Hormone regulation of microglial cell activation: relevance to multiple sclerosis (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 10.
Abstract
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of proteins. The role of PPARs in regulating the transcription of genes involved in glucose and lipid metabolism has been extensively characterized. Interestingly, PPARs have also been demonstrated to mediate inflammatory responses.
Microglia participate in pathology associated with multiple sclerosis (MS). Upon activation, microglia produce molecules including NO and TNF-α that can be toxic to CNS cells including myelin-producing oligodendrocytes and neurons, which are compromised in the course of MS.
Previously, we and others demonstrated that PPAR-γ agonists including 15d-PGJ2 are effective in the treatment of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. PPAR-γ modulation of EAE may occur, at least in part, by inhibition of microglial cell activation. Here, we indicate that 15d-PGJ2 is a more potent inhibitor of microglial activation than thiazolidinediones, which are currently used to treat diabetes. Furthermore, 15d-PGJ2 acts cooperatively with 9-cis retinoic acid, the ligand for the retinoid X receptor (RXR), in inhibiting microglial cell activation. This suggests that 15d-PGJ2 and 9-cis RA inhibit cell activation through the formation of PPAR-γ/RXR heterodimers. Interestingly, PGA2, which like 15d-PGJ2 is a cyclopentenone prostaglandin, but which unlike 15d-PGJ2 does not bind PPAR-γ, is a potent inhibitor of microglial cell activation. Collectively, these studies suggest that 15d-PGJ2 inhibits microglial cell activation by PPAR-γ-dependent as well as PPAR-γ-independent mechanisms. The studies further suggest that the PPAR-γ agonist 15d-PGJ2 in combination with retinoids may be effective in the treatment of MS.
Keywords: Microglia, Hormone, PPAR, Cytokine, Nitric oxide, Multiple sclerosis
1. Introduction
Multiple sclerosis (MS) is one of the most common causes of non-traumatic neurological disability in young adults. The disease commonly initiates in approximately the third decade of life, and most patients confront the devastating effects of this progressive disease for many years. The cause of MS is unknown. However, an autoimmune etiology is presumed since MS is characterized by perivascular mononuclear-cell inflammatory infiltrates and demyelination, features also characteristic of the autoimmune animal disorder experimental autoimmune encephalomyelitis (EAE) [32]. Genetics and environment, particularly viral infection, also are believed to play a role in the pathogenesis of MS [32]. MS occurs more frequently in females, which has suggested that hormones play a significant role in modulating the disease [15]. Multiple drugs have been approved for use in the treatment of MS, but these agents are not a cure for the disease, so the need remains for the development of better treatment strategies in MS.
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily of proteins that regulate the expression of genes involved in reproduction, metabolism, development, and immune responses. The role of PPARs in regulating the transcription of genes involved in glucose and lipid metabolism has been extensively characterized [11]. Three PPAR subtypes designated α, δ (also referred to as β), and γ exist, and these receptors demonstrate differential ligand specificities and cellular expression patterns [11,45]. PPAR-γ ligands include the synthetic thiazolidinediones which are commonly prescribed for the treatment of type II diabetes, certain high-affinity tyrosine derivatives, nonsteroidal anti-inflammatory drugs (NSAIDS), and fibrates, as well as the naturally occurring eicosanoids, polyunsaturated fatty acids, and the cyclopentenone prostaglandin 15d-PGJ2. The fact that PPAR-γ is activated by ligands including the NSAID indomethacin and is expressed in monocytes/macrophages, suggested that this receptor might regulate the activation of these immune cells. Indeed, studies demonstrating that PPAR-γ agonists are capable of suppressing immune activation of monocyte/macrophages were seminal to this field of investigation [25,42].
2. PPAR-γ-signaling
PPAR-γ agonists are believed to regulate the immune response through both receptor-dependent and receptor-independent mechanisms. For example, ligand binding can activate this receptor resulting in binding to conserved DNA sequences termed peroxisome-proliferator response elements (PPREs) present in the promoter of target genes. PPAR-γ regulates the expression of these genes by binding to PPREs as a heterodimer in association with a 9-cis retinoic acid receptor (RXR) [11]. In addition, PPAR-γ receptors are also capable of regulating gene expression independent of binding to PPREs through a mechanism termed receptor-dependent transrepression. The molecular mechanisms resulting in transrepression are not completely understood, but are believed to result from physical interaction of the receptor with other transcription factors. Transrepression may also result from receptor interaction with transcriptional co-activator/co-repressor molecules that are in limited supply, or by altering interactions with the basal transcription machinery [26,27]. PPAR-γ is believed to inhibit transcription factors including NF-κB, AP-1, and STAT-1 from activating gene expression through receptor-dependent transrepression. The PPAR-γ agonist 15d-PGJ2 has been demonstrated to regulate gene expression through receptor-independent mechanisms. For example, this prostaglandin inhibits NF-κB activity by suppressing IKK activation in response to inflammatory stimuli, which blocks I-κB degradation, thus preventing the nuclear translocation of NF-κB [3,43], and also by directly inhibiting NF-κB binding to NF-κB DNA-response elements [46].
3. PPAR-γ: effects on peripheral leukocytes
T cells autoreactive to myelin antigens are likely to contribute to the initiation of MS. T cells express PPAR-γ, and PPAR-γ ligands inhibit T-cell proliferation and IL-2 secretion [7]. Elimination of autoreactive T cells by apoptosis protects CNS tissue from immune-mediated destruction and likely contributes to the resolution of disease (reviewed in [39]). Interestingly, PPAR-γ ligands have been demonstrated to be capable of inducing T-cell apoptosis [19,20], suggesting that these agonists may facilitate the resolution of T-cell-mediated autoimmune diseases. The role of B cells in modulating MS is becoming more appreciated. Interestingly, B cells express PPAR-γ and the PPAR-γ ligands 15d-PGJ2 and thiazolidinediones inhibit B-cell proliferation and induce apoptosis of these cells [37,38]. PPAR-γ agonists can also potentiate the apoptosis of macrophages [23]. Collectively, these studies suggest that PPAR-γ may modulate MS in part by stimulating apoptosis of peripheral immune cells.
The entry of cells into the CNS from the periphery is regulated, in part, by the expression of chemotactic cytokines (chemokines), matrix metalloproteinases (MMPs), and adhesion molecules. Chemokines are synthesized at sites of inflammation and establish a concentration gradient to which target cell populations migrate. A variety of lymphocyte and monocyte chemokines are elevated in MS and EAE (reviewed in [18,47]). The effects of PPAR-γ on chemokine expression have not been thoroughly investigated. These receptor agonists attenuate the expression of the monocyte chemoattractant MCP-1 [29,49], as well as IP-10 (CXCL3), Mig (CXCL3), and I-TAC (CXCL3) which are chemotactic for T cells [33]. Collectively, these studies suggest that PPAR-γ agonists may play an important role in mediating the migration of monocyte/macrophages and T cells into the CNS as well as retaining these cells and microglia at sites of CNS inflammation. Matrix metalloproteinases (MMP) are digestive enzymes, which function in the formation and reorganization of the extracellular matrix. MMPs are believed to play a critical role in immune cell extravasation into the CNS through the blood–brain barrier (BBB). Interestingly, the expression of various MMPs is increased in MS [10,31] and EAE [8]. Inhibitors of MMP also block the development of EAE [4,17,22]. The effects of PPAR-γ agonists on MMP expression in the CNS have not been thoroughly investigated. However, PPAR-γ agonists including thiazolidinediones and 15d-PGJ2 inhibited MMP-9 expression by human monocyte/macrophages [34,44]. Adhesion molecules present on the cerebral vascular endothelium facilitate immune cell extravasation across the BBB into the CNS. PPAR-γ agonists have been shown to selectively modulate the expression of various adhesion molecules [5,6,24], which may block the extravasation of immune cells through an endothelial barrier. Additional studies are required to determine the effects of PPAR-γ agonists on the expression of specific chemokines, MMPs, and adhesion molecules, and how these agonists regulate immune cell extravasation into the inflamed CNS.
4. Effects of PPAR-γ agonists on microglial activation
Microglia are derived from bone marrow precursors that enter the CNS during development where they serve as resident CNS macrophages. Microglia function as antigen presenting cells and are phagocytic. Normally, microglia are quiescent in the CNS, but upon bacterial or viral infection, these cells become activated and initiate a protective immune response against these pathogens. This immune response results in increased production of NO, and proinflammatory cytokines and chemokines, by activated microglia. In addition to the beneficial effects microglia have in initiating protective immunity against pathogens, when chronically activated, microglia have been implicated in contributing to neuronal cell death associated with neuroinflammatory and neurodegenerative disorders. Petrova et al. [41] were the first to report that the PPAR-γ agonist 15d-PGJ2 inhibited microglial activation. They demonstrated that 15d-PGJ2 inhibited NO production by the BV-2 mouse microglial cell line. Subsequently, they demonstrated that this prostaglandin also inhibited LPS induction of IL-1β and TNF-α production by these cells [30]. These studies also suggested that 15d-PGJ2 does not affect NF-κB nuclear translocation and DNA binding, but does affect the activation of NF-κB-responsive genes, which was proposed to occur through a PPAR-γ-independent mechanism since troglitazone did not have similar effects [41]. Bernardo et al. [2] demonstrated that 15d-PGJ2 suppressed TNF-α, NO, and MHC class II expression by primary microglia. This was proposed to occur through a PPAR-γ-dependent manner since ciglitazone had similar effects. The basis for the discrepancy between these studies concerning whether 15d-PGJ2 influences microglial cell function through PPAR-γ-dependent or -independent mechanisms may be explained by the fact that the Bernardo studies were conducted using primary microglia while the Petrova studies were conducted with BV-2 cells which express little or no PPAR-γ. On the contrary, primary microglia constitutively express this receptor, and the expression is increased in response to 15d-PGJ2 and suppressed by LPS [2]. Furthermore, PPAR-γ agonists activated the receptor in microglia, resulting in receptor binding to PPREs [1].
Cytokines such as IL-12 play a critical role in the differentiation of T lymphocytes toward the T helper 1 (Th1) subset. Th1 cells are believed to be encephalitogenic and contribute to the development of EAE. Therefore, attenuating IL-12 expression may have beneficial effects on the clinical course of EAE. We have reported that 15d-PGJ2 represses NO, TNF-α, and IL-12 production in both the mouse N9 microglial cell line and primary microglia [14]. Importantly, this study revealed that 15d-PGJ2 was capable of attenuating microglial activation in response to TNF-α and IFN-γ, cytokines that are believed to contribute to the development of EAE. This study further suggests that 15d-PGJ2 may modulate EAE progression by inhibiting IL-12 production by microglia, which may inhibit Th1 cell differentiation.
Recently, we have demonstrated that 15d-PGJ2 is a more potent inhibitor of microglial activation than thiazolidinediones, which are commonly used to treat type II diabetes. Furthermore, 15d-PGJ2 acts cooperatively with 9-cis retinoic acid, the ligand for RXR, in inhibiting microglial cell activation. This suggests that 15d-PGJ2 and 9-cis RA inhibit cell activation through the formation of PPAR-γ/RXR heterodimers. Interestingly, PGA2, which like 15d-PGJ2 is a cyclopentenone prostaglandin, but which unlike 15d-PGJ2 does not bind PPAR-γ, is a potent inhibitor of microglial cell activation. Collectively, these studies suggest that 15d-PGJ2 inhibits microglial cell activation by PPAR-γ-dependent as well as PPAR-γ-independent mechanisms.
5. PPAR-γ: effects on neuronal cell viability
Neuroinflammatory diseases including MS and Alzheimer’s disease are characterized by neuron cell death. Products of activated microglia including NO, TNF-α, and reactive oxygen species may contribute to the loss of neurons in these diseases. A variety of PPAR-γ agonists have been demonstrated to indirectly protect neurons in neuron–glial co-cultures by inhibiting glial cell activation [9,28]. PPAR-γ agonists also directly protect cerebellar granule neurons [21,48]. MS is also characterized by the death of myelin-producing oligodendrocytes. The effects of PPAR-γ agonists on oligodendrocyte viability have not been extensively examined. However, our preliminary studies indicate that these agonists protect these cells from cytotoxic insults including hydrogen peroxide (data not shown). Collectively, these studies suggest that PPAR-γ agonists may be effective in protecting neurons and oligodendrocytes in neuroinflammatory and neurodegenerative disorders.
6. PPAR-γ: role in EAE
EAE is an autoimmune disorder characterized by CNS inflammation and demyelination, together with remittent paralysis—features consistent with MS [32]. Evidence to suggest that PPAR-γ agonists may potentially ameliorate MS was initially provided by Niino et al. [36] who demonstrated that troglitazone inhibited the development of EAE. Diab et al. [12] reported that the PPAR-γ agonist 15d-PGJ2 inhibited the proliferation of neural antigen-specific T cells from the spleen of myelin basic protein Ac1–11 TCR-transgenic mice, which spontaneously develop EAE. This PPAR-γ agonist also suppressed IFN-γ, IL-10, and IL-4 production by activated lymphocytes [12]. Administration of 15d-PGJ2 at the onset of clinical disease significantly reduced EAE severity, supporting the potential clinical relevance of this treatment paradigm [12]. Feinstein et al. [16] demonstrated that oral administration of thiazolidinediones including pioglitazone reduced the clinical severity of monophasic EAE, but it did not delay disease onset. In a relapsing EAE model, pioglitazone had no effect on the onset or severity of the initial disease attack, but rather reduced the severity of subsequent relapses and resulted in an overall decrease in mortality. Natarajan and Bright [35] demonstrated that the PPAR-γ agonists 15d-PGJ2 and ciglitazone were capable of reducing the severity of both active and passive EAE. Both 15d-PGJ2 and ciglitazone reduced IL-12-induced differentiation of Th1 cells in these studies. Recently, the Racke laboratory has demonstrated that retinoids and 15d-PGJ2 act cooperatively to inhibit the development of EAE [13]. This suggests the intriguing possibility that PPAR-γ and RXR ligands could act cooperatively in the treatment of MS. A recent clinical study supports the idea that PPAR-γ may be effective in the treatment of MS [40].
7. Conclusions
Activated microglia likely contribute to the pathology associated with chronic inflammatory diseases of the CNS. PPAR-γ agonists potently inhibit microglial cell activation, which may protect neural cells that are compromised in these diseases. These receptor agonists also affect the function of peripheral monocytes and lymphocytes and may control the extravasation of these immune cells into the CNS. PPAR-γ agonists have been demonstrated to block the development of EAE, suggesting that these molecules may be efficacious in the treatment of MS. The potential mechanisms by which PPAR-γ agonists may modulate EAE/MS are summarized in Fig. 1. Future clinical trials regarding the efficacy of PPAR-γ agonists in the treatment of MS may be facilitated by the fact that several of the thiazolidinedione group of PPAR-γ agonists are currently prescribed for the treatment of type II diabetes. PPARγ agonists may also be effective in the treatment of other chronic inflammatory disorders of the CNS including Alzheimer’s disease, AIDS, dementia, Parkinson’s disease, and stroke. Future studies designed to determine the molecular mechanisms by which PPAR-γ agonists modulate CNS inflammation will facilitate determination of the potential clinical efficacy of PPAR-γ agonists in the treatment of these CNS disorders.
Fig. 1.
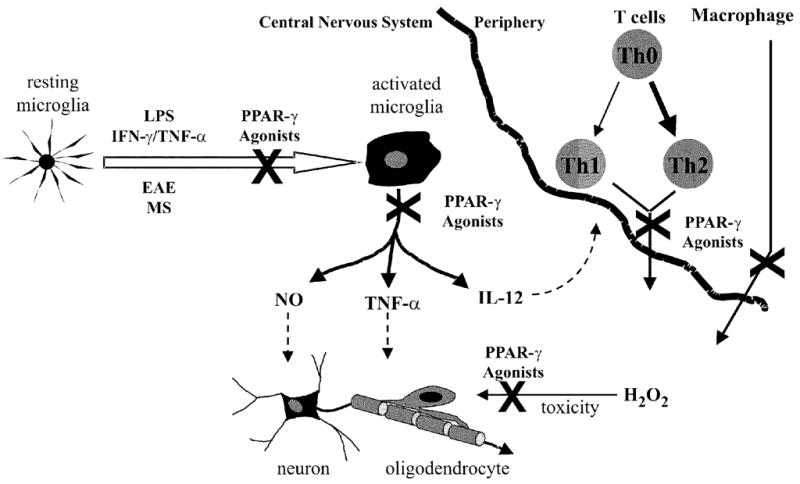
PPAR-γ agonist modulation of MS: potential mechanisms. PPAR-γ agonists block the activation of microglial cells resulting in repression of NO and TNF-α expression. This likely protects oligodendrocytes and neurons from the toxic effects of NO and TNF-α. These agonists may also directly protect oligodendrocytes from toxic agents including hydrogen peroxide. PPAR-γ agonists also inhibit IL-12 production by microglia. Since IL-12 plays a critical role in Th1 differentiation, these receptor agonists may induce skewing toward a protective Th2 phenotype, which is believed to protect against EAE/MS. PPAR-γ agonists may also regulate the extravasation of peripheral immune cells into the CNS.
Acknowledgments
This work was supported by grants from the National Multiple Sclerosis Society (RG 3198A1) and the NIH (NS 042860) to P.D. Drew.
References
- 1.Bernardo A, Ajmone-Cat MA, Levi G, Minghetti L. 15-deoxy-Δ12,14-prostaglandin J2 regulates the functional state and the survival of microglial cells through multiple molecular mechanisms. J Neurochem. 2003;87:742–751. doi: 10.1046/j.1471-4159.2003.02045.x. [DOI] [PubMed] [Google Scholar]
- 2.Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-γ (PPAR-γ) and its natural ligand 15-deoxy-Δ12,14-prostaglandin J2 in the regulation of microglial functions. Eur J Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- 3.Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Inhibition of IκB kinase and IκB phosphorylation by 15-deoxy-Δ12,14-prostaglandin J2 in activated murine macrophages. Mol Cell Biol. 2000;20:1692–1698. doi: 10.1128/mcb.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandler S, Miller KM, Clements JM, Lury J, Corkill D, Anthony DC, Adams SE, Gearing AJ. Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: an overview. J Neuroimmunol. 1997;72:155–161. doi: 10.1016/s0165-5728(96)00179-8. [DOI] [PubMed] [Google Scholar]
- 5.Chen NG, Han X. Dual function of troglitazone in ICAM-1 gene expression in human vascular endothelium. Biochem Biophys Res Commun. 2001;282:717–722. doi: 10.1006/bbrc.2001.4628. [DOI] [PubMed] [Google Scholar]
- 6.Chen NG, Sarabia SF, Malloy PJ, Zhao XY, Feldman D, Reaven GM. PPAR-gamma agonists enhance human vascular endothelial adhesiveness by increasing ICAM-1 expression. Biochem Biophys Res Commun. 1999;263:718–722. doi: 10.1006/bbrc.1999.1437. [DOI] [PubMed] [Google Scholar]
- 7.Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol. 2000;164:1364–1371. doi: 10.4049/jimmunol.164.3.1364. [DOI] [PubMed] [Google Scholar]
- 8.Clements JM, Cossins JA, Wells GM, Corkill DJ, Helfrich K, Wood LM, Pigott R, Stabler G, Ward GA, Gearing AJ, Miller KM. Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumor necrosis factor-alpha inhibitor. J Neuroimmunol. 1997;74:85–94. doi: 10.1016/s0165-5728(96)00210-x. [DOI] [PubMed] [Google Scholar]
- 9.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPAR-gamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J. The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol. 1996;55:1194–1204. doi: 10.1097/00005072-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 12.Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-γ agonist 15-deoxy-Δ12,14-prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- 13.Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK. Ligands for the peroxisome proliferator-activated receptor-γ and the retinoid receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148:116–126. doi: 10.1016/j.jneuroim.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Drew PD, Chavis JA. The cyclopentanone prostaglandin 15-deoxy-Δ12,14 prostaglandin J2 represses nitric oxide, TNF-α, and IL-12 production by microglial cells. J Neuroimmunol. 2001;115:28–35. doi: 10.1016/s0165-5728(01)00267-3. [DOI] [PubMed] [Google Scholar]
- 15.Duquette P, Girard M. Hormonal factors in susceptibility to multiple sclerosis. Curr Opin Neurol Neurosurg. 1993;6:195–201. [PubMed] [Google Scholar]
- 16.Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT. Peroxisome proliferator-activated receptor-γ agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 17.Gijbels K, Galardy RE, Steinman RE. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J Clin Invest. 1994;94:2177–2182. doi: 10.1172/JCI117578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godessart N, Kunkel SL. Chemokines in autoimmune disease. Curr Opin Immunol. 2001;13:670–675. doi: 10.1016/s0952-7915(01)00277-1. [DOI] [PubMed] [Google Scholar]
- 19.Harris SG, Phipps RP. The nuclear receptor PPAR-gamma is expressed by mouse T lymphocytes and PPAR-gamma agonists induce apoptosis. Eur J Immunol. 2001;31:1098–1105. doi: 10.1002/1521-4141(200104)31:4<1098::aid-immu1098>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 20.Harris SG, Smith RS, Phipps RP. 15-deoxy-Delta 12,14-PGJ2 induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. J Immunol. 2002;168:1372–1379. doi: 10.4049/jimmunol.168.3.1372. [DOI] [PubMed] [Google Scholar]
- 21.Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol. 1999;100:156–168. doi: 10.1016/s0165-5728(99)00192-7. [DOI] [PubMed] [Google Scholar]
- 22.Hewson AK, Smith T, Leonard JP, Cuzner ML. Suppression of experimental allergic encephalomyelitis in the Lewis rat by the matrix metalloproteinase inhibitor Ro31-9790. Inflamm Res. 1995;44:345–349. doi: 10.1007/BF01796266. [DOI] [PubMed] [Google Scholar]
- 23.Hortelano S, Castrillo A, Alvarez AM, Bosca L. Contribution of cyclopentenone prostaglandins to the resolution of inflammation through the potentiation of apoptosis in activated macrophages. J Immunol. 2000;165:6525–6531. doi: 10.4049/jimmunol.165.11.6525. [DOI] [PubMed] [Google Scholar]
- 24.Jackson SM, Parhami F, Xi XP, Berliner JA, Hsuch WA, Law RE, Demer LL. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler, Thromb Vasc Biol. 1999;19:2094–2104. doi: 10.1161/01.atv.19.9.2094. [DOI] [PubMed] [Google Scholar]
- 25.Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 26.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 27.Kerppola TK, Luk D, Curran T. Fox is a preferential target of glucocorticoid receptor inhibition of AP-1 activity in vitro. Mol Cell Biol. 1993;13:3782–3791. doi: 10.1128/mcb.13.6.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim EJ, Kwon KJ, Park JY, Lee SJ, Moon CH, Baik EJ. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: associated with iNOS and COX-2. Brain Res. 2002;941:1–10. doi: 10.1016/s0006-8993(02)02480-0. [DOI] [PubMed] [Google Scholar]
- 29.Kintscher U, Goetze S, Wakino S, Kim S, Nagpal S, Chandraratna RA, Graf K, Fleck E, Huseh WA, Law RE. Peroxisome proliferator-activated receptor and retinoid X receptor ligands inhibit monocyte chemotactic protein-1-directed migration of monocytes. Eur J Pharmacol. 2000;401:259–270. doi: 10.1016/s0014-2999(00)00461-1. [DOI] [PubMed] [Google Scholar]
- 30.Koppal T, Petrova TV, Van Eldik LJ. Cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 acts as a general inhibitor of inflammatory responses in activated BV-2 microglial cells. Brain Res. 2000;867:115–121. doi: 10.1016/s0006-8993(00)02270-8. [DOI] [PubMed] [Google Scholar]
- 31.Maeda A, Sobel RA. Matrix metalloproteinases in the normal human central nervous system, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol. 1996;55:300–309. doi: 10.1097/00005072-199603000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 33.Marx N, Mach F, Sauty A, Leung JH, Sarafi MN, Ransohoff RM, Libby P, Plutzky J, Luster AD. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN-gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J Immunol. 2000;164:6503–6508. doi: 10.4049/jimmunol.164.12.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma contain PPARgamma: differentiation-dependent peroxisomal proliferator-activated receptor gamma (PPARgamma) expression and reduction of MMP-9 activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153:17–23. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 36.Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-γ. J Neuroimmunol. 2001;116:40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 37.Padilla J, Kaur K, Cao HG, Smith TJ, Phipps RP. Peroxisome proliferator activator receptor-gamma agonists and 15-deoxy-Delta(12,14)-PGJ(2) induce apoptosis in normal and malignant B-lineage cells. J Immunol. 2000;12:6941–6948. doi: 10.4049/jimmunol.165.12.6941. [DOI] [PubMed] [Google Scholar]
- 38.Padilla J, Leung E, Phipps RP. Human B lymphocytes and B lymphomas express PPAR-gamma and are killed by PPAR-gamma agonists. Clin Immunol. 2002;103:22–33. doi: 10.1006/clim.2001.5181. [DOI] [PubMed] [Google Scholar]
- 39.Pender MP, Rist MJ. Apoptosis in inflammatory cells in immune control of the nervous system. Glia. 2001;36:137–144. doi: 10.1002/glia.1103. [DOI] [PubMed] [Google Scholar]
- 40.Pershadsingh HA, Heneka MT, Saini R, Amin NM, Broseske DJ, Feinstein DL. Effect of pioglitazone in a patient with secondary multiple sclerosis. J Neuroinflammation. 2004;1:3–6. doi: 10.1186/1742-2094-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Δ12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 43.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 44.Shu H, Wong B, Zhou G, Li Y, Berger J, Woods JW, Wright SD, Cai TQ. Activation of PPARα or γ reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267:345–349. doi: 10.1006/bbrc.1999.1968. [DOI] [PubMed] [Google Scholar]
- 45.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 46.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κ B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trebst C, Ransohoff RM. Investigating chemokines and chemokine receptors in patients with multiple sclerosis. Arch Neurol. 2001;58:1975–1980. doi: 10.1001/archneur.58.12.1975. [DOI] [PubMed] [Google Scholar]
- 48.Uryu S, Harada J, Hisamoto M, Oda T. Troglitazone inhibits both post-glutamate neurotoxicity and low-potassium-induced apoptosis in cerebellar granule neurons. Brain Res. 2002;924:229–236. doi: 10.1016/s0006-8993(01)03242-5. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Wang JM, Gong WH, Mukaida N, Young HA. Differential regulation of chemokine gene expression by 15-deoxy-Δ12,14 prostaglandin J2. J Immunol. 2001;166:7104–7111. doi: 10.4049/jimmunol.166.12.7104. [DOI] [PubMed] [Google Scholar]