouting of the lower plexus. Red arrows denote blood vessels in the ganglion cell layer (GCL) and toward the inner nuclear layer (INL). (B,C) TUNEL and lectin costaining of such sections in FTI-treated animals. TUNEL-positive cells (C) colocalized with lectin (B) are only found in the region toward the INL. (green) TUNEL; (red) endothelial cells; (blue) nuclei. (D) Anti-PCNA staining (pink) of a large vessel. The nuclei are labeled with Hoechst and shown in blue. (e) Erythrocytes.
FTI mediates its affects on the vasculature via RhoB and Akt
To investigate how FTI triggered apoptosis in sprouting endothelial cells, we turned to a second in vivo model system for angiogenesis that is more tractable to biochemical studies. In the developing mouse, the embryonic yolk sac is a source of highly enriched vascular tissue that undergoes the initial stages of vessel formation between embyronic days E7.5 and 8.5, before beginning a subsequent phase of remodeling and continued angiogenesis. Although early stages of vasculogenesis in the yolk sac are distinct from the retina, the weblike vasculature that forms at later stages in the yolk sac closely recapitulates that in the retina that involves extensive sprouting angiogenesis. Prompted by evidence that FTI and RhoB can influence Akt signaling (Jiang et al. 2000; Liu and Prendergast 2000), we investigated the ability of FTI to regulate RhoB and Akt in vivo by injecting pregnant mice at day E8 and harvesting protein from yolk sac endothelial cells 24 h later. It should be noted that this relatively short period of drug treatment is sufficient to effectively deplete farnesylated RhoB, which is relatively unstable (t1/2 = 2-4 h; Lebowitz et al. 1995).
In these yolk sacs, the steady-state levels of RhoB were dramatically reduced by FTI treatment (Fig. 4A). Levels of H-Ras but not RhoA were also reduced in yolk sacs cells from FTI-treated animals (Fig. 4A). In addition, there was a reduction in the steady-state levels of Akt, phospho-Akt, and the phosphorylated isoform of the key Akt substrate forkhead (FKHR). Other signaling pathways were unaffected, as illustrated by the lack of effect on Erk-1 status in these cells (Fig. 4B). RhoB was the essential target of FTI for mediating Akt loss, because Akt loss was not elicited by FTI in yolk sacs derived from RhoB nullizyous mice. In contrast, H-Ras was irrelevant to Akt loss, because whereas Akt was differentially affected, H-Ras was similarly affected in endothelial cells from wild-type or RhoB nullizygous animals (Fig. 4C). Taken together, these results established that RhoB mediates the loss of Akt levels elicited by FTI in developing blood vessels.
Figure 4.
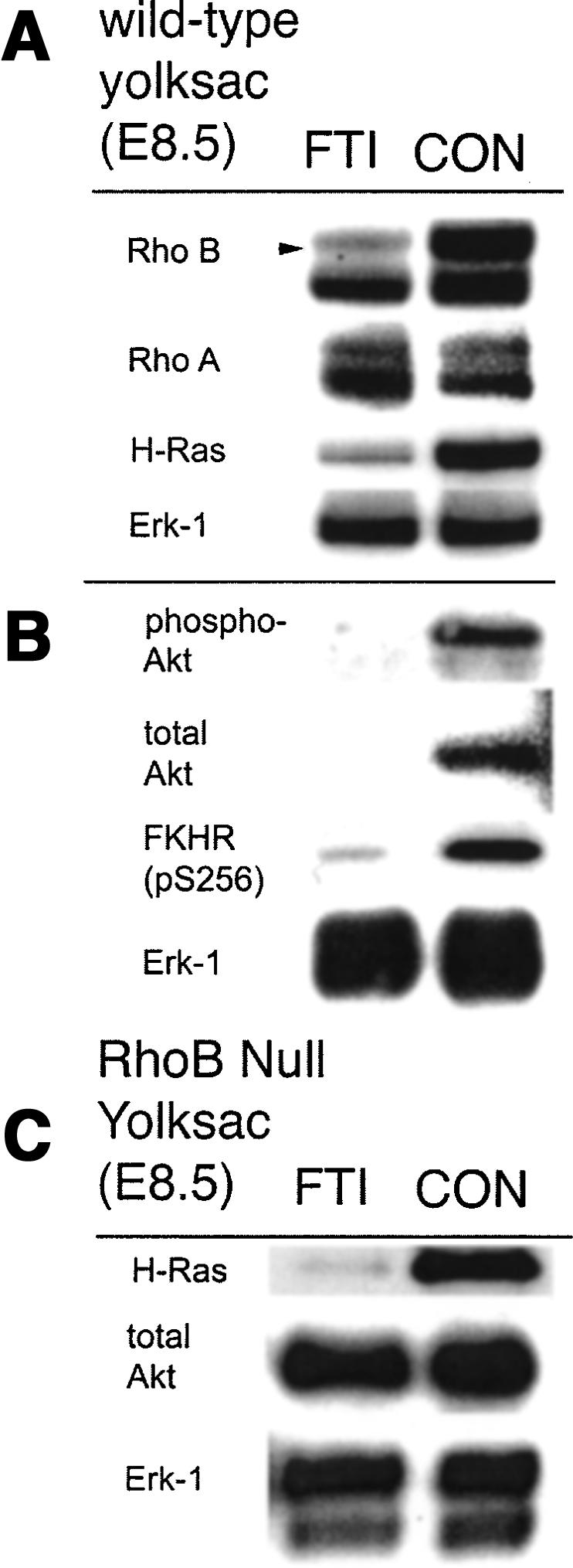
Akt signaling is RhoB dependent in vivo. FTI treatment of wild-type pregnant mice leads to reduction of RhoB, H-ras but not RhoA or Erk-1 in embryonic yolk sac (A) and reduced levels of activated and total Akt protein, and blocking of the phosphorylation of the Akt target gene, FKHR (B). (C) FTI treatment of pregnant RhoB null mice leads to loss of H-ras but not Akt levels in embryonic yolk sac, showing that RhoB is the essential target of FTI that mediates effects on Akt.
RhoB regulates cell survival and Akt function in endothelial cells
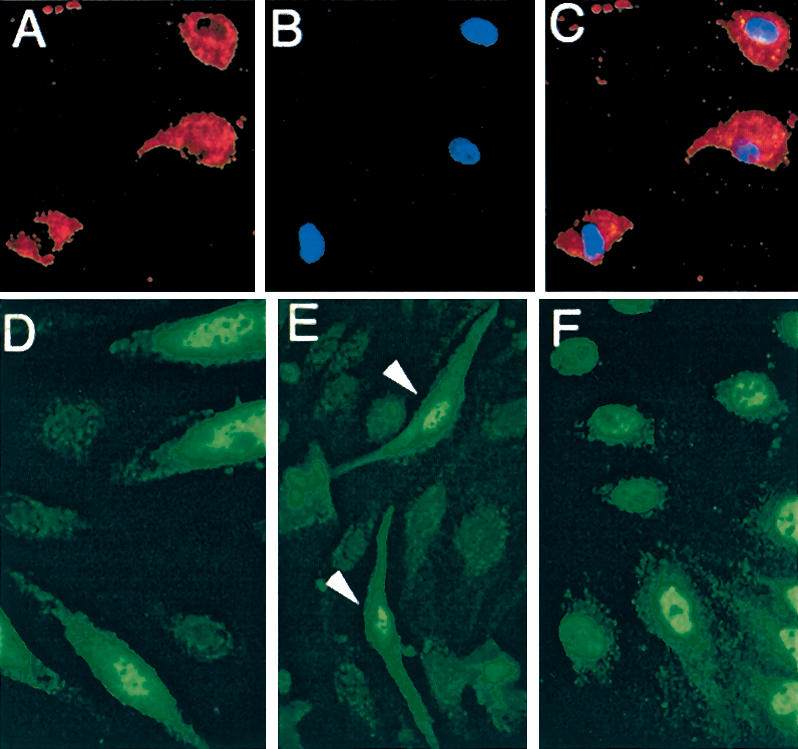
To determine whether loss of RhoB could explain the morphological defects in the retina and the reduced Akt expression observed in FTI-treated animals, we used an antisense strategy to specifically reduce RhoB expression. This strategy addressed whether RhoB loss acted in combination with the loss of other targets to mediate FTI-induced loss of Akt signaling, or whether loss of RhoB alone elicited by FTI was sufficient. FTI has complex effects on prenylation of various targets in cells (Prendergast and Oliff 2000). Because in endothelial cells FTI elicited a loss in steady-state levels of RhoB protein, we wished to confirm directly that loss of RhoB expression was a sufficient mechanism for mediating loss of Akt protein. Antisense morpholino oligonucleotides that are highly stable in cells were used to block expression of RhoB protein in primary endothelial cell cultures. The sequence of the specific rhoB antisense oligonucleotides used overlapped the ATG start codon (Fig. 5A). The oligonucleotides was also labeled with a fluorophore to document transfection efficiency (Morcos 2001), which approached 90% as judged by cell fluorescence for both the scrambled control oligonucleotides or the rhoB antisense oligonucleotides used in primary cell cultures (Fig. 5B,C). Control oligonucleotides tested included both a scrambled sequence and an inverted sequence for the RhoB. Relative to the control oligonucleotides, the rhoB antisense oligonucleotides strongly reduced RhoB protein levels in cells (Fig. 5D). This effect was specific insofar as the levels of the structurally related but functionally distinct RhoA and RhoC proteins were unaffected by the same treatment. To determine whether RhoB loss could lead to altered vessel morphology, we used an assay that mimics aspects of tube formation in vitro. When endothelial cells in a confluent monolayers are plated in a collagen gel matrix, the cells are stimulated to reorganize into cords. Endothelial cells treated with an antisense RhoB oliogonucleotide failed to properly realign when plated in three-dimensional collagen. They became highly refractile and appeared to be dying (Fig. 5F) even before cord formation was completed in the control cells (Fig. 5E). The same cells grown on top of a collagen matrix remained in a monolayers and demonstrated significantly less cell death but still failed to align into cords like the control cells (Fig. 5G,H). To investigate whether the refractile cells may indeed represent apoptosis following RhoB depletion, we used both the RhoB antisense and a RhoB(N19) dominant-negative construct in Western blot assays to measure PARP cleavage. For PARP analysis, cells were harvested 38 h after transfection and total cell lysates was prepared for Western blot. Both means to block RhoB function led to increased in PARP cleavage (Fig. 5I). Erk-1 was used as a loading control. In addition, cell-cycle analysis by FACS on the antisense morpholino and control oligonucleotides transfected cells showed a doubling of the number of cells in the sub-G1 fraction after 42 h, further suggesting that loss of RhoB leads to cell death (Fig. 5J).
Figure 5.

RhoB is critical for survival in endothelial cells. Acute knock-down of RhoB expression was performed in primary endothelial cell culture using FITC-labeled antisense morpholino oligos. (A) Antisense morpholino sequence. The transfection efficiency was determined by comparing FITC-labeled cells (B) 48 h after transfection cells with nuclei of the same field (C). Western blot of these cultures following antisense treatment showed loss of RhoB (D) without loss of RhoA, RhoC, or Actin expression. Tube formation assays were performed in three-dimensional collagen on cells transfected with control (E) or morpholino antisense-treated (F) cells. Less cell death but failed tube formation was also seen in two-dimensional cultures with control (G) or antisense-treated (H) cells. (I) In cultures transfected with either RhoB DN or antisense RhoB morpholino oligos, PARP cleavage was increased. (J) DNA content analysis revealed increased cell death quantitated as a sub-G1 fraction in morpholino transfected cultures.
RhoB colocalizes with the Akt at the nuclear margin of primary endothelial cells and partially colocalizes with phospho-Akt inside the nucleus
A specific physiological function in intracellular receptor trafficking has been defined previously for RhoB (Gampel et al. 1999). To ascertain whether RhoB influenced the trafficking of Akt in endothelial cells, we used confocal immunofluorescence microscopy to examine the localization of RhoB, Akt, and phosphorylated Akt in growing cultures of primary human microvascular endothelial cells. Consistent with observations made in other cell types, the endogenous RhoB protein in endothelial cells was localized primarily at the nuclear margin but to a lesser degree also inside the nucleus and at the vesicular and plasma membranes (Fig. 6A,E; Adamson et al. 1992; Lebowitz et al. 1995; Zalcman et al. 1995; Lebowitz and Prendergast 1998b; Michaelson et al. 2001). Whether RhoB was integral or proximal to the nuclear membrane was unclear. The monoclonal antibody used gives a single band in Western blots and was tested against extracts from the RhoB null mice to ensure its specificity (Fig. 6D). The endogenous Akt protein in endothelial cells was localized throughout the cell in both the cytoplasm and nucleus with slightly increased staining also on the nuclear margin (Fig. 6G). Notably, visualization of just the active phosphorylated isoform of Akt showed it to be localized chiefly to the nucleus and more weakly at the cell membrane in these cultures (Fig. 6C). The same results were obtained with a phosphospecific Akt antibody obtained from a different source and has been previously reported (Andjelkovic et al. 1997). Cytosolic Akt was visualized by an antibody that recognized both unphosphorylated and phosphorylated isoforms (antitotal Akt), but not by phosphospecific Akt antibodies (Fig. 6, cf. C and G). The secondary antibodies for the Akts (rabbit) and RhoB (mouse) were also tested and showed no signals (Fig. 6H shows secondary control for both secondary antibodies used, costained with DAPI to show that cells are present). Taken together, the results supported the conclusion that the active phosphorylated isoform of Akt is localized primarily to the nucleus of actively growing endothelial cells. The compartmentalization of phospho- and total Akt and the partial colocalization with RhoB at the nuclear margin and nucleoplasm supported the hypothesis that RhoB may play a role in nuclear trafficking of Akt as it does for other proteins (Gampel et al. 1999).
Figure 6.

RhoB colocalizes with phospho-Akt in endothelial cell nuclei. Double immunofluorescence with RhoB and phospho-Akt (A) and total Akt (E) were analyzed by confocal microscopy. Single staining for each antibody is shown as follows: (B) RhoB. (C) Phospho-Akt. (F) RhoB. (G) Total Akt. RhoB is clearly localized to cell membrane and nuclei, phospho-Akt is most abundant in the nucleus, and total Akt is most abundant in the cytoplasm but also on the nuclear margin. D shows the specificity of the RhoB antibody on extracts from wild-type and null mice and H shows secondary controls with Hoescht staining to demonstrate the presence of endothelial cells in the field. The antibodies used were the same anti-rabbit FITC and anti-mouse TRITC used in confocal analysis. (red) RhoB; (green) total or phospho-Akt.
Acute gene silencing or inhibition of RhoB function abolishes the nuclear accumulation of Akt in primary endothelial cells
To show that RhoB was required for the stability and nuclear trafficking of Akt, we documented the localization of Akt in cells where RhoB function was acutely disrupted by two distinct strategies. Gene silencing of RhoB in endothelial cells was done using morpholino antisense oligonucleotides and a dominant-negative (N19) RhoB construct lacking GTPase activity. These treatments markedly and specifically reduced the steady-state levels of phosphorylated and total Akt in nuclear and cytosolic fractions fractions, as documented by Western analysis (Fig. 7A). The nuclear levels were most significantly depleted. These results phenocopy the effect on Akt produced in FTI-treated yolk sacs. Loss of nuclear Akt was confirmed and extended in experiments where cells were transfected with the RhoB(N19) dominant-negative construct. The nuclear and cytosolic fractions were checked by confirming that tubulin was only found in the cytosolic fraction and that histone 4 was found in the nuclear fraction (Fig. 7B). Thus, the identification of Akt in nuclear fractions does not appear due to contamination of our nuclear fractions by cytosolic proteins. These data and the confocal analysis shown in Figure 6 clearly demonstrate that phosphorylated Akt is found in the nucleus of endothelial cells. Immunocytochemistry to total Akt also clearly shows the loss of total Akt from the cell nucleus, along with a rounding of the cells transfected with the HA-tagged RhoB(N19) compared to empty vector transfected controls (Fig. 7C). We noted that the HA-tagged RhoB also was primarily cytoplasmic, suggesting that perhaps the GTPase function is required for proper RhoB localization in the nucleus. The loss of phospho-Akt is usually used to indicate a block in Akt activation; however, total Akt levels usually remain constant, and total Akt is often used as a loading control in cytokine activation experiments. Following RhoB loss, however, loss of phospho-Akt may simply mirror the loss of total Akt levels without reflecting a specific block in Akt activation. To further address the mechanism of total Akt loss, we demonstrated that the observed reductions in total Akt were protesome dependent (Fig. 7D). This observation further supports the hypothesis that, in the absence of RhoB, Akt is improperly degraded. In summary, the results of these genetic and pharmacological experiments supported the conclusion that RhoB loss was a sufficient cause in eliciting Akt loss and cellular mislocalization in primary endothelial cells.
Figure 7.

RhoB inhibition blocks nuclear Akt accumulation. (A) Nuclear and cytosolic extracts made from human dermal primary endothelial cell culture treated with antisense morpholino RhoB and transfected with RhoB N19 were analyzed for phospho- and total Akt, with Actin for a loading control. (B) Controls for fractionation showed tubulin and histone 4 localization. (C) Transfection of primary endothelial cells with dominant-negative RhoB (N19) blocks Akt accumulation in transfected cells as evidenced by staining for HA-tag to identify transfected cells and total Akt to see nuclear exclusion. (D) Cells treated with antisense morpholino oligos were treated with vehicle or proteosome inhibitor and analyzed for total Akt levels to show proteosome-dependent degradation.
Akt nuclear localization is not dependent on phosphorylation
To determine whether Akt phosphorylation was essential to nuclear trafficking, we examined the localization of Akt proteins that were activated or inactivated at the levels of membrane recruitment or phosphorylation. PI-3′kinase is an upstream activator of Akt that operates by recruiting Akt to cellular membranes [via the pleckstrin homology (PH) membrane-binding domain present in Akt]. To probe the role of membrane binding in nuclear trafficking, we asked how trafficking was affected by inactivation of PI-3′kinase (which prevents membrane association) or N-myristoylation (which provides membrane association independent of PI-3′kinase activity). Inhibiting PI-3′kinase by serum deprival and the addition of wortmannin, a potent inhibitor of PI-3′kinase, caused endogenous Akt to be excluded from the nucleus of endothelial cells (Fig. 8A-C). Conversely, exogenous Akt that was localized to membranes by N-myristoylation was fully competent to localize to the nucleus of endothelial cells (Fig. 8D). To probe the role of Akt phosphorylation in nuclear trafficking, we asked how trafficking was affected by alanine or aspartate mutations at the key phosphoamino acid residues T308 and S473. These mutants retain wild-type PH domains and are therefore competent for membrane recruitment. Aspartate mutations, which mimic Akt phosphorylation, did not affect nuclear trafficking, as expected (Fig. 8E). However, alanine mutations, which prevent Akt phosphorylation and therefore its activation, also did not affect nuclear trafficking (Fig. 8F). This result argued that phosphorylation was coupled to nuclear trafficking, but not essential for it to occur, in contrast to membrane recruitment, which was essential. Thus, the mechanism for nuclear trafficking of Akt was distinct from the mechanism that mediated its phosphorylation. These data were compatible with the evidence that RhoB enabled nuclear trafficking of phospho-Akt through a mechanism that was coupled to Akt phosphorylation, but separable from it. In summary, the results indicated that the survival signal delivered by phospho-Akt in endothelial cells was coupled to a RhoB-dependent mechanism for nuclear trafficking of Akt in endothelial cells, rather than a block in Akt activation.
Figure 8.
Akt nuclear accumulation is linked but not dependent on phosphorylation. Serum-starved endothelial cells treated with wortmannin show exclusion of total Akt from the nuclei (A), which are labeled by Hoescht staining (B). (C) The fused image of Akt staining and Hoescht. Antibody staining of the HA tag of membrane-tethered myr-Akt (D), constitutively active Akt-DD (E), and phosphorylation-incompetent Akt-AAA (F) revealed that all of these forms of Akt were competent to enter the nucleus.
Discussion
Organ morphogenesis depends on stage-specific signaling pathways that govern the correct movement, division, and survival of cells. This study offers the first evidence of a survival signal that discriminates the earliest stages of blood vessel assembly from those used for subsequent blood vessel enlargement. Anti-angiogenesis agents that block proliferation and survival of endothelium have been described, but these agents do not similarly discriminate the survival signals required for vessel assembly and enlargement. For example, one of the best-studied angiogenic factors, VEGF-A, an important cytokine that has gained acceptance as a therapeutic target, is required for endothelial cell proliferation but also survival, migration, and permeability (Ferrara 2000). All forming and newly formed blood vessels depend on VEGF-A signaling. Thus, loss of the signaling from the VEGF-A pathway not only blocks new blood vessel formation, but induces regression and apoptosis of pre-formed blood vessels that have not established a stable interaction with perivascular pericytes (Benjamin et al. 1998, 1999). The stage-specific survival signal defined here is both unique and distinct from other survival signal(s) that act more universally during vascular development.
We linked this stage-specific process to a novel mechanism in the control of Akt, a key survival kinase, at the level of nuclear trafficking. Vascular development requires the integration of signals contributed by mechanical stresses, cytokines, cell-cell contacts, survival controls, glucose uptake, cell movement, and production of intercellular messengers. Akt has been shown to be a central player in survival, migration, and other processes critical to endothelial cell homeostasis controlled by these factors (Dimmeler et al. 1998; Gerber et al. 1998; Carmeliet et al. 1999; Fulton et al. 1999; Michell et al. 1999; Hermann et al. 2000; Kim et al. 2000; Morales-Ruiz et al. 2000; Edinger and Thompson 2002). In this study, we showed that stability and nuclear trafficking of activated Akt requires RhoB, a small GTPase that functions in intracellular vesicle trafficking (Gampel et al. 1999). Although Akt is likely to be important at multiple stages of vascular development, our findings suggest that survival signaling by Akt during the initial stages of blood vessel assembly is greatly influenced by RhoB. Previous studies have offered evidence that nuclear trafficking of Akt in other cell types is important for controlling cell survival, perhaps best illustrated by the regulation of the Forkhead/FOXO family of transcription factors in neuronal cells by Akt. For example, a recent study elegantly corroborated the hypothesis that phosphorylation of Forkhead by activated Akt leads to the active export of Forkhead from the nucleus. Neither Forkhead nor Akt encode canonical sequences that direct nuclear import or export, but the 14-3-3 chaperone was shown in the same study to be critical for nuclear export of Fork-head (Brunet et al. 2002). These observations strongly imply that Akt must first move to the nucleus so that it can phosphorylate Forkhead there. In primary endothelial cells, the nuclear localization of Akt is stimulated by growth factors, and the active phosphorylated form of Akt is virtually exclusively nuclear. In support of the evidence that RhoB was crucial to mediate nuclear trafficking of Akt, there was a close correlation in the localization of RhoB and Akt at the nuclear periphery and to a smaller extent, inside the nucleoplasm of serum-stimulated cells. As a novel mechanism of Akt control, nuclear trafficking may find relevance beyond primary endothelial cells, especially as a mechanism for influencing stage-specific cell survival during development.
RhoB may promote the delivery of phosphorylated Akt to the nucleus, or RhoB may impede the export of phosphorylated Akt from the nucleus after its delivery. Notably, phosphorylation of Akt was coupled to trafficking, but distinct from it: Mutations in Akt that affected phosphorylation did not affect its competence for nuclear trafficking. Membrane tethering of Akt was also coupled to nuclear trafficking, insofar as N-myristoylated Akt was competent to localize to the nucleus. Small GTPases act by recruiting complexes to membranes, so it is tempting to speculate that RhoB may recruit a complex containing Akt to an appropriate trafficking vesicle. However, further work is needed to discern the mechanism of nuclear accumulation of Akt, particularly given evidence that RhoB can antagonize the action of other Rho family GTPases that may influence vesicle trafficking (Ridley et al. 2001; Zeng et al. 2003).
The environment of endothelial cells undergoing sprouting angiogenesis differs markedly from the environment of endothelial cells present in mature blood vessels. For example, the endothelial cell in a sprouting segment is exposed to the surrounding tissue even before a basement membrane is laid, before lumens are formed, before perivascular support cells are recruited, and before blood circulation is initiated in that segment. Studies of the Akt signaling pathway in blood vessels have shown that Akt mediates cell-matrix or cell-cell survival signals provided by the engagement of integrins or VE-cadherins, respectively (Frisch and Ruoslahti 1997; Carmeliet et al. 1999; Fujio and Walsh 1999; Byzova et al. 2000) and also mediates survival signals triggered by the mechanical stresses produced by blood flow (Garcia-Cardena et al. 2000) and cell morphological changes consistent with sprouting phenotypes (Morales-Ruiz et al. 2000). However, many of these signals are not yet firmly established in endothelial cells actively sprouting. Thus, at the earliest stages of vessel assembly, other pathways are needed to overcome the relative deficiency in Akt stimulation. In this manner, RhoB's ability to increase Akt trafficking and stability may become rate limiting for survival of the sprouting cells. Once blood vessels have established basement membrane-integrin engagement and blood flow, the increase in quantitative inputs to Akt may relieve a need for RhoB. We sensed this dependency gradient in the tube formation assays: Cells growing in two-dimensional collagen without RhoB resulted in failed morphological rearrangements and some cell death, but in three dimensions, a much more stressful environment, lack of RhoB resulted in massive cell death before tube formation was even complete in the controls. This assay is very similar to a sprouting environment where an endothelial cell must leave the basement membrane and venture into a rich collagen environment (a situation relevant to both the developing retina and endocardial cushion; Gampel et al. 1999). Thus, increasing the efficiency of nuclear trafficking may compensate for the quantitatively poorer activation of Akt that occurs in the relatively barren survival environment of the sprouting endothelial cell. Because of the high degree of complexity inherent to subcellular trafficking, it seems highly likely that the mechanisms available to amplify (or attenuate) the Akt signal in the nucleus will be highly degenerate. If so, then plasticity or compensation in any one mechanism may be common during development, as appears to be the case in RhoB nullizygous mice that overcome their initial retinal retardation. Acute deprivation of Akt trafficking mechanisms may be required to identify their potential roles in stage-specific cell survival during development. Given the central, nodal quality of Akt in cell survival signaling, identifying additional elements that modulate Akt trafficking may lead to additional insights into how complex organisms confer developmental specificity on what is a ubiquitous signaling pathway.
There may be significant clinical benefits from molecularly separating the cell survival pathways needed during blood vessel assembly versus enlargement. For example, macular degeneration and diabetic retinopathy, two of the most common causes of age-associated blindness, are characterized by inappropriate sprouting angiogenesis in the retina. The greatest damage to vision in these diseases is caused by the unstable nature of angiogenic vessels that sprout into the vitreous fluid or neural retina causing bleeding and edema. Existing therapies are surgical in nature, but anti-angiogenic drugs have been suggested as a method to relieve morbidity (Duh and Aiello 1999). Drugs that ablate sprouting endothelial cells, yet permit existing blood vessels to enlarge, may be particularly effective in this regard, because they will kill the endothelial cells in new vessels while promoting blood flow through existing vessels to relieve retinal ischemia. On the basis of our observations, we suggest that FTIs may be useful to treat these common types of age-associated blindness. Although originally developed to target Ras proteins, the function of the K-Ras and N-Ras proteins are not targeted as planned because of alternate prenylation in drug-treated cells (possibly explaining the nontoxic nature of many FTIs tested; Prendergast and Oliff 2000). Consistent with other studies of the particular FTI used above (e.g., Kohl et al. 1995), we found that systemic application was highly effective in reducing RhoB levels in endothelial cells in the absence of apparent side effects. Phase I trials of other clinically tested FTIs have shown them to be similarly well tolerated (Prendergast and Rane 2001). FTIs may be useful to block sprouting angiogenesis in other settings, including cancer, where the role for various FTI target proteins has remained a source of debate (Cox and Der 2002). The demonstration that FTIs can modulate RhoB and Akt signaling in endothelial cells supports the notion that RhoB is an important target for understanding FTI action. It may be useful to identify other pathophysiological settings where selective anti-angiogenesis via RhoB targeting is desirable.
Materials and methods
Endothelial cell culture
Primary human dermal microvascular endothelial cells were isolated as previously described (Richard et al. 1998) and grown in EGM-2MV (Cambrex), treated at passage 4-6, and used for antisense and transfection studies.
Subcellular localization
Endothelial cells were grown on coverslip precoated with collagen in complete medium. While the cells were 80% subconfluent they were fixed with 4% paraformaldehyde or cold methanol for 10 min, followed by incubation with 1% Triton X-100 for 10 min at room temperature and blocking with 1% FCS and 0.1% donkey serum for 30 min at room temperature. Primary antibodies used were: Rabbit anti-PKB/Akt (pT473; Biosource), mouse-anti-RhoB (Santa Cruz), Rabbit anti-Akt (Santa Cruz), and rat anti HA-taq (Roche). These were incubated at room temperature for 30 min. After washing three times with cold PBS, the cells were incubated with appropriate secondary antibody conjugated to FITC or TRITC. Coverslips were washed and mounted in a mixture (9:1) of glycerol and PBS containing 1% propyl gallate. Slides were examined by confocal microscopy.
Morpholino antisense transfection
While the cells were confluent, 1 µM of Morpholino antisense RhoB oligo or standard control Morpholino (Gene Tools) was incubated with transfection medium for 20 min at room temperature. The nucleotide sequences of the hRhoB oligo is shown in Figure 5, and the sequences of the control oligos were 5′-CCTCTTACCTCAGTTACAATTTATA-3′ (scrambled) and 5′-CCGCTCGCCCTCCCGTTACCGCCG-3′ (inverted hRhoB). Transfection was performed for 3 h in serum-free medium and then the cells were washed and returned to complete medium for 24-48 h (Morcos 2001).
Nucleofector transfection
Dermal microvascular endothelial cells were grown on flasks precoated with collagen-1 in EGM-2-MV complete medium. After 4 d, the cells were harvested with trypsin-EDTA and washed. For each 1 × 106 cells, 100 µL Nucleofector with 2 µg DNA was used. The samples were transferred into Amaxa certified cuvettes (Amaxa Biosystems) and program S-05 was used. The cells were then removed from the cuvette and plated into a precoated six-well tissue culture plate for 48 h.
Western blots
For studies on nuclear and cytosolic expression of Akt, fractions were isolated using hypotonic lysis buffer (10 mM HEPES, 10 mM KCl, 3 mM MgCl, 0.05% NP-40, 1 mM EDTA) and followed by low-speed centrifugation and further extraction of supernatant with 2× GSLB (cytosol) and pellet with 1× GSLB (nuclear). All buffers contain protease inhibitors. GSLB contains 50 mM HEPES, 250 mM KCl, 0.1% NP-40, 0.1 mM EDTA, 0.1 mM EGTA, 10% glycerol. Protein was quantitated and 40 µg were run on 10% PAGE, blotted, and incubated with Rabbit anti-phospho-AKT p-Ser472/473/474 (Pharmingen) and Rabbit antitotal Akt (Santa Cruz). Other antibodies used for Western blot analysis included: PKB/Akt (pT473; Cell Signaling), antitotal Akt (H-136; Santa Cruz Biotechnology), phospho-forkhead (FKHR; ser 256; Cell Signaling), antiPARP (Oncogene), anti-HRas, mouse-anti-RhoB and anti-RhoA (Santa Cruz Biotechnology) anti-β-actin (clone 15; Sigma), anti-alpha tubulin (Sigma), anti-histone 4 (Santa Cruz Biotechnology), and ERK-1 (K-23; Santa Cruz Biotechnology). Pierce detection reagents were used to visualize HRP-labeled secondary antibodies.
Yolk sac analysis
Timed pregnant mice were injected with FTI as described below one time at E8. Embryos were harvested 24 h later and carefully dissected in cold PBS. Yolk sacs were put into protein lysis buffer with protease inhibitors. Proteins were quantitated and run on standard Western blots.
Retina whole mounts
Following daily I.P. injections of 35 mg/kg FTI L-744, 832 treatments or DMSO vehicle to 10 newborn rats (Wistar), animals were sacrificed and eyes removed into formalin. Eyes from RhoB null mice and strain-matched controls were similarly taken into formalin. Retinas were dissected and incubated with Lectin BS-1 TRITC (Sigma) overnight at 4°C. Four to five 1-h washes in PBS were performed before making incisions into the cup for flattening on a glass slide. Conventional fluorescent microscopy in coordination with a Leica DC200 digital camera was used to capture the images.
Immunohistochemistry
Eyes were fixed in formalin overnight and embedded in paraffin. Sections (4 µm) were depariffinated 2 × 15 min using xylene-free solvent (Citrisolv, Fisher). TUNEL was performed according to protocol for paraffin sections from Clontech (apoalert DNA fragmentation kit). Blood vessels were visualized following overnight incubation in Lectin BS-1 conjugated to TRITC (Sigma). PCNA antibody (Zymed) was used to demarcate proliferating cells. Nuclei were labeled by including Hoechst in the mounting medium (Dako, antifade). Images were captured using the Leica DC200 digital camera and imported into Adobe Photoshop 5.0.
Proteosome inhibition
Microvascular endothelial cells were treated with Morpholino as described previously. After 24 h, 0.01 ng/mL proteasome inhibitor (Lactacystin, Synthetic Boston Biochem) were added for overnight at 4°C. As control we added DMSO in the same volume. Then the cells were lysed with RIPA buffer containing protease inhibitor cocktail (Sigma) on ice for 30 min and then centrifuged for 10 min at high speed.
DNA content analysis
Microvascular endothelial cells were treated with Morpholino as described previously. After 40 h, the cells were trypsinized and 5.0 × 106 cells were fixed in 100% ethanol overnight at -20°C. Then the cells were centrifuged and resuspended in DNA staining buffer [PBS with 0.1% Triton X-100, 0.1 mM EDTA at pH 7.4, 0.05 mg/mL RNaseA (50 U/mL)] and incubated for 1 h at room temperature. The 10,000 cells were counted by FACS (FACSSCalibur; Becton Dickinson) and analyzed by Cell Quest.
Collagen gel assays
Microvascular endothelial cells were treated with Morpholino antisense and control oligonucleotides as described previously. For three-dimensional assays, the cells were seeded on 1 mg/mL Collagen plugs (type 1 rat tail; BD Biosciences) and overlaid with a second plug of 1 mg/mL Collagen I after 1.5 h. For two-dimensional assays, cells were plated on top of 0.5 mg/mL Collagen I. Cell images were captured following tube formation using the Leica DC200 digital camera and imported into Adobe Photoshop 5.0.
Acknowledgments
We thank R. Kerbel for discussions about the effects of FTIs on angiogenesis, A. Toker for Akt reagents, and C. Perruzzi for technical assistance. This work was supported by research grants to L.E.B. from the American Diabetes Association and NIH (HL071049), and to G.C.P. from the NIH (CA82222).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1134603.
References
- Adamson P., Paterson, H.F., and Hall, A. 1992. Intracellular localization of the P21rho proteins. J. Cell Biol. 119**:** 617-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aepfelbacher M., Essler, M., Huber, E., Sugai, M., and Weber, P.C. 1997. Bacterial toxins block endothelial wound repair. Evidence that Rho GTPases control cytoskeletal rearrangements in migrating endothelial cells. Arterioscler. Thromb. Vasc. Biol. 17**:** 1623-1629. [DOI] [PubMed] [Google Scholar]
- Alon T., Hemo, I., Itin, A., Pe'er, J., Stone, J., and Keshet, E. 1995. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1**:** 1024-1028. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M., Alessi, D.R., Meier, R., Fernandez, A., Lamb, N.J., Frech, M., Cron, P., Cohen, P., Lucocq, J.M., and Hemmings, B.A. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272**:** 31515-31524. [DOI] [PubMed] [Google Scholar]
- Benjamin L.E., Hemo, I., and Keshet, E. 1998. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF- B and VEGF. Development 125**:** 1591-1598. [DOI] [PubMed] [Google Scholar]
- Benjamin L.E., Golijanin, D., Itin, A., Pode, D., and Keshet, E. 1999. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal [see comments]. J. Clin. Invest. 103**:** 159-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J., Arden, K.C., Blenis, J., and Greenberg, M.E. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96**:** 857-868. [DOI] [PubMed] [Google Scholar]
- Brunet A., Datta, S.R., and Greenberg, M.E. 2001. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11**:** 297-305. [DOI] [PubMed] [Google Scholar]
- Brunet A., Kanai, F., Stehn, J., Xu, J., Sarbassova, D., Frangioni, J.V., Dalal, S.N., DeCaprio, J.A., Greenberg, M.E., and Yaffe, M.B. 2002. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 156**:** 817-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byzova T.V., Goldman, C.K., Pampori, N., Thomas, K.A., Bett, A., Shattil, S.J., and Plow, E.F. 2000. A mechanism for modulation of cellular responses to VEGF: Activation of the integrins. Mol. Cell 6**:** 851-860. [PubMed] [Google Scholar]
- Camper-Kirby D., Welch, S., Walker, A., Shiraishi, I., Setchell, K.D., Schaefer, E., Kajstura, J., Anversa, P., and Sussman, M.A. 2001. Myocardial Akt activation and gender: Increased nuclear activity in females versus males. Circ. Res. 88**:** 1020-1027. [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Lampugnani, M.G., Moons, L., Breviario, F., Compernolle, V., Bono, F., Balconi, G., Spagnuolo, R., Oostuyse, B., Dewerchin, M., et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98**:** 147-157. [DOI] [PubMed] [Google Scholar]
- Cox A.D. and Der, C.J. 2002. Farnesyltransferase inhibitors: Promises and realities. Curr. Opin. Pharmacol. 2**:** 388-393. [DOI] [PubMed] [Google Scholar]
- Dimmeler S., Assmus, B., Hermann, C., Haendeler, J., and Zeiher, A.M. 1998. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: Involvement in suppression of apoptosis. Circ. Res. 83**:** 334-341. [DOI] [PubMed] [Google Scholar]
- Duh E. and Aiello, L.P. 1999. Vascular endothelial growth factor and diabetes: The agonist versus antagonist paradox. Diabetes 48**:** 1899-1906. [DOI] [PubMed] [Google Scholar]
- Edinger A.L. and Thompson, C.B. 2002. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 13**:** 2276-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. 2000. VEGF: An update on biological and therapeutic aspects. Curr. Opin. Biotechnol. 11**:** 617-624. [DOI] [PubMed] [Google Scholar]
- Frisch S.M. and Ruoslahti, E. 1997. Integrins and anoikis. Curr. Opin. Cell. Biol. 9**:** 701-706. [DOI] [PubMed] [Google Scholar]
- Fujio Y. and Walsh, K. 1999. Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 274**:** 16349-16354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D., Gratton, J.P., McCabe, T.J., Fontana, J., Fujio, Y., Walsh, K., Franke, T.F., Papapetropoulos, A., and Sessa, W.C. 1999. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399**:** 597-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampel A., Parker, P.J., and Mellor, H. 1999. Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr. Biol. 9**:** 955-958. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G., Anderson, K.R., Mauri, L., and Gimbrone Jr., M.A. 2000. Distinct mechanical stimuli differentially regulate the PI3K/Akt survival pathway in endothelial cells. Ann. NY Acad. Sci. 902**:** 294-297. [DOI] [PubMed] [Google Scholar]
- Gerber H.P., McMurtrey, A., Kowalski, J., Yan, M., Keyt, B.A., Dixit, V., and Ferrara, N. 1998. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 273**:** 30336-30343. [DOI] [PubMed] [Google Scholar]
- Henderson D.J., Ybot-Gonzalez, P., and Copp, A.J. 2000. RhoB is expressed in migrating neural crest and endocardial cushions of the developing mouse embryo. Mech. Dev. 95**:** 211-214. [DOI] [PubMed] [Google Scholar]
- Hermann C., Assmus, B., Urbich, C., Zeiher, A.M., and Dimmeler, S. 2000. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler. Thromb. Vasc. Biol. 20**:** 402-409. [DOI] [PubMed] [Google Scholar]
- Jiang K., Coppola, D., Crespo, N.C., Nicosia, S.V., Hamilton, A.D., Sebti, S.M., and Cheng, J.Q. 2000. The phosphoinositide 3-OH kinase/AKT2 pathway as a critical target for farnesyltransferase inhibitor-induced apoptosis. Mol. Cell. Biol. 20**:** 139-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Kim, H.G., So, J.N., Kim, J.H., Kwak, H.J., and Koh, G.Y. 2000. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ. Res. 86**:** 24-29. [DOI] [PubMed] [Google Scholar]
- Kohl N.E., Omer, C.A., Conner, M.W., Anthony, N.J., Davide, J.P., deSolms, S.J., Giuliani, E.A., Gomez, R.P., Graham, S.L., Hamilton, K., et al. 1995. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat. Med. 1**:** 792-797. [DOI] [PubMed] [Google Scholar]
- Lebowitz P.F. and Prendergast, G.C. 1998a. Functional interaction between RhoB and the transcription factor DB1. Cell Adhes. Commun. 6**:** 277-287. [DOI] [PubMed] [Google Scholar]
- ____. 1998b. Non-Ras targets of farnesyltransferase inhibitors: Focus on Rho. Oncogene 17**:** 1439-1445. [DOI] [PubMed] [Google Scholar]
- Lebowitz P.F., Davide, J.P., and Prendergast, G.C. 1995. Evidence that farnesyltransferase inhibitors suppress Ras transformation by interfering with Rho activity. Mol. Cell. Biol. 15**:** 6613-6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebowitz P.F., Casey, P.J., Prendergast, G.C., and Thissen, J.A. 1997. Farnesyltransferase inhibitors alter the prenylation and growth-stimulating function of RhoB. J. Biol. Chem. 272**:** 15591-15594. [DOI] [PubMed] [Google Scholar]
- Liu A. and Prendergast, G.C. 2000. Geranylgeranylated RhoB is sufficient to mediate tissue-specific suppression of Akt kinase activity by farnesyltransferase inhibitors. FEBS Lett. 481**:** 205-208. [DOI] [PubMed] [Google Scholar]
- Liu A., Du, W., Liu, J.P., Jessell, T.M., and Prendergast, G.C. 2000. RhoB alteration is necessary for apoptotic and antineoplastic responses to farnesyltransferase inhibitors. Mol. Cell. Biol. 20**:** 6105-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay D.J. and Hall, A. 1998. Rho GTPases. J. Biol. Chem. 273**:** 20685-20688. [DOI] [PubMed] [Google Scholar]
- Michaelson D., Silletti, J., Murphy, G., D'Eustachio, P., Rush, M., and Philips, M.R. 2001. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell. Biol. 152**:** 111-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell B.J., Griffiths, J.E., Mitchelhill, K.I., Rodriguez-Crespo, I., Tiganis, T., Bozinovski, S., de Montellano, P.R., Kemp, B.E., and Pearson, R.B. 1999. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 9**:** 845-848. [DOI] [PubMed] [Google Scholar]
- Morales-Ruiz M., Fulton, D., Sowa, G., Languino, L.R., Fujio, Y., Walsh, K., and Sessa, W.C. 2000. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ. Res. 86**:** 892-896. [DOI] [PubMed] [Google Scholar]
- Morcos P.A. 2001. Achieving efficient delivery of morpholino oligos in cultured cells. Genesis 30**:** 94-102. [DOI] [PubMed] [Google Scholar]
- Prendergast G.C. 2001. Actin' up: RhoB in cancer and apoptosis. Nat. Rev. Cancer 1**:** 162-168. [DOI] [PubMed] [Google Scholar]
- Prendergast G.C. and Oliff, A. 2000. Farnesyltransferase inhibitors: Antineoplastic properties, mechanisms of action, and clinical prospects. Semin. Cancer Biol. 10**:** 443-452. [DOI] [PubMed] [Google Scholar]
- Prendergast G.C. and Rane, N. 2001. Farnesyltransferase inhibitors: Mechanism and applications. Expert Opin. Investig. Drugs 10**:** 2105-2116. [DOI] [PubMed] [Google Scholar]
- Richard L., Velasco, P., and Detmar, M. 1998. A simple immunomagnetic protocol for the selective isolation and long-term culture of human dermal microvascular endothelial cells. Exp. Cell. Res. 240**:** 1-6. [DOI] [PubMed] [Google Scholar]
- Ridley A.J. 2001. Rho proteins: Linking signaling with membrane trafficking. Traffic 2**:** 303-310. [DOI] [PubMed] [Google Scholar]
- Risau W. 1997. Mechanisms of angiogenesis. Nature 386**:** 671-674. [DOI] [PubMed] [Google Scholar]
- Schaper W. and Buschmann, I. 1999. Arteriogenesis, the good and bad of it. Cardiovasc. Res. 43**:** 835-837. [DOI] [PubMed] [Google Scholar]
- Zalcman G., Closson, V., Linares-Cruz, G., Lerebours, F., Honore, N., Tavitian, A., and Olofsson, B. 1995. Regulation of Ras-related RhoB protein expression during the cell cycle. Oncogene 10**:** 1935-1945. [PubMed] [Google Scholar]
- Zeng P.Y., Rane, N., Du, W., Chintapalli, J., and Prendergast, G.C. 2003. Role for RhoB and PRK in the suppression of epithelial cell transformation by farnesyltransferase inhibitors. Oncogene 22**:** 1124-1134. [DOI] [PubMed] [Google Scholar]