Leukocytes navigate by compass: roles of PI3Kγ and its lipid products (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 10.
Abstract
Morphologic polarity is necessary for the motility of mammalian cells. In leukocytes responding to a chemoattractant, this polarity is regulated by activities of small Rho guanosine triphosphatases (Rho GTPases) and the phosphoinositide 3-kinases (PI3Ks). Moreover, in neutrophils, lipid products of PI3Ks appear to regulate activation of Rho GTPases, are required for cell motility and accumulate asymmetrically to the plasma membrane at the leading edge of polarized cells. By spatially regulating Rho GTPases and organizing the leading edge of the cell, PI3Ks and their lipid products could play pivotal roles not only in establishing leukocyte polarity but also as compass molecules that tell the cell where to crawl.
Chemotaxis – the directed movement of cells in a gradient of chemoattractant1 – allows leukocytes to seek out sites of inflammation and infection, neurons to send projections to specific regions of the brain and amoebas of Dictyostelium discoideum to locate prey. In each case, chemoattractant-induced activation of spatially localized cellular signals causes protrusion of the cell plasma membrane towards the highest concentration of chemoattractant1,2. During chemotaxis, filamentous actin (F-actin) is polymerized asymmetrically at the up-gradient edge of the cell, providing the necessary force to thrust projections of the plasma membrane in the proper direction1,2. In this way, highly motile cells such as neutrophilic leukocytes (neutrophils) respond to chemoattractants by adopting a typical polarized morphology, with an F-actin-enriched leading edge facing up the gradient (Fig. 1).
FIGURE 1.
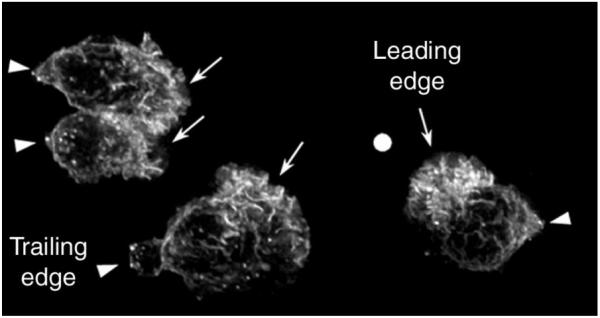
F-actin accumulation in chemoattractant-stimulated human neutrophils. When exposed to a chemotactic gradient, neutrophils polymerize F-actin asymmetrically to their pole facing the highest concentration of chemoattractant. In this case, human neutrophils were exposed to a gradient of f-Met-Leu-Phe (fMLP) delivered from a micropipette tip (shown by a white dot), allowed to polarize, fixed and their F-actin was revealed by fluorescence microscopy. These neutrophils exhibit a typical polarized morphology with a trailing edge (arrow heads) and an F-actin-enriched leading edge (arrows) facing up the gradient.
To adopt this highly polarized polarity and migrate up the chemoattractant gradient, cells must know where to establish a leading edge. Neutrophils, for instance, can polarize and move up very shallow gradients – that is, with a chemoattractant concentration ~2% higher at the front than the back1. To restrict actin polymerization to the leading edge in such a shallow gradient, the compass mechanism must create a much steeper internal gradient of regulatory signals – but not by altering the local abundance of chemoattractant receptors, which remain uniformly distributed on surfaces of neutrophils and Dictyostelium3,4. Here, we propose models to explain the compass mechanism, focusing on recent evidence that the internal signalling gradient is controlled by spatially specific regulation of the metabolism of phosphoinositide 3′-phosphates at the plasma membrane. This topic has also been reviewed in greater detail elsewhere5.
Chemotactic signals regulate Rho GTPases and PI3Ks
In neutrophils, chemoattractants stimulate G-protein-coupled receptors (GPCRs), which in turn activate a trimeric G protein, Gi, releasing the Gi βγ heterodimer from inhibition by αi (Fig. 2a). Gβγ regulates most of the known pathways activated by chemoattractants in leukocytes. Experimental manipulations that inactivate Gβγ6 or inhibit activities of several downstream effectors of Gβγ impair leukocyte motility in response to chemoattractants (Fig. 2a); these effectors include: PI3Ks7,8, guanosine triphosphatases of the Rho family (Rho GTPases)9,10, p38-mitogen activated protein (MAP) kinase11, protein kinase C ζ (PKCζ)12, cytosolic tyrosine kinases13 and cytosolic phospholipase A2 (cPLA2)14. In most cases, we do not know whether these signalling elements regulate polarity per se or are required more generally for normal cell motility.
FIGURE 2.

Overview of chemotactic signalling in neutrophils. (a) Several signalling pathways could regulate neutrophil motility. Chemoattractants, such as f-Met-Leu-Phe (fMLP), trigger signalling by activating their specific G-protein-coupled receptors (GPCRs) and Gi proteins at the surface of neutrophils, leading to release of the Gβγ subunit, which in turn activates directly or indirectly numerous signalling pathways in neutrophils, several of which have been suggested to regulate neutrophil motility (dotted arrows). (b) Putative signalling pathway regulating chemoattractant-induced activation of Rho GTPases in neutrophils. The Gβγ subunit, freed from the activated heterotrimeric G protein, activates phosphoinositide 3-kinase gamma (PI3Kγ) and cytosolic tyrosine kinases. The combined actions of tyrosine kinases and the PI3Kγ lipid products, phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)_P_3] and/or phosphatidylinositol (3,4)-bisphosphate [PtdIns(3,4)_P_2], increase the activity of a specific GDP–GTP exchange factor (GEF; e.g. Vav) for the Rho GTPases. The activated GEF in turn activates the Rho GTPases Rac and Cdc42 that ultimately transmit signals to the actin-polymerization machinery. Abbreviations: cPKC, classical protein kinase C; MAPK, mitogen-activated protein kinase; PLA2, phospholipase A2; PLCβ, phospholipase C β.
Because morphologic polarity directly reflects local increases in F-actin at the leading edge, some of the key signalling pathways involved probably regulate polymerization or organization of actin assemblies. Among these, the Rho GTPases Rac and Cdc42 play especially important roles in relaying signals to the actin cytoskeleton in leukocytes and many other cells15,16. Mutational inactivation of Rac2, the predominant Rac isoform in neutrophils, impairs chemoattractant-induced cell migration, F-actin polymerization and development of polarity17,18, and dominant-negative forms of Rac or Cdc42 impair the same functions in leukocyte cell lines9,10.
Recent studies8,19-22 of signalling to the actin cytoskeleton point also to key roles (Fig. 2b) for PI3Ks – a family of regulated phosphoinositide kinases that synthesize specific 3′-phosphoinositides at the inner leaflet of the plasma membrane (Box 1). Pharmacological inhibitors or dominant-negative mutants of PI3Ks decrease the fraction of neutrophils that polarize in response to chemoattractants8, attenuate chemoattractant-induced activation of Rac and Cdc42 in neutrophils19,20 and reduce Rac and Cdc42-dependent remodelling of actin assemblies in cultured cell lines expressing recombinant chemoattractant receptors21,22. Moreover, PI3K inhibitors reduce chemoattractant-induced motility of four kinds of leukocyte, including neutrophils7,8, macrophages23, T cells24 and natural killer cells25.
BOX 1 – CLASS I PHOSPHOINOSITIDE 3-KINASES.
The main phosphoinositide 3-kinase (PI3K) lipid product, phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)_P_3] is produced at the plasma membrane by two classes of PI3K in neutrophils, class 1A (PI3K α, PI3K β and PI3K δ) and class 1B (PI3K γ). In response to stimulation by different agonists, all class I PI3Ks phosphorylate the 3′ position of the inositol ring of phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)_P_2] to create PtdIns(3,4,5)_P_3a. PI3Kγ, the only known member of class 1B, is mainly activated by G-protein-coupled receptors (GPCRs)a, through the release of Gβγ from the heterotrimeric G protein. This PI3K isoform is essential for PI3K lipid product formation in neutrophilsb-d. PI3Kγ consists of a 110-kDa catalytic subunit and an adaptor molecule, p101, which is thought to confer Gβγ sensitivity to p110γ (Fig. I)a. Class 1A PI3Ks, consisting of a regulatory subunit (p85) and catalytic subunit (p110), are indirectly activated by tyrosine kinase receptors and cytosolic tyrosine kinases through recruitment of their regulatory p85 subunit to phosphorylated tyrosine residues on activated receptor or kinase substrates at the plasma membranea. Recent evidence has demonstrated that one member of class 1A, PI3Kβ, might also be activated by Gβγ liberated from GPCR-activated heterotrimeric G-proteinse.
Once formed, the PI3K lipid product PtdIns(3,4,5)_P_3 can be dephosphorylated by inositol polyphosphate 5′-phosphatases (SHIP) to form PtdIns(3,4)_P_2, which also acts as a second messenger, sharing some targets with PtdIns(3,4,5)_P_3, including the protein kinase Aktf,g. Other PI3K effectors, such as the Bruton tyrosine kinase (Btk) or the GDP–GTP exchange factor (GEF) Grp1, bind only to PtdIns(3,4,5)_P_3h. Another lipid phosphatase, PTEN, removes the 3′ phosphate added by PI3K and therefore inhibits PI3K-dependent signalling pathwaysi.
FIGURE I.

Class I phosphoinositide 3-kinases and the fate of their lipid product.
References
- a.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim. Biophys. Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- b.Sasaki T, et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- c.Li Z, et al. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- d.Hirsch E, et al. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- e.Maier U, et al. Roles of non-catalytic subunits in Gβγ-induced activation of class I phosphoinositide 3-kinase isoforms β and γ. J. Biol. Chem. 1999;274:29311–29317. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- f.Helgason CD, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- g.Coffer PJ, et al. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- h.Blomberg N, et al. The PH superfold: a structural scaffold for multiple functions. Trends Biochem. Sci. 1999;24:441–445. doi: 10.1016/s0968-0004(99)01472-3. [DOI] [PubMed] [Google Scholar]
- i.Nakashima N, et al. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3- L1 adipocytes. J. Biol. Chem. 2000;275:12889–12895. doi: 10.1074/jbc.275.17.12889. [DOI] [PubMed] [Google Scholar]
Although it is clear that inhibition of PI3Ks reduces activation of Rho GTPases19,20, the underlying molecular mechanisms are understood only in broad outline. A proposed regulatory network (Fig. 2b) relates lipid products of PI3Ks to activities of cytosolic tyrosine kinases and GDP–GTP exchange factors (GEFs). The activity of Vav, a specific GEF for Rho GTPases, is greatly increased upon its tyrosine phosphorylation by a cytosolic tyrosine kinase and upon addition, in vitro, of the PI3K lipid products phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)_P_3] and phosphatidylinositol (3,4)-bisphosphate [PtdIns(3,4)_P_2 (Ref. 26)]. Pharmacological inhibition of tyrosine kinases in intact neutrophils substantially reduces activation of Rac and Cdc4219.
PI3K lipid products: indicators of signalling polarity in motile cells
Both neutrophils and Dictyostelium amoebas can create gradients of PI3K-generated signals across their surfaces that are steeper than the extracellular gradient of chemoattractant and that closely parallel cell polarity and accumulation of F-actin. The steep intracellular gradients were detected in experiments that used recombinant green-fluorescent protein (GFP)-tagged pleckstrin homology (PH) domains – small modules of ~120 amino acids that associate with specific membrane-bound phospholipids27. Several of these fluorescent probes accumulate asymmetrically in the plasma membrane at leading edges of polarized HL-60 cells, a neutrophil-like cell line28 (Fig. 3a-b), and of Dictyostelium amoebas29,30 responding to chemoattractants. One such PH domain, that of the protein kinase Akt/PKB, binds to the PI3K lipid products PtdIns(3,4,5)_P_3 or PtdIns(3,4)_P_2 with high affinity, so that a GFP-tagged version of this PH domain (PHAKT–GFP) probably serves as an accurate probe for localized agonist-dependent accumulation of phospholipids generated by PI3Ks31. Indeed, like PHAKT–GFP (Fig. 3a-b), PtdIns(3,4,5)_P_3 detected by a specific antibody localizes at the leading edge of chemoattractant-stimulated HL-60 cells (Fig. 3c). Thus, it is likely that accumulation of PtdIns(3,4,5)_P_3 at the leading edge of motile cells reflects a signal amplification mechanism similar to the mechanism that localizes F-actin.
FIGURE 3.

Localization of lipid products of PI3Ks in chemoattractant-stimulated HL-60 cells. (a) Cells expressing the pleckstrin-homology domain of the protein kinase AKT/PKB fused to green-fluorescent protein (PHAKT–GFP) migrate in a gradient of f-Met-Leu-Phe (fMLP) delivered from a micropipette tip (located outside of the field of view in the top left corner). Fluorescence microscopy shows PHAKT–GFP predominantly localized at the plasma membrane at the leading edge (arrows). Since PHAKT–GFP binds selectively to the phosphoinositide 3-kinase (PI3K) lipid products phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)_P_3] and/or phosphatidylinositol (3,4)-bisphosphate [PtdIns(3,4)_P_2] at the plasma membrane, this indicates that one or both of these phospholipids accumulates at the leading edge of polarized neutrophils. (b) Even under conditions in which all the receptors are detecting the same ambient concentration of chemoattractant, after a uniform stimulation with fMLP, the cells polarize and show asymmetric accumulation of PHAKT–GFP at the leading edge (arrow). (c) PtdIns(3,4,5)_P_3 distribution on a chemoattractant-stimulated cell is revealed with the use of a specific antibody (from Echelon Research Laboratories, UT, USA) that does not bind to PtdIns(3,4)_P_2. In parallel to results obtained with PHAKT–GFP, this phospholipid is seen only at the leading edge of polarized cells (arrows). Bars, 15 μm.
In principle, PtdIns(3,4,5)_P_3 could be localized asymmetrically to the leading edge of the cell by creating asymmetric distributions of PI3K substrates, PI3K activities or activities of proteins that degrade PtdIns(3,4,5)_P_3 (e.g. the PTEN or SHIP phosphatases; see Box 1). An asymmetric distribution of PtdIns(4,5)_P_2, the principal PI3K substrate, is not responsible for asymmetric signalling, as indicated by the uniform distribution throughout the cell membrane (G. Servant, unpublished) of a GFP-tagged probe for this phospholipid, the PH domain of phospholipase Cδ (Ref. 32). We do not know whether asymmetric distribution of PtdIns(3,4,5)_P_3 reflects spatially controlled regulation of synthesis or degradation of the phospholipid. Overexpression of PTEN reduces fibroblast motility in a wound-healing assay by decreasing the concentration of PtdIns(3,4,5)_P_3 at the plasma membrane33; conversely, fibroblasts and myeloid cells from mice deficient in PTEN and SHIP, respectively, exhibit enhanced motility34,35.
The PI3Kγ isozyme is key – but not the whole story
Three laboratories recently reported36-38 that genetic disruption of the PI3Kγ isozyme produces mice with neutrophils that show defective migration responses to chemoattractant in vitro and impaired accumulation at sites of inflammation in vivo. PI3Kγ-deficient mouse neutrophils produced little or no PtdIns(3,4,5)_P_3 after stimulation with chemoattractants such as the tripeptide formyl-Met-Leu-Phe (fMLP), interleukin 8, or complement factor 5a36-38. These neutrophils showed 50–70% reductions in their capacity for migration in models of inflammation, in vitro or in vivo36-38. Thus, despite the apparent loss of PtdIns(3,4,5)_P_3 synthesis in PI3Kγ-deficient neutrophils, mechanisms independent of PI3Kγ must compensate to a substantial degree in regulating the motility response to chemoattractants. Similarly, one of these studies reported37 that the PI3Kγ knockout does not prevent chemoattractant-induced polymerization of actin or activation of Rac in neutrophils. The latter result appears to contradict several reports19,20 that pharmacological inhibitors of PI3K can dramatically inhibit chemoattractant-induced Rac and Cdc42 activation in human neutrophils.
At least three possibilities, alone or in combination, could account for this contradiction and for the incomplete loss of motility in PI3Kγ knockout mice. First, it might be that the PtdIns(3,4,5)_P_3 concentration in PI3Kγ-deficient neutrophils treated with chemoattractant is below the level of detection by ordinary assays, but basal (unstimulated) PtdIns(3,4,5)_P_3 synthesis or chemoattractant-activated synthesis by class 1A PI3K isozymes (Fig. 4; see also Box 1) activates Rho GTPases and exerts marginally adequate control over actin polymerization and assembly. An alternative explanation is that neutrophil motility might be regulated by signalling pathways that are quite independent of any PI3K isozyme. One such mechanism in neutrophils involves activation of classical PKCs (cPKC; Fig. 4), which leads to activation of Rac and Cdc42 that is not blocked by pharmacological inhibitors of PI3K20. In addition, Gβγ might activate Rho GEFs directly, without using a PI3K as an intermediary (Fig. 4); in the yeast Saccharomyces cerevisiae, Gβγ activates Cdc42p in this way by recruiting and activating an adaptor molecule, Far1p, and a specific GEF for Cdc42p, Cdc24p (Ref. 39). Such alternative signalling pathways could partially mask the effect of the PI3Kγ knockout if they are relatively more important for motility in mouse than in human neutrophils.
FIGURE 4.

Alternative pathways for activation of Rho GTPases by chemoattractants in neutrophils. Pathways in addition to the phosphoinositide 3-kinase gamma (PI3Kγ) pathway (red arrows) could mediate activation of Rho GTPases in neutrophils. The activated cytosolic tyrosine kinases (black arrows) could induce activation of class 1A PI3Ks (α, β and δ; see Box 1). Combined effects of tyrosine kinases and PI3K lipid products on a GDP–GTP exchange factor (GEF) could induce Rac or Cdc42 activation. Alternatively, released Gβγ could also activate a GEF directly (yellow arrows). Gβγ also activates the phospholipases PLCβ2 and PLCβ3 in neutrophils (blue arrows). Activation of these phospholipases results in an increase in cytosolic Ca2+ and formation of diacylglycerol at the plasma membrane37. These two events lead to the activation of classical protein kinase C (cPKC) enzymes. These protein kinases have been shown to activate Rac and Cdc42 independently of PI3K in neutrophils20. Abbreviations: PtdIns(3,4,5)_P_3, phosphatidylinositol (3,4,5)-trisphosphate; PtdIns(3,4)_P_2, phosphatidylinositol (3,4)-bisphosphate.
A qualitatively different possibility is that chemoattractants induce relatively normal neutrophil motility in PI3Kγ knockout mice, but the cells move in random directions – that is, the chemokinetic response to chemoattractants is preserved, but chemotaxis (directed migration) is impaired. Indeed, reported assays of residual neutrophil motility in PI3Kγ knockout mice36-38 did not clearly distinguish chemokinesis from chemotaxis. Thus, it is possible that loss of PI3Kγ selectively impairs the compass mechanism. A reasonable guess, however, is that lipid products of PI3Kγ probably regulate both chemokinetic and chemotactic motility. In human neutrophils, two PI3K inhibitors, wortmannin and LY294002, prevented both kinds of motility response to chemoattractant7. Moreover, adding PtdIns(3,4,5)_P_3 directly to neutrophils induces cell polarization and chemokinesis40.
It is unlikely that the regulatory role of PI3Kγ is confined to control of actin assemblies. Cell motility requires not only actin polymerization but also the coordinated control of cell adhesion at the leading edge and contraction of the trailing edge1,16. Future experiments will test the potential effects of PI3K products and their effectors on these pathways. One strong candidate for such an effector role is the serine/threonine kinase Akt/PKB. In Dictyostelium, genetic deletion of pkbA, the homologue of mammalian Akt, produced major defects in polarization and directional movement of cells in a gradient of chemoattractant30. Although a role for Akt in mammalian chemotaxis has not been investigated, its known substrates in other cells transmit signals that block apoptosis and regulate transcription, protein synthesis and cell proliferation. Akt can phosphorylate in vitro the Rho GTPase Rac1, inhibiting its GTP binding activity41; the relevance of this observation to cell motility is not yet established. PtdIns(3,4,5)_P_3 and PtdIns(3,4,)_P_2 activate Akt by a rather complex mechanism, involving recruitment of Akt to the plasma membrane via its PH domain, activation of an upstream kinase, 3′-phosphoinositide-dependent kinase 1 (PDK1) and a conformational change that allows Akt to be phosphorylated and thus activated by PDK142.
Roles for PI3K lipid products in cell polarity and pointing the compass
Lipid products of PI3Kγ emerge as strong candidates for a role intimately related to the cellular compass that directs chemotaxis, based on several lines of evidence: the motility defect of PI3Kγ-deficient neutrophils36-38, the PI3K requirement for chemoattractant-induced activation of Rho GTPases19,20 and observations that PI3K lipid products accumulate at the leading edge of migrating cells (Fig. 3)28-30. As compass molecules, lipid products of PI3Kγ would organize the leading edge of the cell by, at least in part, asymmetrical activation of Rho GTPases and consequent de novo polymerization of actin.
As noted above, the concentration gradient of an internal polarity determinant or compass molecule must be much steeper than the gradient of chemoattractant outside the cell. A plausible biochemical mechanism for achieving a disproportionately higher concentration of a compass molecule at the front of the cell would incorporate positive feedback – that is, the compass molecule would somehow stimulate its own accumulation. Indeed, recent experiments suggest that PtdIns(3,4,5)_P_3 can serve as the initiator of a positive-feedback loop: exogenous PtdIns(3,4,5)_P_3 (supplied as an analogue that allowed entry into the cells) induced neutrophils to polarize morphologically and to activate p42/p44 MAP kinase, and these effects were blocked by two PI3K inhibitors, wortmannin and LY294002 (Ref. 40).
How might PtdIns(3,4,5)_P_3 stimulate its own accumulation? Suggestive evidence indicates two potential mechanisms (Fig. 5a), either or both of which could mediate positive-feedback between PI3Kγ and other PI3Ks. The first scenario (Fig. 5a, top) involves a pathway in which PtdIns(3,4,5)_P_3 somehow causes tyrosine phosphorylation of membrane proteins and recruitment of the p85 regulatory subunit of tyrosine kinase-dependent PI3Ks (α, β and δ; see Box 1). In this scenario, the tyrosine kinase-dependent PI3K(s) act as effectors downstream of PI3Kγ. This is plausible, in that PI3K-dependent phosphorylation of tyrosine residues of cellular proteins has already been identified in neutrophils43. Two other observations in neutrophils are in keeping with this idea. First, PI3K activity can be detected in immunoprecipitates of tyrosine-phosphorylated proteins derived from chemoattractant-stimulated neutrophils but not from control cells44-46. Second, pharmacological inhibition of tyrosine kinases abolished chemoattractant-induced PtdIns(3,4,5)_P_3 accumulation45.
FIGURE 5.

Amplification of the accumulation of phosphoinositide 3-kinase (PI3K) lipid products relative to the gradient of chemoattractant determines cell polarity. (a) Amplification could occur in part through positive-feedback loops involving PI3Kγ and PI3Ks of class 1A. In the top scheme, chemoattractant-induced activation of cytosolic tyrosine kinases links activation of PI3Kγ to that of PI3Ks of class 1A. In this scenario, Gβγ activates PI3Kγ, which in turn produces phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)_P_3]. This lipid product then somehow induces the activation of cytosolic tyrosine kinases that then relay the signal to PI3Ks of class 1A (α, β and δ). These PI3Ks then further increase the accumulation of PI3K lipid products. The increased production of PI3K lipid products and the effect of tyrosine kinases then increase the activity of a GDP–GTP exchange factor (GEF) and the Rho GTPases that induce cell polarity. In the bottom scheme, Rho GTPases link PI3Kγ to activation of class1A PI3Ks. Gβγ-induced activation of PI3Kγ and cytosolic tyrosine kinases lead to activation of the Rho GTPases, just as described in the legend of Fig. 2. These Rho GTPases not only induce cell polarity but also activate class 1A PI3Ks, which in turn further increase the level of PI3K lipid product formation. This increase in PI3K lipid products then leads to a greater increase in activity of Rho GTPases, which reinforces polarity. (b) Asymmetric accumulation of PI3K lipid products [including PtdIns(3,4,5)_P_3 and phosphatidylinositol (3,4)-bisphosphate (PtdIns(3,4)_P_2)] in polarized neutrophils also requires inhibition of signalling at the trailing edge. The positive-feedback loops increasing the level of formation of PI3K lipid products cannot explain their absence in the trailing edge of polarized cells. A model53,54 suggests that activated receptors might also generate a long-range inhibitor (–) of signalling, in addition to local enhancers (+) such as the postulated PtdIns(3,4,5)_P_3-dependent positive feedback. The hypothetical inhibitor would initially prevent PI3K lipid product accumulation everywhere, but proportionally more at the pole of the cell facing the lowest concentration of chemoattractant in a gradient (I: initially). The combined effects of global inhibitors and local activators of PI3K lipid product formation would then result in accumulation of PI3K lipid products exclusively at the maximally stimulated area of the neutrophil plasma membrane. This local accumulation of PI3K lipid products would lead to cell polarization through a local activation of the Rho GTPases (II: after amplification).
In the second scenario, Rho GTPases play a pivotal role in the positive feedback loop. While considerable evidence19,20 has indicated that PI3K activity is required for chemoattractants to activate Rho GTPases (see above, and Fig. 4), in this positive-feedback scenario, Rho GTPases also play a role upstream of PtdIns(3,4,5)_P_3 accumulation (Fig. 5a, bottom panel). An upstream role of Rho GTPases in neutrophils is supported by an observation28 in neutrophils produced by differentiated cells of the HL-60 neutrophil precursor line: treatment with Clostridium difficile toxin B, which glucosylates and inactivates all three principal Rho GTPases (Rac, Rho and Cdc42)28, prevented chemoattractants from inducing translocation to the plasma membrane of PHAKT–GFP, the fluorescent probe for lipid products of PI3Ks. The toxin also prevented chemoattractants from increasing activity of endogenous Akt itself (G. Servant, O. Weiner, D. Stokoe and H.R. Bourne, unpublished). In this scenario, PtdIns(3,4,5)_P_3, initially formed through the Gβγ-dependent increase in PI3Kγ activity, increases its own synthesis through Rho GTPases, which in turn activate PI3Ks of class 1A (Fig. 5a, bottom scheme). In keeping with this model, mounting evidence in various cell types indicates that Rac and Cdc42 can act in signalling pathways both downstream and upstream of PI3Ks47-49. Although we do not know how Rho GTPases regulate PI3K activity in vivo, activated Rac and Cdc42 can bind to the p85 regulatory subunit of class 1A PI3Ks in vitro and stimulate their activity50.
In this hypothesis, Rho GTPases mediate positive feedback that creates asymmetric accumulation of PI3K products, and this amplified asymmetry polarizes the cell. Achieving such polarity is necessary for pointing a cell towards the source of chemoattractant, but polarity and choice of the correct direction of polarity need not be identical. Indeed, neutrophils polarize and migrate robustly upon exposure to a uniform concentration of chemoattractant1-3,28,40. Thus a built-in capacity for asymmetry points the cell quite unambiguously in a single, albeit random, direction, even in the absence of a chemoattractant gradient.
How do the Rho GTPases control the subtle relation between cell polarity and the direction of this polarity? Observations in other cell types suggest that Rac and Cdc42 play related but different roles. In T cells, at least, Rac appears both necessary and sufficient to maintain the putative positive-feedback loop that amplifies PtdIns(3,4,5)_P_3 concentrations48: either Rac or Cdc42 led to activation of Akt, but activation of this enzyme by the T-cell receptor was blocked by dominant-negative Rac and unaffected by dominant-negative Cdc42; moreover, as expected for a positive-feedback loop, and as previously demonstrated in many cell types, Rac was shown to act downstream of PI3K as well. We imagine, then, that Rac suffices to establish and maintain the amplified asymmetry of PtdIns(3,4,5)_P_3 that allows a neutrophil to polarize, even in a uniform concentration of chemoattractant.
Similar evidence suggests that Cdc42 might play a key role in creating the correct bias in polarity that points a cell in the right direction. Thus, mutations in Cdc42 or its GEF cause budding yeast51, T lymphocytes52 and macrophages9 to lose their normal ability to polarize selectively towards extracellular stimuli. Dominant-negative Cdc42 does not prevent macrophages from migrating in response to chemoattractant gradients, but they migrate in random directions9. If Cdc42 does bias neutrophil polarity in response to a gradient, we shall want to know how it cooperates with Rac to control feedback among PI3Ks, asymmetric distribution of PI3K lipid products across the surface of leukocytes in gradients and the final ‘readout’ – directional motility.
Positive feedback might not suffice to establish polarity
It is unlikely that localized positive feedback by itself accounts for the extreme difference in concentrations of PI3K lipid products at the leading versus the trailing edges of chemotaxing leukocytes. An attractive model (Fig. 5b) to explain creation of steep signalling gradients incorporates, in addition to localized enhancement of the extracellular signal (positive feedback), a rapidly diffusible global inhibitor of the signal29,53. According to this notion, upon exposure to a concentration gradient of chemoattractant, each activated receptor at the surface of a neutrophil would generate not only a short-range local activator of PI3K activity (e.g. activated Cdc42 and/or tyrosine phosphorylation) but also a long-range inhibitor of the accumulation of PI3K lipid products (Fig. 5b). Rapid diffusion of the inhibitor, resulting in uniform concentration throughout the cell (cell I, initially), would globally elevate the threshold for chemoattractant stimulation of PtdIns(3,4,5)_P_3 accumulation; regions of the cell (that is, those on its up-gradient side) where activation exceeds this threshold would preferentially stimulate PtdIns (3,4,5)_P_3 accumulation, thus synergistically increasing localization of the positive-feedback signals to the leading edge (cell II, afteramplification).
The existence of a globally acting inhibitor is suggested by comparing spatial distribution of the activated state in cells exposed to a gradient, as opposed to a uniform concentration, of chemoattractant. We (O. D. Weiner, G. Servant and H.R. Bourne, unpublished) have observed, in cells exposed to a gradient, that PtdIns(3,4,5)_P_3 or filamentous actin accumulates selectively at the up-gradient edge; in a uniform concentration of chemoattractant, however, cellular activation is first apparent throughout the periphery of the cell and only later is remodelled to form a polarized leading edge. The biochemical nature of the putative inhibitor, however, remains unknown. In addition to rapidly diffusible inhibitors of PI3K activities, possible mechanisms might reduce activities of the chemoattractant receptor, the heterotrimeric G-protein, phosphoinositide phosphatases or Rho GTPases.
Full steam ahead
Taken together, a body of observations and experiments strongly suggests that lipid products of PI3Ks play significant roles as compass molecules in neutrophil polarity and chemotaxis. Many questions remain to be explored. First, and probably most important, experiments must be designed to ask whether local accumulation of PI3K lipid products at the plasma membrane is necessary or sufficient to dictate the spatially specific activation of key Rho GTPases and consequently cell polarity.
Other questions abound. Are chemoattractant receptors or heterotrimeric G-proteins at the leading edge more active than those at the trailing edge? Are activities of PI3K γ and other PI3Ks, as well as phosphoinositide phosphatases (SHIP or PTEN) spatially regulated during polarization and chemotaxis? Can we identify the postulated global inhibitor that prevents accumulation of PI3K lipid products at the trailing edge of polarized cells? What effectors of PI3K lipid products mediate establishment of polarity and organization of the leading edge? Is Akt one of these? Do other cells, including neurons, use the same compass mechanism for directional migration?
Neutrophils interpret extremely shallow gradients and navigate quite accurately towards the source of chemoattractants. We already know that loss of PI3K γ fails to prevent a significant percentage of neutrophils from crawling into sites of inflammation36-38. Thus, it remains possible that neutrophils, like ocean liners, navigate by more than one compass. If so, investigators will discover PI3K-independent mechanisms for creating and amplifying the asymmetry of polarizing signals. Although it will not be easy to devise a general scheme for explaining polarity and chemotaxis of eukaryotic cells, we think we are heading in the right direction.
Footnotes
P.R. is currently at: Incyte Genomics, 3160 Porter Drive, Palo Alto, CA 94304, USA
G.S. is currently at: Senomyx, Inc., 11099 N. Torrey Pines Drive, Suite 160, La Jolla, CA 92037, USA
References
- 1.Devreotes PN, Zigmond SH. Chemotaxis in eukaryotic cells: a focus on leukocytes and Dictyostelium. Annu. Rev. Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- 2.Weiner OD, et al. Spatial control of actin polymerization during neutrophil chemotaxis. Nat. Cell Biol. 1999;1:75–81. doi: 10.1038/10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Servant G, et al. Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Mol. Biol. Cell. 1999;10:1163–1178. doi: 10.1091/mbc.10.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao Z, et al. Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 1997;139:365–374. doi: 10.1083/jcb.139.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firtel RA, Chung CY. The molecular genetics of chemotaxis: sensing and responding to chemoattractant gradients. BioEssays. 2000;22:603–615. doi: 10.1002/1521-1878(200007)22:7<603::AID-BIES3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 6.Arai H, et al. Chemotaxis in a lymphocyte cell line transfected with C–C chemokine receptor 2B: evidence that directed migration is mediated by βγ dimers released by activation of Galphai-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 1997;94:14495–14499. doi: 10.1073/pnas.94.26.14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knall C, et al. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3052–3057. doi: 10.1073/pnas.94.7.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niggli V, Keller H. The phosphatidylinositol 3-kinase inhibitor wortmannin markedly reduces chemotactic peptide-induced locomotion and increases in cytoskeletal actin in human neutrophils. Eur. J. Pharmacol. 1997;335:43–52. doi: 10.1016/s0014-2999(97)01169-2. [DOI] [PubMed] [Google Scholar]
- 9.Allen WE, et al. A role for Cdc42 in macrophage chemotaxis. J. Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber KS, et al. Chemokine-induced monocyte transmigration requires cdc42-mediated cytoskeletal changes. Eur. J. Immunol. 1998;28:2245–2251. doi: 10.1002/(SICI)1521-4141(199807)28:07<2245::AID-IMMU2245>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 11.Zu YL, et al. p38 Mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-alpha or FMLP stimulation. J. Immunol. 1998;160:1982–1989. [PubMed] [Google Scholar]
- 12.Laudanna C, et al. Evidence of ζ protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. J. Biol. Chem. 1998;273:30306–30315. doi: 10.1074/jbc.273.46.30306. [DOI] [PubMed] [Google Scholar]
- 13.Welch H, et al. Nonreceptor protein-tyrosine kinases in neutrophil activation. Methods. 1996;9:607–618. doi: 10.1006/meth.1996.0067. [DOI] [PubMed] [Google Scholar]
- 14.Locati M, et al. Inhibition of monocyte chemotaxis to C–C chemokines by antisense oligonucleotide for cytosolic phospholipase A2. J. Biol. Chem. 1996;271:6010–6016. doi: 10.1074/jbc.271.11.6010. [DOI] [PubMed] [Google Scholar]
- 15.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez-Madrid F, del Pozo MA. Leukocyte polarization in cell migration and immune interactions. EMBO J. 1999;18:501–511. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts AW, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 18.Ambruso DR, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benard V, et al. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 20.Akasaki T, et al. Phosphoinositide 3-kinase-dependent and -independent activation of the small GTPase Rac2 in human neutrophils. J. Biol. Chem. 1999;274:18055–18059. doi: 10.1074/jbc.274.25.18055. [DOI] [PubMed] [Google Scholar]
- 21.Belisle B, Abo A. N-formyl peptide receptor ligation induces Rac dependent actin reorganization through Gbg subunits and class Ia phosphoinositide 3-kinases. J. Biol. Chem. 275:26225–26232. doi: 10.1074/jbc.M002743200. [DOI] [PubMed] [Google Scholar]
- 22.Ma AD, et al. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase γ, a Rac guanosine exchange factor, and Rac. Mol. Cell. Biol. 1998;18:4744–4751. doi: 10.1128/mcb.18.8.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanhaesebroeck B, et al. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat. Cell Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 24.Vicente-Manzanares M, et al. Involvement of phosphatidylinositol 3-kinase in stromal cell-derived factor-1 α-induced lymphocyte polarization and chemotaxis. J. Immunol. 1999;163:4001–4012. [PubMed] [Google Scholar]
- 25.al-Aoukaty A, et al. Recruitment of pleckstrin and phosphoinositide 3-kinase gamma into the cell membranes, and their association with Gβγ after activation of NK cells with chemokines. J. Immunol. 1999;162:3249–3255. [PubMed] [Google Scholar]
- 26.Han J, et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. doi: 10.1126/science.279.5350.558. [DOI] [PubMed] [Google Scholar]
- 27.Blomberg N, et al. The PH superfold: a structural scaffold for multiple functions. Trends Biochem. Sci. 1999;24:441–445. doi: 10.1016/s0968-0004(99)01472-3. [DOI] [PubMed] [Google Scholar]
- 28.Servant G, et al. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parent CA, et al. G protein signaling events are activated at the leading edge of chemotactic cells. Cell. 1998;95:81–91. doi: 10.1016/s0092-8674(00)81784-5. [DOI] [PubMed] [Google Scholar]
- 30.Meili R, et al. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 1999;18:2092–2105. doi: 10.1093/emboj/18.8.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watton SJ, Downward J. Akt/PKB localisation and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell–cell interaction. Curr. Biol. 1999;9:433–436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- 32.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell. Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura M, et al. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 34.Liliental J, et al. Genetic deletion of the pten tumor suppressor gene promotes cell motility by activation of rac1 and cdc42 GTPases. Curr. Biol. 2000;10:401–404. doi: 10.1016/s0960-9822(00)00417-6. [DOI] [PubMed] [Google Scholar]
- 35.Helgason CD, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasaki T, et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, et al. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 38.Hirsch E, et al. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 39.Butty AC, et al. The role of Far1p in linking the heterotrimeric G protein to polarity establishment proteins during yeast mating. Science. 1998;282:1511–1516. doi: 10.1126/science.282.5393.1511. [DOI] [PubMed] [Google Scholar]
- 40.Niggli V. A membrane-permeant ester of phosphatidylinositol 3,4,5-trisphosphate (PIP(3)) is an activator of human neutrophil migration. FEBS Lett. 2000;473:217–221. doi: 10.1016/s0014-5793(00)01534-9. [DOI] [PubMed] [Google Scholar]
- 41.Kwon T, et al. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J. Biol. Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- 42.Coffer PJ, et al. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naccache PH, et al. Inhibition of tyrosine phosphorylation by wortmannin in human neutrophils. Dissociation from its inhibitory effects on phospholipase D. Lab. Invest. 1993;69:19–23. [PubMed] [Google Scholar]
- 44.Coffer PJ, et al. Comparison of the roles of mitogen-activated protein kinase kinase and phosphatidylinositol 3-kinase signal transduction in neutrophil effector function. Biochem. J. 1998;329:121–130. doi: 10.1042/bj3290121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ptasznik A, et al. A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J. Biol. Chem. 1996;271:25204–25207. doi: 10.1074/jbc.271.41.25204. [DOI] [PubMed] [Google Scholar]
- 46.Ptasznik A, et al. A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J. Biol. Chem. 1995;270:19969–19973. doi: 10.1074/jbc.271.41.25204. [DOI] [PubMed] [Google Scholar]
- 47.Keely PJ, et al. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 48.Genot EM, et al. The T-cell receptor regulates akt (protein kinase B) via a pathway involving Rac1 and phosphatidylinositide 3-kinase. Mol. Cell. Biol. 2000;20:5469–5478. doi: 10.1128/mcb.20.15.5469-5478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishida K, et al. Anti-apoptotic function of Rac in hematopoietic cells. Oncogene. 1999;18:407–415. doi: 10.1038/sj.onc.1202301. [DOI] [PubMed] [Google Scholar]
- 50.Tolias KF, et al. Rho family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- 51.Nern A, Arkowitz RA. A GTP-exchange factor required for cell orientation. Nature. 1998;391:195–198. doi: 10.1038/34458. [DOI] [PubMed] [Google Scholar]
- 52.Stowers L, et al. Regulation of the polarization of T cells toward antigen-presenting cells by Ras-related GTPase CDC42. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5027–5031. doi: 10.1073/pnas.92.11.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meinhardt H. Orientation of chemotactic cells and growth cones: models and mechanisms. J. Cell Sci. 1999;112:2867–2874. doi: 10.1242/jcs.112.17.2867. [DOI] [PubMed] [Google Scholar]
- 54.Parent CA, Devreotes PN. A cell’s sense of direction. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]