Identification of small molecule inhibitors of pyruvate kinase M2 (original) (raw)
. Author manuscript; available in PMC: 2011 Apr 15.
Published in final edited form as: Biochem Pharmacol. 2009 Dec 11;79(8):1118–1124. doi: 10.1016/j.bcp.2009.12.003
Abstract
A common feature of tumors arising from diverse tissue types is a reliance on aerobic glycolysis for glucose metabolism. This metabolic difference between cancer cells and normal cells could be exploited for therapeutic benefit in patients. Cancer cells universally express the M2 isoform of the glycolytic enzyme pyruvate kinase (PKM2), and previous work has demonstrated that PKM2 expression is necessary for aerobic glycolysis and cell proliferation in vivo. Because most normal tissues express an isoform of pyruvate kinase other than PKM2, selective targeting of PKM2 provides an opportunity to target cell metabolism for cancer therapy. PKM2 has an identical catalytic site as the related M1 splice variant (PKM1). However, isoform selective inhibition is possible as PKM2 contains a unique region for allosteric regulation. We have screened a library of greater than 100,000 small molecules to identify such inhibitors. The inhibitors identified for PKM2 fell primarily into three distinct structural classes. The most potent PKM2 inhibitor resulted in decreased glycolysis and increased cell death following loss of growth factor signaling. At least part of this effect was due to on-target PKM2 inhibition as less cell death was observed in cells engineered to express PKM1. These data suggest that isoform selective inhibition of PKM2 with small molecules is feasible and support the hypothesis that inhibition of glucose metabolism in cancer cells is a viable strategy to treat human malignancy.
Keywords: pyruvate kinase M2, glycolysis, cellular metabolism, small molecule inhibitors, high-throughput screening
1. Introduction
One of the first distinctions noted between cancer tissues and normal tissues was a difference in metabolism [1, 2]. Cancer cells, unlike their normal counterparts, metabolize glucose by aerobic glycolysis. This phenomenon, known as the Warburg effect, is characterized by increased glycolysis with lactate production and decreased oxidative phosphorylation [3, 4]. The propensity for tumor cells to rely on increased glucose uptake which accompanies the Warburg effect has been exploited for diagnostic purposes and forms the basis of 18F-deoxyglucose Positron Emission Tomography (FDG-PET) as a tool for detecting and staging malignancy [2, 4]. However, although this observation was made over 90 years ago, the fundamental difference in cellular metabolism exhibited by cancer cells has not yet been exploited for therapeutic benefit [5].
Achieving specificity has been a challenge for targeting cell metabolism. All cells rely on intermediary metabolism for their survival, and glucose is a major source of energy for many cell types in humans. This raises the possibility that using a small molecule to interfere with glucose metabolism would have a detrimental effect on both cancer and normal tissues. However, cancer cells have distinct metabolic needs that are required for proliferation and there is growing evidence that the genetic alterations that drive malignancy also cause cells to become addicted to high levels of flux through specific metabolic pathways [4, 6]. The metabolism of glucose via aerobic glycolysis may be a consequence of selection for cells that reprogram glucose metabolism to meet the distinct metabolic needs of cell proliferation [4].
An important determinant of how glucose is metabolized in cells depends on the glycolytic enzyme pyruvate kinase [7, 8]. Pyruvate kinase has four isoforms in mammalian cells encoded by two separate genes [9]. The L gene is alternatively spliced to produce the L and R isoforms of pyruvate kinase, which are expressed exclusively in the liver and red blood cells respectively [10]. The M gene is alternatively spliced by mutual exclusion of a single exon to generate either the M1 or M2 isoform of pyruvate kinase [11]. Most tissues in the adult animal express pyruvate kinase M1 (PKM1), while pyruvate kinase M2 (PKM2) is expressed during embryonic development [7, 12, 13]. Tumor cells, including those tumors derived from tissues that normally express PKL or PKR, express exclusively PKM2 [12, 13]. Knockdown of PKM2 using RNA interference significantly impairs cell growth in tissue culture, inhibition of PKM2 with peptide aptamers inhibits cell proliferation, and PKM2 expression is necessary for both aerobic glycolysis and tumor growth in vivo [7, 14]. Furthermore, PKM2 selection for tumor growth in vivo suggests that reversion to another isoform of pyruvate kinase might be incompatible with cell proliferation in the setting of an established tumor. These observations identify PKM2 as an attractive target for disruption of aerobic glycolysis in tumors.
One potential challenge for identifying a small molecule that selectively inhibits PKM2 is the similarity between PKM2 and the other isoforms of pyruvate kinase. PKM2 and PKM1 are identical in size and differ only in the 56 amino acid region encoded by the alternatively spliced exon [13]. In comparison to PKM1, PKL and PKR are less similar to PKM2, but still exhibit significant sequence conservation. The unique portion of PKM2 encoded by the alternatively spliced exon does not contribute to the active site of the enzyme, but rather allows PKM2, but not PKM1, to be allosterically activated by the upstream glycolytic intermediate, fructose-1,6-bisphosphate (FBP) [13]. The unique region of PKM2 also allows for enzymatic regulation by interaction with tyrosine-phosphorylated proteins [15]. Targeting the allosteric site of PKM2 may allow for isoform selective small molecule inhibitors of pyruvate kinase.
Here we describe a screen designed to identify inhibitors with selectivity for PKM2 over PKM1. This screen identified three classes of molecules that inhibit PKM2 with minimal effect on PKM1. These molecules can mimic some aspects of PKM2 knockdown using RNAi, including inhibition of glycolysis. These data demonstrate that selective targeting of PKM2 with a drug-like molecule is possible and suggest that efforts to target PKM2 may yield compounds suitable for targeting cancer metabolism for cancer therapy.
2. Materials and Methods
2.1. Purification of recombinant pyruvate kinase isoforms
The human cDNA for PKM2, PKM1 and PKL were cloned into pET28a with a N-terminal 6x-His tag and purified from E. coli using Ni-Agarose beads (Qiagen) as described previously [15]. Briefly, E. coli grown to an OD(600nm) of 0.7 were induced with 0.5 mM IPTG at room temperature for 6 hours. Cells were collected and lysed by freeze/thaw cycles and sonication. Lysate was passed over an Ni-NTA agarose column and pyruvate kinase eluted with 250 mM Imidazole in 1 mL fractions. Fractions with high concentration of pyruvate kinase were determined using SDS-PAGE and coomassie staining according to standard protocol.
2.2. Characterization of enzyme activity
Phosphoenolpyruvate (PEP), ADP, Fructose-1,6-bisphosphate (FBP), Lactate Dehydrogenase, DDT, Glycerol, and NADH were purchased from Sigma-Aldrich (St Louis, MO). To assess pyruvate kinase activity, a reaction mixture containing 8 units of LDH with NADH, ADP, and PEP at the indicated concentrations was added to purified pyruvate kinase in the presence or absence of FBP. The reaction buffer contained 100 mM KCl, 50 mM Tris pH 7.5, 10 mM MgCl2, 1 mM DTT, and 5% Glycerol. To assess activity in cell lysates, cells were treated with compound for the indicated time and lysed in NP40 lysis buffer immediately before measuring pyruvate kinase activity as described previously [15].
2.3. Compound Library
The compound library consisted of 107,360 small molecules, including compounds approved by the Food and Drug Administration (FDA), a purified natural products library, compounds purchased from Peakdale (High Peak, UK), Maybridge plc. (Cornwall, UK), Cerep (Paris, France), Bionet Research Ltd. (Cornwall, UK), Prestwick (Ilkirch, France), Specs and Biospecs (CP Rijswijk, the Netherlands), ENAMINE (Kiev, Ukraine), Life Chemicals, Inc. (Burlington, Canada), MicroSource Diversity System’s NINDS custom collection (Gaylordsville, CT) and ChemDiv (San Diego, CA), and from various academic institutions. Compounds were selected from the different vendors by applying a series of filters, including clogP and predicted solubility. All of the small molecules in the library generally adhere to Lipinski’s rules (i.e. molecular weight < 500, H-bond donors ≤ 5, H-bond acceptors ≤ 10 and logP < 5), contain a low proportion of known toxicophores (i.e. Michael acceptors and alkylating agents) and unwanted functionalities (i.e. imines, thiols, and quaternary amines), and have been optimized for maximization of molecular diversity.
2.4 Primary screening assay
A CRS CataLyst Express robotic arm (Thermo Fisher Scientific, Waltham, MA), a μFill microplate dispenser (BioTek/Labtech International LTD, Ringmer, East Sussex, UK) and a Cybi-well 384 channel simultaneous pipettor (CyBio AG, Jena, Germany) were used to carry out the high throughput screening of the small molecule library. PKM2 reactions were performed in 50 mM Tris, pH 7.5, 1 mM DTT, 0.02% Brij-35 and 5% Glycerol using Costar black 384 assay plates (Corning Inc. Corning, NY). In the final HTS conditions, 5 pg recombinant PKM2, 125 μM FBP and 1.25 mM PEP (final concentration at 0.5 mM) in a volume of 10 μl kinase buffer were added to the assay plate containing 0.4 μl of 1.67 mM compound (final concentration 27 μM) and pre-incubated for 25 minutes. The reaction was initiated by addition of 15 μl of 1 mM ADP (final concentration at 0.6 mM), 400 μM NADH, and 2 units of lactate dehydrogenase per reaction. The reaction was incubated for 20 minutes at room temperature. Then the NADH amount was measured using a PHERAstar HTS microplate reader (BMG Labtech, Offenberg, Germany) with 340 nm excitation and 460 nm emissions. PKM2 activity for each well was calculated by following formula: percent inhibition (I%) = (reading-AVElow control)/(AVEhigh control-AVElow control)*100, in which AVEhigh control is the average reading of 16 wells of high control (no enzyme) and AVElow control is the average reading of 16 wells of low control (no compound with enzyme).
2.5 Confirmation and counter screening assays
All primary hits with >50% inhibition were searched for their background fluorescent signals using previous experimental data stored in our internal database. Any compounds with >5 fold increase in fluorescence over normal background fluorescence were eliminated. The remaining potential small molecule inhibitors were confirmed manually in the same 384-well assay plate under the same assay conditions as the primary screening. Time dependent values of fluorescence were measured at 60 sec intervals for 60 min as described above for each compound. Two concentrations (27 and 10 μM) for each compound were tested for confirmation.
An ATP luminescence assay was used to eliminate potential lactate dehydrogenase inhibitors. 5 pg PKM2, 125 μM FBP and 1.25 mM PEP (final concentration at 0.5 mM) in a volume of 10 μl kinase buffer were added to the assay plate containing compound at different concentrations and pre-incubated for 25 minutes, then the reaction was initiated by addition of 15 μl of 1 mM ADP (Final concentration at 0.6 mM). The reaction was incubated for 20 minutes at room temperature. 5 μl of ATP detection reagent (Promega (Madison, WI)) was added to the well and luminescence signal measured using a PHERAstar HTS microplate reader.
All the confirmed hits were then tested against recombinant PKM1 under the same conditions.
2.6 IC50 determinations
For the most promising compounds, IC50 values were determined using the same 384-well assay plate under the same assay conditions as the primary screening. Time dependent values of fluorescence were measured at 60 sec intervals for 60 min as described above for each compound. Each compound was assayed in triplicate for each inhibitor concentration. The inhibition for each well was calculated by the following formula: percent inhibition (I%) = (Slope of well-Slope of AVElow control)/(Slope of AVEhigh control-Slope of AVElow control)*100, in which Slope of AVEhigh control is the average slopes of 16 wells of high control (no enzyme) and Slope of AVElow control is the average slopes of 16 wells of low control (no compound with enzyme). IC50 values were determined by fitting measurements of percent inhibition (I%) to a sigmoidal dose-response curve using Prism software.
2.7 Testing of related available compounds
All compounds were tested for activity against fresh recombinant PKM2, PKM1 and PKL protein as indicated using the NADH-coupled assay described above. Compounds 1, 2 and 3 were purchased from ChemDiv (San Diego, CA). Compound 3 was also independently obtained as a gift from Agios Pharmaceticals. Compounds shown in Supplemental Figure 5 were purchased from ChemDiv or ChemBridge (San Diego, CA). Additional thiazolidinediones tested included Pioglitazone obtained from Sigma (St. Louis, MO), and several Compound 2 analogs available from ChemDiv and ChemBridge (San Diego, CA).
2.8 Cell lines and cell culture experiments
H1299 and HCC827 cells were grown in RPMI supplemented with 10% FCS using standard tissue culture conditions. H1299 cells were engineered to express mouse PKM1 or mouse PKM2 as described previously [7]. Knockdown of endogenous PKM2 was confirmed by Western blot using a commercially available anti-PK antibody (Abcam). FL5.12 cells were grown in RPMI supplemented with 10% FCS and 0.35 ng/mL of recombinant mouse IL-3 as described previously [16]. For IL-3 withdrawal, cells were washed 3 times in RPMI and cultured in the original media minus IL-3. Cell viability was measured by PI exclusion using flow cytometry as described previously at the indicated time following drug treatment and/or IL-3 withdrawal [16].
2.9 Measurement of glycolysis
The rate of glucose metabolism by glycolysis was measured by following conversion of 5-3H-glucose to 3H2O as described previously [16]. Briefly, cells were washed with PBS prior to incubation in Krebs buffer without glucose for 30 minutes at 37 degrees. The buffer was then replaced with Krebs buffer containing 10 μCi 5-3H-glucose in the presence of 10 mM cold glucose and cells incubated for 1 hour at 37 degrees. Samples of media were then transferred to tubes containing 0.2 N HCl and the amount of 3H2O generated was determined by diffusion.
3. Results
Pyruvate kinase catalyzes the transfer of phosphate from phosphoenolpyruvate (PEP) to adenosine-5′-diphosphate (ADP), generating pyruvate and adenosine-5′-triphosphate (ATP). Pyruvate kinase activity can be measured via coupled assays to detect production of either pyruvate or ATP (Figure 1). Pyruvate generation can be monitored kinetically by coupling the pyruvate kinase reaction to the lactate dehydrogenase (LDH) reaction. LDH oxidizes NADH to NAD+ while reducing pyruvate to lactate. Because the reaction catalyzed by LDH is more efficient than the PK reaction, the rate of NADH fluorescence loss corresponds to PK activity.
Figure 1. Schematic representation of the assays used to measure pyruvate kinase activity.
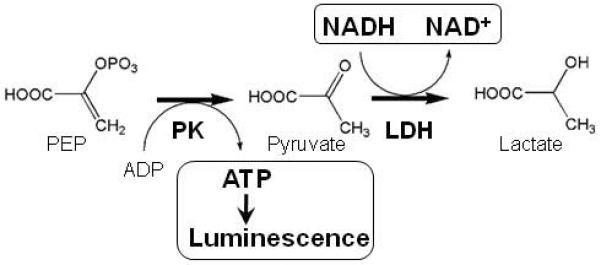
The pyruvate kinase (PK) reaction can be coupled to the faster lactate dehydrogenase (LDH) reaction, and pyruvate kinase activity followed by measuring a decrease in NADH fluorescence. Alternatively, ATP produced by the pyruvate kinase reaction can be measured by luminescence produced during the ATP-dependent firefly luciferase reaction.
To generate PKM2 enzyme suitable for inhibitor screening, N-terminal 6-His tagged human PKM2 was produced in E. coli and purified using Ni-affinity chromatography (Figure 2A). Kinetic characterization of rPKM2 generated was in general agreement with published values. rPKM2 demonstrated robust activation by FBP (Figure 2B). Given that FBP has a slow off rate from PKM2 and does not activate according to classic Michaelis-Menton kinetics [15], a precise Ka was not determined. However FBP activated PKM2 in the μM range as expected from previously published studies [17], and FBP decreased the Km of PKM2 for PEP (Figure 2C). The Km for both ADP and PEP of the rPKM2 used for screening were consistent with published values in the range of 100 μM (Figure 2D, E) [17].
Figure 2. Characterization of recombinant PKM2.
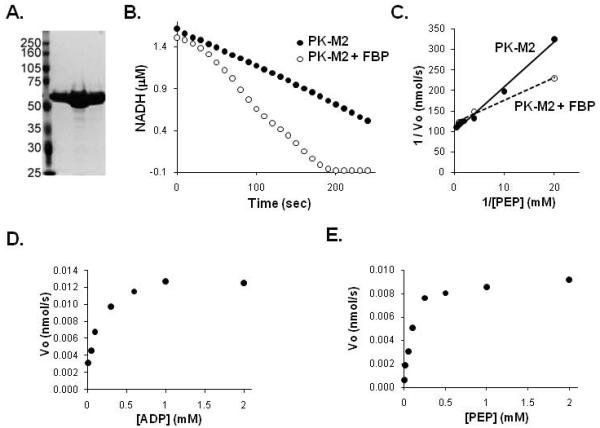
A. PKM2 was expressed and purified from E. coli. A coomassie stained SDS-PAGE gel of the purified PKM2 fractions used for subsequent assays is shown. B. Activity of PKM2 protein was assessed by following loss of NADH fluorescence in an LDH coupled assay. PKM2 enzyme activity in the absence or presence of 50 μM FBP is shown. C. Lineweaver-Burk plot of enzyme velocity measured for PKM2 at various concentrations of PEP in the presence or absence of 50 μM FBP as shown. D. Enzyme velocity is plotted at various concentrations of ADP as shown. E. Enzyme velocity is plotted at various concentrations of PEP as shown.
To identify inhibitors of PKM2, the LDH-coupled kinetic assay was adapted to 384 well format for high-throughput screening (HTS). We modified the assay buffer conditions such that the enzyme retained activity even at room temperature for 2 hours, which is the time period needed to facilitate HTS (Supplemental Figure 1). The screen was carried out in the presence of 125 μM FBP, which was sufficient to completely activate the enzyme at the beginning of the reaction. Reproducible results were observed between plates included in a pilot screen and the same plates included in the HTS (Supplemental Figure 2). The Z’-factor ranged from 0.6-1.0 for all the plates screened (Figure 3A). Of the 107,360 compounds included in the HTS, approximately 7.4% of the compounds scored as greater than 50% inhibition (Table 1). These included many compounds which were subsequently excluded from further study because of high background fluorescent signal. 515 remaining compounds were cherry picked and retested. 41.5% scored as positive upon retesting at 30 μM compound concentration with 17.8% of those scoring positive at a compound concentration of 10 μM.
Figure 3. A screen for small molecule inhibitors identified compounds selective for PKM2.
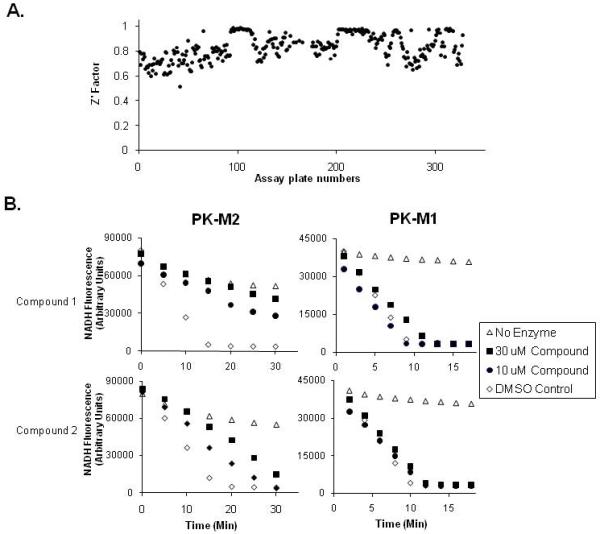
A. The Z’-factor for each plate in the HTS is plotted as shown. B. The effect of two representative small molecules identified in the HTS as PKM2 selective inhibitors on PKM2 and PKM1 activity using the LDH coupled assay is shown. The data presented is from Compound 1 and Compound 2 as labeled (see Figure 4). Differences in NADH scale shown for PKM1 and PKM2 assays reflect optimization of the assay to compensate for inherent differences in specific activity of the two enzymes.
Table 1. HTS results for PKM2.
| Total compound screened | 107,360 |
|---|---|
| PKM2 Inhibition >50% | 5661* |
| PKM2 Inhibition >60% and <120% | 2334** |
| Non-fluorescent inhibitors | 519*** |
| Compounds retested | 515 |
| Confirmed hits at 30 μM | 214 |
| Confirmed hits at 10 μM | 92 |
| Confirmed hits also hit PKM1 at 30 μM | 128 |
| Confirmed hits also hit PKM1 at 10 μM | 18 |
| PKM2 specific inhibitors at 10 μM | 74 |
To identify selective inhibitors of PKM2, we next screened those compounds that demonstrated confirmed activity against PKM2 for activity against PKM1. Recombinant PKM1 was generated in bacteria and exhibited a high specific activity that was insensitive to FBP (Supplemental Figure 3A, B). 59.8% of the confirmed hits against PKM2 at 30 μM compound concentration also demonstrated significant inhibition of PKM1 (Table 1). However, compounds were identified which exhibited selective, dose-dependent inhibition of PKM2 with less inhibition of PKM1 (Figure 3B).
Given the assay used to identify inhibitors was coupled to LDH, it is possible that compounds with activity against LDH could score as hits in the screen. These compounds would not be expected to score as specific inhibitors of PKM2 because LDH is used for both the PKM2 and PK-M1 assays; however, to further eliminate this possibility a luminescent end-point assay to measure ATP production was used (Figure 1). Using this assay, compounds identified as selective PKM2 inhibitors were tested for the ability to inhibit PKM2- or PKM1-dependent ATP production (Supplemental Figure 3C). The majority of compounds tested demonstrated selective PKM2 inhibition by this assay.
46% of the compounds with selective activity against PKM2 at 10 μM fell into three distinct structural classes. The most potent inhibitors in each class are shown in Figure 4. Compound 1 is a thiazolidinedione. Several thiazolidinediones scored as hits in the HTS, and many thiazolidinedione-like compounds are commercially available. However we were unable to identify a more potent inhibitor of PKM2 either in terms of IC50 or % inhibition (not shown). Compound 2 is a dye derivative and was insoluble in aqueous solution even at low μM concentrations. This insolubility made calculation of a precise IC50 impossible and inhibited further characterization of Compound 2. Compound 3 demonstrated the lowest IC50 among the compounds identified from the screen and was selected for further studies.
Figure 4. Three distinct structural classes of small molecules were identified that exhibit selective inhibition of PKM2 over PKM1.
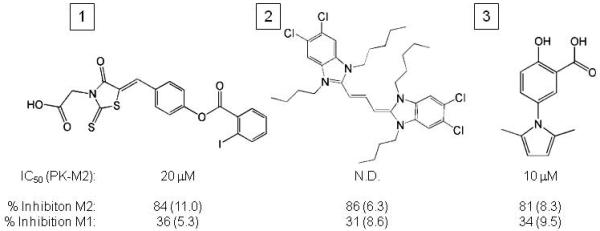
The structure of the most potent compound as judged by both IC50 and % inhibition in each class is shown. The IC50 against PKM2 as well as the percent inhibition of both PKM2 and PKM1 at 30 μM is shown for each compound. The standard error of the mean from three independent determinations of percent inhibition is shown in parentheses. The IC50 for compound 2 was not determined (n.d.).
Two other compounds related to Compound 3 also demonstrated greater than 60% inhibition of enzyme activity (Supplemental Figure 4). Neither compound demonstrated increased potency or selectivity for PKM2 when compared with Compound 3. Several commercially available analogues of Compound 3 were also obtained and tested for activity against PKM2. All of these compounds proved to be inactive as PKM2 inhibitors.
To confirm that Compound 3 and not a contaminant present in the library used for HTS was responsible for PKM2 inhibition, Compound 3 was obtained from two independent sources and retested using the LDH-coupled kinetic assay. Compound 3 from both sources demonstrated inhibition of PKM2 with an IC50 in the 50 μM range (Figure 5A, not shown). While this value was slightly higher than that obtained from the screening compound, the selectivity of Compound 3 for PKM2 over PKM1 was confirmed (Figure 5B). At least some of the variability in activity could be accounted for by assay configuration as the concentration of PKM2 influences maximal enzyme activity presumably via affecting amounts of highly active tetramer [14]. Compound 3 was also tested for an ability to inhibit PKL. Compound 3 was found to inhibit PKL with similar potency to PKM2 (Figure 5B). Analogous to Compound 3, Compound 1 was also found to inhibit PKL to a similar degree as PKM2 (Supplemental Figure 5).
Figure 5. Compound 3 inhibits PKM2 and PKL activity.
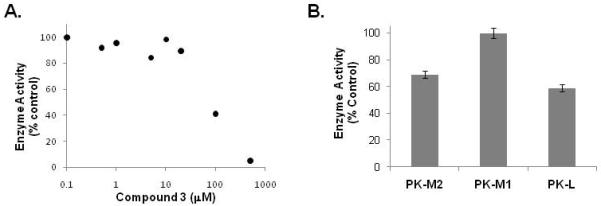
A. Independently obtained Compound 3 was re-tested for inhibition of PKM2 activity in the LDH-coupled assay at various concentrations as shown. B. The inhibition of PKM2, PKM1 and PKL activity by 50 μM of Compound 3 is shown.
To test the effects of PKM2 inhibition on cancer cells, Compound 3 was added to H1299 human lung cancer cells. H1299 cells are known to express PKM2 as their only pyruvate kinase isoform [7]. Consistent with an IC50 of 10-50 μM, high μM concentrations of Compound 3 were required to impact cellular glucose utilization or cell proliferation. Overnight treatment of H1299 cells with 100 μM of Compound 3 resulted in a 18.5% (+/− 3.5% (SEM)) decrease in glycolysis. For comparison, a 57.7% (+/− 5.5% (SEM)) decrease in glycolysis is observed upon RNA interference mediated knockdown of PKM2 to levels that result in inhibition of cell proliferation [7]. Consequently, greater than 100 μM of Compound 3 is needed for cytotoxicity (Figure 6A).
Figure 6. Compound 3 inhibits PKM2 and is toxic to cells in culture.
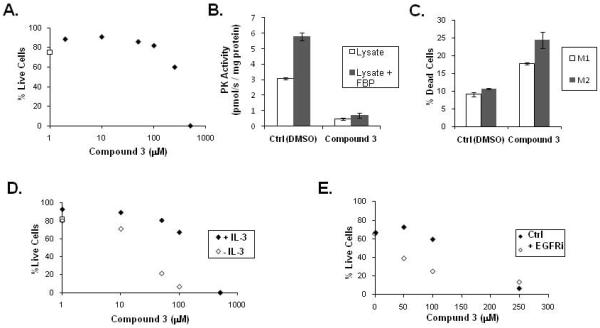
A. Viability of H1299 cells was assessed by propidium iodide exclusion using flow cytometry after two days of treatment with Compound 3 at the indicated concentration (solid diamonds) or DMSO control (open square) as shown. The Compound 3 used for in vivo experiments was independently purchased and not obtained from the screening library.
B. H1299 cells were treated for six hours with DMSO (Ctrl) or 250 μM Compound 3 prior to lysis. The pyruvate kinase activity in the lysates was assessed by following loss of NADH fluorescence in the LDH coupled assay in the presence or absence of exogenously added FBP (125 μM) as shown.
C. H1299 cells engineered to express either PKM1 or PKM2 were treated with DMSO (Ctrl) or 250 μM Compound 3 for 36 hours and viability assessed by propidium idodide exclusion and flow cytometry. The increased cell death observed in PKM2-expressing cells compared with PKM1-expressing cells in the presence of Compound 3 was statistically significant by Student’s t-test (p < 0.05).
D. Viability of IL-3-dependent FL5.12 cells was assessed by propidium iodide exclusion using flow cytometry. Viability was assessed following treatment with Compound 3 at the indicated concentration in the presence or absence of IL-3 for 20 hours as shown. The effect of vehicle only (DMSO) is plotted as an open square on the Y axis.
E. Viability of HCC827 cells was assessed by propidium iodide exclusion using flow cytometry. Viability was assessed after 2 days of treatment with Compound 3 at the indicated concentration in the presence or absence of 1 μM of the EGFR inhibitor Gefitinib (EGFRi) as shown.
To determine if pyruvate kinase inhibition was observed in cells at the concentrations needed for cytotoxicity, the activity of pyruvate kinase in cell lysates was measured following treatment with 250 μM Compound 3 or vehicle control. 250 μM Compound 3 was the lowest concentration where a significant decrease in viability was observed after two days (Figure 6A). No difference in viability was apparent after 6 hours of drug treatment; however lysates from Compound 3 treated cells had significantly less pyruvate kinase activity (Figure 6B). In addition, treatment of cells with Compound 3 prevented the ability of exogenous FBP to activate pyruvate kinase in the lysates. These data are consistent with Compound 3 inhibiting pyruvate kinase activity in cells at concentrations where cytotoxicity is seen.
Despite the correlation between PKM2 inhibition in lysates and cytotoxicity, it is possible that the toxicity results from an off target effect of Compound 3. To evaluate this possibility, we generated PKM1 expressing cell by expressing the highly conserved mouse versions of PKM1 or PKM2, and then subjecting those cells to shRNA-mediated knockdown of the endogenous pyruvate kinase (Supplemental Figure 6, [7]). PKM1 has a higher IC50 for Compound 3 than PKM2, so PKM1-expressing cells should be less susceptible to toxicity mediated by on-target pyruvate kinase inhibition. Thirty-six hour exposure to Compound 3 resulted in increased cell death in the PKM2-expressing cells compared with the PKM1-expressing cells (Figure 6C). These data show that inhibition of PKM2 activity is responsible for at least a portion of the cytotoxicity caused by Compound 3, and suggest that a window for selective inhibition of PKM2 can be achieved in cells.
Growth factor signaling has been shown to increase the rate of glucose utilization, and cell death following growth factor withdrawal correlates with decreases in glycolysis [16]. To test if inhibition of glycolysis can sensitize cells to growth factor withdrawal, we used the FL5.12 hematopoetic cell line, which is dependent on the growth factor IL-3 for survival. Addition of Compound 3 to FL5.12 cells increased the amount of cell death observed at 20 hours following growth factor withdrawal (Figure 6D). To test if PKM2 inhibition can increase cell death following inhibition of growth factor signaling in cancer cells, we utilized HCC827 lung cancer cells that carry a mutation in EGFR and are sensitive to the EGFR tyrosine kinase inhibitor Gefitinib [18]. Similar to results observed in FL5.12 cells, cell death from Compound 3 was increased in the presence of Gefitinib (Figure 6E). These data suggest that PKM2 inhibition might have a role in cancer therapy even in the absence of full inhibition of glycolysis.
4. Discussion
Identification of targets that provide a sufficient therapeutic window to selectively target glucose metabolism in cancer cells has been a major hurdle to the development of drugs to exploit cancer cell metabolism. PKM2 is such a metabolic target; however its similarity to PKM1 raised questions as to whether specific targeting was feasible. Here we demonstrate that selective inhibition of PKM2 with a small molecule, at least in relation to PKM1, is possible. The unique portion of PKM2 allows for interaction with FBP raising the possibility that the compounds identified target the allosteric FBP binding site of PKM2. The finding that PKM2 inhibition by Compound 3 in cell lysates is not reversible by FBP further supports the possibility that Compound 3 interferes with the FBP activation of PKM2. PKL also binds FBP, and the finding that both Compound 1 and Compound 3 inhibit PKL is consistent with the hypothesis that these compounds target the allosteric FBP binding site.
Like PKL and PKM2, PKR is also allosterically activated by FBP [10, 13]. PKR is a splice variant of PKL and contains the same FBP binding site as PKL. Thus, it is likely that the molecules we identified will also inhibit PKR. Inhibition of pyruvate kinase activity in liver or red blood cells could present toxicity issues for human cancer therapy. However, it is possible that additional efforts could identify compounds that inhibit PKM2 but do not inhibit PKL or PKR. The FBP binding property alone of PKM2 does not appear to confer an advantage for tumor growth. Tumors arising from the liver and red blood cells (which normally express PKL and PKR respectively) also switch expression to PKM2 [12, 13]. A unique property of PKM2 that distinguishes it from all other isoforms of pyruvate kinase is the ability of PKM2 to be regulated by binding to proteins that are phosphorylated on tyrosine in response to cell growth signals [15]. This phosphotyrosine interaction with PKM2 results in release of FBP from the protein suggesting that it may also involve the allosteric site [15]. This property of PKM2 to interact with phosphotyrosine-containing proteins could be leveraged to find molecules with relative selectively for PKM2 over PKL and PKR as well as PKM1.
More potent inhibitors would be beneficial for consideration as a cancer therapeutic. Using ATP production as an endpoint in screening would avoid elimination of fluorescent compounds and is one approach to screen for more potent inhibitors. RNA interference experiments suggest that significant knockdown of PKM2 expression is required to inhibit cell proliferation in culture [7]. In addition, a large decrease in glycolysis in response to 2-deoxyglucose is required for cell death [19, 20]. Peptide aptamers which selectively target PKM2 compared to PKM1, and promote the low activity dimeric form of the enzyme, have been shown to inhibit cell proliferation under conditions where glucose metabolism is disrupted [8, 14]. Therefore, enough PKM2 inhibition to significantly decrease glycolysis will likely be required for cytotoxicity as a single agent.
Partial inhibition of glucose metabolism may be a viable strategy for cancer therapy even in the absence of inducing cell death as a single agent. Other active anti-tumor drugs also decrease glycolysis [21] and there is emerging data that metabolic response to therapy as measured by FDG-PET is a more reliable indicator of patient survival than decreased tumor size [22]. The ability to inhibit glucose metabolism as assessed by FDG-PET scanning is a powerful predictor of therapeutic efficacy across many tumor types and anti-neoplastic agents [23]. The degree by which glycolysis is inhibited appears to contribute to apoptosis sensitivity in cell culture [16, 21]. The ability of Gefitinib to induce apoptosis in EGFR positive lung cancer correlates with the ability to inhibit glucose utilization (MVH, JE, LCC unpublished). In support of these findings, Compound 3 was more toxic to cells dependent on mutant EGFR in the presence of an EGFR inhibitor. Inhibition of glycolysis using a PKM2 inhibitor may synergize with other therapeutic agents to increase cell death.
PKM2 remains an attractive target for cancer therapy. We demonstrate that selective targeting of PKM2 with drug-like molecules is feasible and suggest that inhibition of PKM2 could be synergistic with other targeted therapies. These compounds will provide an opportunity to explore targeting of cancer cell metabolism. Development of more potent PKM2 inhibitors, as well as the optimal strategy to use these molecules for cancer therapy remains an exciting and active area of cancer drug development.
Supplementary Material
01
Supplemental Figure 1: Stability of PKM2 enzyme for HTS. Recombinant PKM2 was diluted to 3 pg and activity in HTS conditions assessed immediately (0 hr) or after 2 hours (2 hr) as shown.
02
Supplemental Figure 2: Reproducibility of the assay. An example of the hits distribution observed for the same plate during pilot screening and HTS is shown.
03
Supplemental Figure 3: Characterization of recombinant PKM1.
A. PKM1 was expressed and purified from E. coli. A coomassie stained SDS-PAGE gel of the purified PKM1 fractions used for subsequent assays is shown. B. Activity of PKM1 protein was assessed by following loss of NADH fluorescence in an LDH coupled assay. PKM1 enzyme activity in the absence or presence of FBP is shown. C. The effect of two representative small molecules indentified as PKM2 selective inhibitors on PKM2 and PKM1 activity as measured by production of ATP in a luciferase assay is shown. Compounds were tested at 10 μM. The data presented is from Compound 1 and Compound 3 as labeled (see Figure 4).
04
Supplemental Figure 4: Two compounds related to Compound 3 also demonstrate selective inhibition of PKM2. The structure of two additional active compounds related to Compound 3 are shown, as are the structures of several compounds related to Compound 3 which were found to not inhibit pyruvate kinase (inactive). The IC50 against PKM2 is shown for each active compound. Both active compounds retained similar selectivity for PKM2 as Compound 3. The standard error of the mean from three independent determinations of percent inhibition is shown in parentheses.
05
Supplemental Figure 5: Compound 1 inhibits PKM2 and PKL activity. The inhibition of PKM2, PKM1 and PKL activity by 50 μM of Compound 1 is shown.
06
Supplemental Figure 6: Generation of PKM1 and PKM2 expressing cells. N-terminal FLAG-epitope tagged versions of the mouse cDNA of PKM1 (mM1) or PKM2 (mM2) were expressed in H1299 cells, and these cells subjected to shRNA-mediated knock-down of endogenous PKM2. The mM1 and mM2 are nearly identical to the human homologs at the amino acid level but differ at the RNA level such that their expression in not targeted by the shRNA. Western blot analysis using an antibody against an epitope shared between PKM1 and PKM2 to demonstrate efficient knockdown of endogenous PK2 is shown. To demonstrate the efficiency of endogenous PKM2 shRNA knockdown the parental mM2 cells are also shown (parental).
Acknowledgements
We thank the Dana-Farber/Harvard Cancer Center Cancer Drug Assays screening facility for supporting this project. We thank J. Engelman for Gefitinib and HCC827 cells, C. Rudin and D. Plas for FL5.12 cells, and Agios Pharmaceuticals for providing independently synthesized Compound 3. MGVH was a fellow supported by the Damon Runyon Cancer Research Foundation when much of this work was performed.
Abbreviations
PK
pyruvate kinase
FDG-PET
18F-deoxyglucose Positron Emission Tomography
FBP
fructose-1,6-bisphosphate
PEP
Phosphoenolpyruvate
LDH
Lactate Dehydrogenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- [2].Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- [3].Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. 2008;134:703–7. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- [4].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- [6].Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- [8].Spoden GA, Rostek U, Lechner S, Mitterberger M, Mazurek S, Zwerschke W. Pyruvate kinase isoenzyme M2 is a glycolytic sensor differentially regulating cell proliferation, cell size and apoptotic cell death dependent on glucose supply. Exp Cell Res. 2009;315:2765–74. doi: 10.1016/j.yexcr.2009.06.024. [DOI] [PubMed] [Google Scholar]
- [9].Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998;6:195–210. doi: 10.1016/s0969-2126(98)00021-5. [DOI] [PubMed] [Google Scholar]
- [10].Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. J Biol Chem. 1987;262:14366–71. [PubMed] [Google Scholar]
- [11].Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986;261:13807–12. [PubMed] [Google Scholar]
- [12].Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- [13].Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–29. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- [14].Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Durr P, et al. Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation. Int J Cancer. 2008;123:312–21. doi: 10.1002/ijc.23512. [DOI] [PubMed] [Google Scholar]
- [15].Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–6. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- [16].Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yamada K, Noguchi T. Nutrient and hormonal regulation of pyruvate kinase gene expression. Biochem J. 1999;337(Pt 1):1–11. [PMC free article] [PubMed] [Google Scholar]
- [18].Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–94. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- [19].Liu H, Hu YP, Savaraj N, Priebe W, Lampidis TJ. Hypersensitization of tumor cells to glycolytic inhibitors. Biochemistry. 2001;40:5542–7. doi: 10.1021/bi002426w. [DOI] [PubMed] [Google Scholar]
- [20].Dwarkanath BS, Zolzer F, Chandana S, Bauch T, Adhikari JS, Muller WU, et al. Heterogeneity in 2-deoxy-D-glucose-induced modifications in energetics and radiation responses of human tumor cell lines. Int J Radiat Oncol Biol Phys. 2001;50:1051–61. doi: 10.1016/s0360-3016(01)01534-6. [DOI] [PubMed] [Google Scholar]
- [21].Zhou R, Vander Heiden MG, Rudin CM. Genotoxic exposure is associated with alterations in glucose uptake and metabolism. Cancer Res. 2002;62:3515–20. [PubMed] [Google Scholar]
- [22].Holdsworth CH, Badawi RD, Manola JB, Kijewski MF, Israel DA, Demetri GD, et al. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol. 2007;189:W324–30. doi: 10.2214/AJR.07.2496. [DOI] [PubMed] [Google Scholar]
- [23].Ben-Haim S, Ell P. 18F-FDG PET and PET/CT in the evaluation of cancer treatment response. J Nucl Med. 2009;50:88–99. doi: 10.2967/jnumed.108.054205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
Supplemental Figure 1: Stability of PKM2 enzyme for HTS. Recombinant PKM2 was diluted to 3 pg and activity in HTS conditions assessed immediately (0 hr) or after 2 hours (2 hr) as shown.
02
Supplemental Figure 2: Reproducibility of the assay. An example of the hits distribution observed for the same plate during pilot screening and HTS is shown.
03
Supplemental Figure 3: Characterization of recombinant PKM1.
A. PKM1 was expressed and purified from E. coli. A coomassie stained SDS-PAGE gel of the purified PKM1 fractions used for subsequent assays is shown. B. Activity of PKM1 protein was assessed by following loss of NADH fluorescence in an LDH coupled assay. PKM1 enzyme activity in the absence or presence of FBP is shown. C. The effect of two representative small molecules indentified as PKM2 selective inhibitors on PKM2 and PKM1 activity as measured by production of ATP in a luciferase assay is shown. Compounds were tested at 10 μM. The data presented is from Compound 1 and Compound 3 as labeled (see Figure 4).
04
Supplemental Figure 4: Two compounds related to Compound 3 also demonstrate selective inhibition of PKM2. The structure of two additional active compounds related to Compound 3 are shown, as are the structures of several compounds related to Compound 3 which were found to not inhibit pyruvate kinase (inactive). The IC50 against PKM2 is shown for each active compound. Both active compounds retained similar selectivity for PKM2 as Compound 3. The standard error of the mean from three independent determinations of percent inhibition is shown in parentheses.
05
Supplemental Figure 5: Compound 1 inhibits PKM2 and PKL activity. The inhibition of PKM2, PKM1 and PKL activity by 50 μM of Compound 1 is shown.
06
Supplemental Figure 6: Generation of PKM1 and PKM2 expressing cells. N-terminal FLAG-epitope tagged versions of the mouse cDNA of PKM1 (mM1) or PKM2 (mM2) were expressed in H1299 cells, and these cells subjected to shRNA-mediated knock-down of endogenous PKM2. The mM1 and mM2 are nearly identical to the human homologs at the amino acid level but differ at the RNA level such that their expression in not targeted by the shRNA. Western blot analysis using an antibody against an epitope shared between PKM1 and PKM2 to demonstrate efficient knockdown of endogenous PK2 is shown. To demonstrate the efficiency of endogenous PKM2 shRNA knockdown the parental mM2 cells are also shown (parental).