Millisecond-Timescale Optical Control of Neural Dynamics in the Nonhuman Primate Brain (original) (raw)
. Author manuscript; available in PMC: 2010 Apr 30.
Summary
To understand how brain states and behaviors are generated by neural circuits, it would be useful to be able to perturb precisely the activity of specific cell types and pathways in the nonhuman primate nervous system. We used lentivirus to target the light-activated cation channel channelrhodopsin-2 (ChR2) specifically to excitatory neurons of the macaque frontal cortex. Using a laser-coupled optical fiber in conjunction with a recording microelectrode, we showed that activation of excitatory neurons resulted in well-timed excitatory and suppressive influences on neocortical neural networks. ChR2 was safely expressed, and could mediate opticalneuromodulation, in primate neocortex over many months. These findings highlight a methodology for investigating the causal role of specific cell types in nonhuman primate neural computation, cognition, and behavior, and open up the possibility of a new generation of ultraprecise neurological and psychiatric therapeutics via cell-type-specific optical neural control prosthetics.
Introduction
The rhesus macaque is an important model species for understanding neural computation, cognition, and behavior, as well as for probing the circuit-level basis of human neurological and psychiatric disorders. To resolve how complex functions emerge from the activity of diverse cell types, ideally one would be able to perturb the activity of genetically specified cell types and neural pathways in the primate brain, in a temporally precise fashion. In one recent study, adeno-associated virus (AAV) was used to deliver the Drosophila allatostatin receptor to neurons in the primate thalamus (Tan et al., 2006), enabling neural silencing via intracranial delivery of the small molecule allatostatin. In general, however, the adaptation of neural control tools to the primate brain has been slow in comparison to the rapid adaptation of such tools for characterizing circuit functions in worms, flies, and mice (reviewed in Luo et al., 2008). Indeed, although molecular techniques have been used to deliver genetic payloads to the primate brain (e.g., Kordower et al., 2000; Liu et al., 2004; Stettler et al., 2006), as well as to make transgenic primates (Chan et al., 2001; Yang et al., 2008), no attempts have been made to target genes to genetically specified neuron types. Here we used channelrhodopsin-2 (ChR2), a genetically encoded molecular sensitizer that enables activation of neurons in response to pulses of blue light (Boyden et al., 2005; Han and Boyden, 2007; Ishizuka et al., 2006; Li et al., 2005; Nagel et al., 2003; Zhang et al., 2007), to assess the impact of selective activation of cortical excitatory neurons on primate cortical dynamics. We used optical fibers in conjunction with microelectrodes to perform simultaneous in vivo optical stimulation and electrical recording in the awake primate. Selectively activating ChR2-positive excitatory neurons resulted in well-timed excitatory and suppressive influences on neural activity, reflecting neural dynamics downstream of excitatory neuron activation. ChR2 was safely expressed and could mediate temporally precise optical neural stimulation of significant volumes of cortical tissue for months after viral injection, opening up the possibility for such technologies to support precise, cell-specific optical control prosthetics for patients with severe neurological and psychiatric disorders.
Results
We targeted ChR2-GFP to neurons in the frontal cortex in two monkeys (denoted N and A), by injecting VSVg-pseudotyped lenti-virus carrying the ChR2-GFP gene behind the 1.3 kb α-CaMKII promoter (Figure 1A; details in Supplemental Experimental Procedures, available online), as used before in mice to target excitatory neurons (Dittgen et al., 2004). To insure repeatable targeting of viruses, optical fibers, and electrodes to the same sites over extended periods of time (Figure 1B), we designed and used a grid to coordinate stereotactic virus injections, photostimulation, and recording (Figure 1C). Histology showed that 1 μl viral injections labeled roughly spherical regions of cortex 1.4 ± 0.5 mm in diameter (mean ± standard deviation [SD]; exemplar in Figure 1D; details in Figure S4, available online). We did not observe GFP-positive cells in thalamic regions that project to injected regions, indicating a lack of retrograde labeling using lentivirus prepared as described. ChR2-GFP appeared to be well localized to the plasma membrane at the cell body and throughout neuronal processes (Figure 1E). To assess the cell-type specificity of ChR2-GFP gene expression driven by the α-CaMKII promoter, we immunostained primate cortical slices with antibodies against the excitatory neuron-specific marker α-CaMKII (Jones et al., 1994; Tighilet et al., 1998), the inhibitory neuron-specific neurotransmitter GABA (Hendry et al., 1989; Houser et al., 1983), and the astrocyte-specific marker glial fibrillary acidic protein (GFAP) (Cahoy et al., 2008; McLendon and Bigner, 1994). Neurons expressing ChR2-GFP were positive for α-CaMKII (Figure 1Fi), but not GABA (Figure 1Fii) or GFAP (Figure 1Fiii). Of the ChR2-GFP-positive neurons examined, all coexpressed α-CaMKII (127/127 cells; Figure 1Gi), but none coexpressed GABA (Figure 1Gii, 0/78 cells) or GFAP (Figure 1Giii, 0/84 cells). In order to gauge the efficiency of viral labeling, we counted the fraction of α-CaMKII-positive cells that expressed ChR2-GFP. Near centers of injection sites, where ChR2-GFP expression peaked, 78% ± 8% of the α-CaMKII-positive cells expressed ChR2-GFP (mean ± SD; n = 3 fields of view; 42 ChR2-GFP neurons counted). Thus, lenti-virus expressing ChR2-GFP under the α-CaMKII promoter enables cell-specific targeting and efficient expression of ChR2-GFP in excitatory neurons of the monkey frontal cortex.
Figure 1. Expression of ChR2-GFP in Excitatory Neurons in Frontal Cortex of Primate Brain.
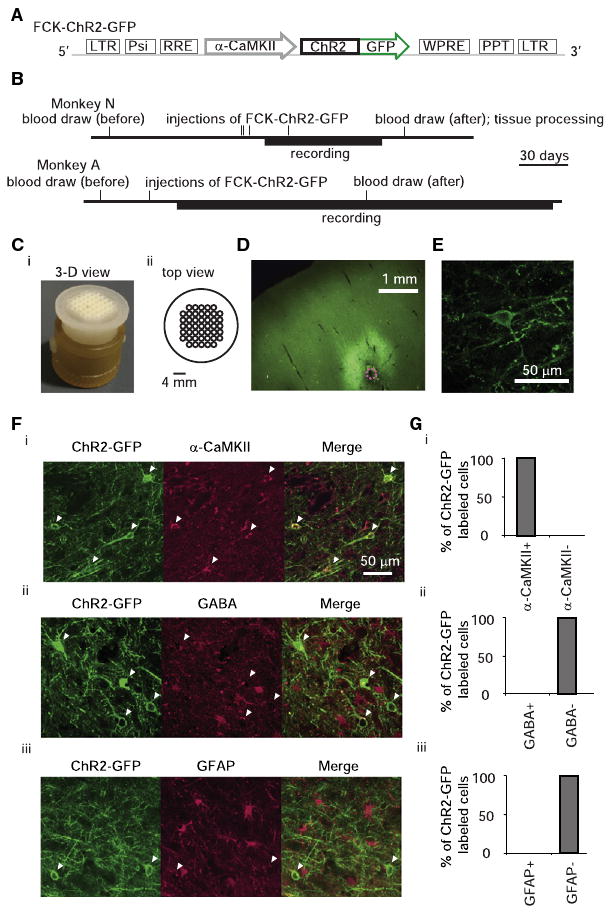
(A) Schematic of lentiviral cassette. (B) Timeline of experiments for monkey N (top) and monkey A (bottom). (C) 3D printed targeting grid, inserted into a recording chamber (Ci) and in a top-view schematic (Cii). (D) Fluorescence image showing ChR2-GFP expression in deep layers of cortex (coronal slice; dotted magenta circle indicates diameter of virus injection cannula). (E) Representative cortical neuron expressing ChR2-GFP. (F) Images of anti-GFP fluorescence (left) as well as immunofluorescence of three cell-type markers: α-CaMKII (Fi), GABA (Fii), and GFAP (Fiii) (middle; right, overlay of the two left images). Arrowheads indicate ChR2-GFP-positive cell bodies. (G) Percent of ChR2-GFP-positive cells coexpressing each of the three markers in (F).
Given the extended duration of nonhuman primate experiments, and the prospect of using cell-specific optical neuroprosthetics for therapy, we assessed the safety of ChR2-GFP expression in primate brain. After months of ChR2-GFP expression, during which time we repeatedly illuminated neurons with blue light and successfully made recordings, we saw widespread expression of ChR2-GFP in healthy-looking neurons, with no histological abnormalities in neurons or glia, and no immune reaction at the cellular or antibody level (Figure 2; see detailed text in Supplemental Data). These multiple lines of evidence together support the safety of ChR2-GFP expression in the brain of the nonhuman primate, and if supported by further and longer-term analyses, may provide the basis for cell-specific neuromodulation therapy in humans.
Figure 2. Analyses of Potential Immune Responses against ChR2-GFP-Expressing Neurons in Primate Cortex.
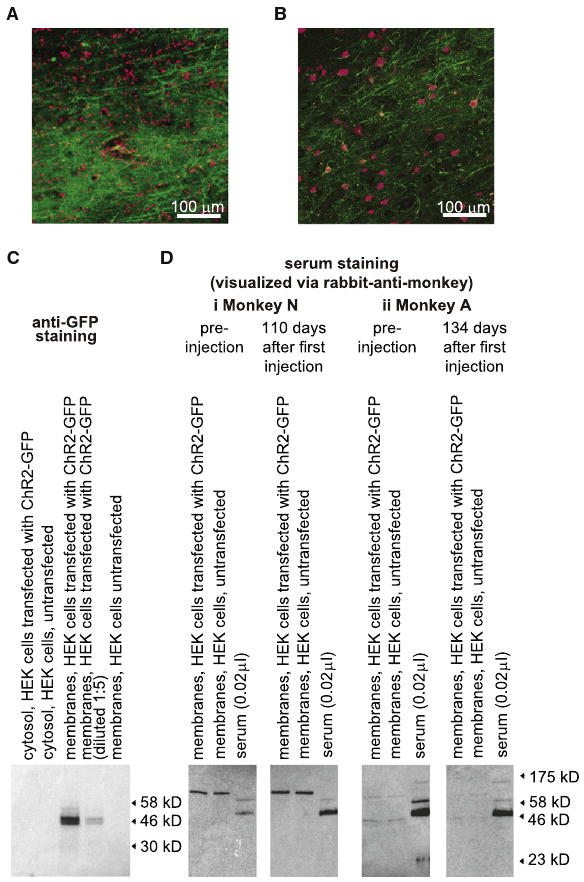
(A) Nuclear DNA staining (red; To-Pro-3 stain) of slices of monkey cortex containing ChR2-GFP-expressing neurons (green). (B) Neuronal staining (red; NeuN antibody) of slices of monkey cortex containing ChR2-GFP-expressing neurons (green). (C) Validation of ChR2-GFP expression in HEK cells via western blotting, using anti-GFP antibody. From left to right, lanes show, immunostained with anti-GFP: cytosolic fraction of HEK cells transfected with ChR2-GFP plasmid, cytosolic fraction of untransfected HEK cells, membrane fraction of HEK cells transfected with ChR2-GFP plasmid, membrane fraction (diluted 1:5) of HEK cells transfected with ChR2-GFP plasmid, and membrane fractions of untransfected HEK cells. (D) Assessment of monkey serum reaction to ChR2-GFP, for monkey N (Di) and monkey A (Dii), via western blotting, comparing preinjection (left) to postinjection (right). Membrane fractions of HEK cells transfected with ChR2-GFP (left lane), membrane fractions of untransfected HEK cells (middle lane), and monkey serum samples (right lane) were incubated with monkey serum (1:50 dilution), followed by rabbit-anti-monkey secondary antibody for visualization.
To assess the effect of optical activation of ChR2-expressing excitatory neurons on frontal cortical neural circuits in awake monkey, we developed a system appropriate for in vivo monkey use, coupling a fiber to a blue 473 nm laser (Bernstein et al., 2008) and assembling multiple electrodes into independently controlled drives (Figures 3Ai and 3Aii), which were then inserted into a single hole within the 3D printed grid (Figure 1C). This setup allowed us to record from neurons while exposing local cortex to pulses of blue light. In regions of cortex that were not virus labeled, we never observed light modulation of neural activity (n = 32 such sites). In regions that were virus labeled, many neurons increased their firing rate during cortical exposure to blue light (Figures 3B and 3C). We called these neurons “excited” units. In addition to these excited units, many neurons decreased their firing rate during cortical exposure to blue light (Figures 3D and 3E). We called these neurons “suppressed” units, because they did not increase firing rate during blue light exposure, but instead decreased firing rate during light delivery, even after brief illumination (i.e., a single light pulse). We hypothesized that since suppressed units decreased their firing rates without having undergone prior increases in spiking, the observed suppression was due to neural network activity, i.e., recruitment of inhibitory neurons downstream of the driven excitatory neurons. For both the excited and suppressed units, action potential waveforms elicited during light exposure were not different from waveforms observed in the dark (p>0.1 for each of n=15 excited single units; Kolmogorov-Smirnov test comparing waveform shapes in light versus dark; exemplars in Figures 3F and 3G). In regions where excited or suppressed units were found, few light-nonmodulated units were observed (Figure S5). These excited and suppressed units were also observed in the cortex of mice, when excitatory neurons expressing ChR2-GFP were activated by light (Figure S3). Light did, however, result in a low-frequency electrical artifact on our tungsten electrodes in the brain, presumably due to the photoelectric effect; this artifact was removed from our data by high-pass filtering (see Figure S1). Light (80 mW/mm2 radiant flux out the tip of the fiber) modulated neurons at distances over 1.2 mm away from the fiber (Figure S2).
Figure 3. Increases and Decreases in Neural Activity Resulting from Optical Stimulation of Excitatory Neurons.
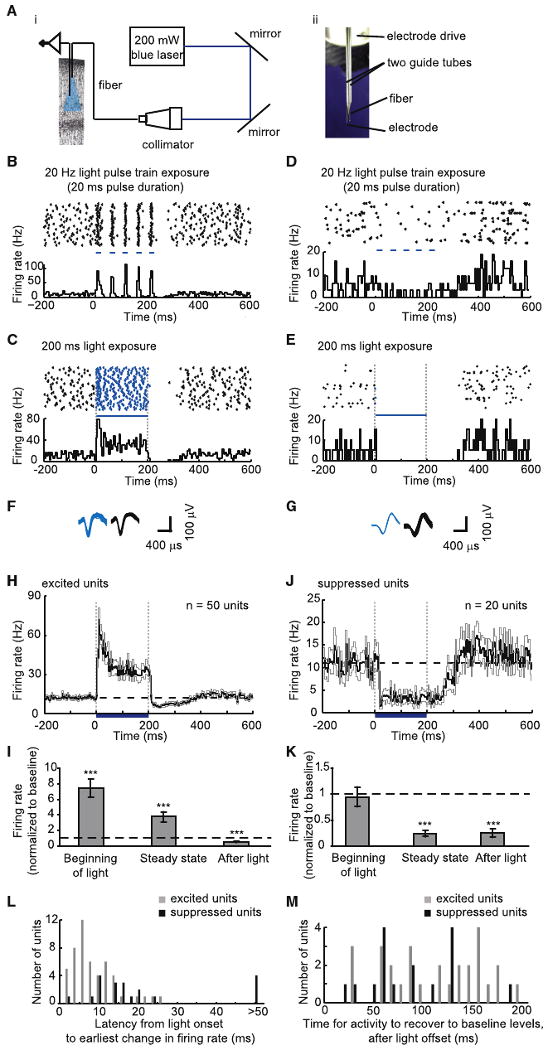
(A) Apparatus for optical activation and electrical recording. (Ai) Schematic. (Aii) Photograph, showing optical fiber (200 μm diameter) and electrode (200 μm shank diameter) in guide tubes. (B and C) Increases in spiking activity in one neuron during blue light illumination (five pulses, 20 ms duration each [B], and 1 pulse, 200 ms duration [C]). In each panel, shown at top is a spike raster plot displaying each spike as a black dot; 40 trials are shown in horizontal rows (in this and subsequent raster plots); shown at bottom is a histogram of instantaneous firing rate, averaged across all trials; bin size, 5 ms (in this and subsequent histogram plots). Periods of blue light illumination are indicated by horizontal blue dashes, in this and subsequent panels. (D and E) Decreases in spiking activity in one neuron during blue light illumination (five pulses, 20 ms duration (D), and one pulse, 200 ms duration [E]). As with (B) and (C), shown at top are spike raster plots and shown at bottom are histograms of instantaneous firing rate. (F and G) Action potential waveforms elicited during light (shown in blue, left) or occurring spontaneously in darkness (shown in black, right), for the neurons plotted in (C) and (E), respectively. (H) Instantaneous firing rate, averaged across all excited units recorded upon 200 ms blue light exposure (black line, mean; gray lines, mean ± SE; n = 50 units). (I) Relative firing rate (i.e., firing rate during the indicated period, divided by baseline firing rate) during the first 20 ms after light onset (“beginning of light”), during the period between 20 ms after light onset and 20 ms after light cessation (“steady state”), and during the 20 ms period starting 20 ms after light cessation (“after light”), for the n = 50 units shown in (H). (***), significantly different (p < 0.0001; paired t test) from baseline rate (shown as dotted line); plotted is mean ± SE. (J) Instantaneous firing rate averaged across all suppressed units upon 200 ms blue light exposure (black line, mean; gray lines, mean ± SE; n = 20 units). (K) Relative firing rate, during the beginning of light, steady state, and after light periods, for the n = 20 units shown in (J). (L) Histogram of latencies between light onset and the earliest change in firing rate, for excited units (gray bars, n = 50 units) and suppressed units (black bars, n = 20 units); latencies longer than 50 ms were plotted in a bin labeled “>50.” (M) Histogram of time elapsed until activity recovery to baseline after light cessation, for excited (gray bars, n = 28 units) and suppressed (black bars, n = 16 units) units that had lower-than-baseline firing rates during the after light period.
In the monkey cortex, we recorded 50 excited and 20 suppressed units during illumination with 200 ms blue light pulses. Out of these 70 units, 31 were single units (15 excited, 16 suppressed) and 39 were multi-units (35 excited, 4 suppressed). We pooled multiunits and single units for analysis unless otherwise indicated. Excited and suppressed units had similar baseline firing rates (p > 0.2, t test; only single units compared) and similar waveform shapes (see Supplemental Experimental Procedures). For excited units, firing rates increased rapidly at light onset, and then settled to a lower steady-state firing level (Figure 3H). For suppressed units, firing rates fell sharply after a short delay, and remained low for the duration of the light pulse (Figure 3J). For both excited and suppressed units, after light cessation the firing rates often dipped below baseline levels for ∼100 ms. We quantified the magnitude of these changes in firing rate during three distinct periods: the first 20 ms of light exposure (“beginning of light”), the period between 20 ms after light onset and 20 ms after light cessation (“steady state”), and during the 20 ms period starting 20 ms after light cessation (“after light”). Excited units fired at 750%, 370%, and 46% of baseline firing rate during these three periods, respectively, in each case significantly different from baseline (p < 0.0001 for each, paired t test; Figure 3I). For single units, which yield absolute values of firing rate, excited neurons fired at 37 ± 7 Hz, 16 ± 4 Hz, and 1.3 ± 1 Hz during these three periods (mean ± standard error [SE]; n = 15 single units); baseline their firing rates was 6.5 ±1.3 Hz. In contrast to the excited units, suppressed units did not change their firing rates relative to baseline during the beginning of light period (p > 0.5, paired t test; Figure 3K), but reduced their firing rates by 76% and 75%, respectively, during the steady state and after light periods (significantly lower than that during baseline [p < 0.0001, paired t test], but not different from each other; p > 0.8). Suppressed single units fired at 7.4 ± 1.7 Hz, 3.1 ± 1.0 Hz, and 2.5 ± 1.2 Hz during these three periods, respectively (mean ± SE; n=16 single units); baseline firing rate was 9.9 ± 2.0 Hz.
We compared the latencies to changes in firing rate between excited versus suppressed units, and found two different, but overlapping, distributions. Excited units rapidly responded to light with latencies of 8.8 ± 0.8 ms (mean ± SE; Figure 3L). This short latency was not different from the first-spike latency of ChR2-positive cultured pyramidal neurons responding to pulses of blue light (p > 0.6, unpaired t test; compared to published data in Boyden et al., 2005), consistent with the idea, but not proving, that excited units were ChR2-positive pyramidal cells. In contrast to the short latencies of excited units, suppressed units began decreasing their firing rates 30.8 ± 8.0 ms after light onset (mean ± SE), a latency significantly longer than the latency for the increases in firing rates of excited units (p < 0.0001, unpaired t test). This difference is consistent with our hypothesis that suppressed units decreased their firing rates through neural network mechanisms involving inhibitory neuron recruitment, whereas excited units were directly activated by light. After light cessation (after light period), the majority of excited and suppressed units exhibited firing rates below baseline levels (28 out of 50 excited units; 16 out of 20 suppressed units). The time for this reduced firing rate to recover to baseline was similar for excited and suppressed units (Figure 3M, p > 0.1, unpaired t test), consistent with the idea that suppressive influences downstream of excitatory neuron activation are mediated by a neural-network-scale phenomenon such as inhibitory neuron recruitment.
To probe the nature of neural suppression further, we examined the activity of single units before, during, and after light exposure. For excited cells, we found that increases in firing rate during optical stimulation were independent of baseline firing rate (R2 = 0.025, p > 0.5; Figure 4A; n = 15 excited single units). In addition, decreases in firing rate after light cessation were independent of the light-induced increases in firing rate (R2 = 0.096, p > 0.2; Figure 4B). Finally, the time for firing rate to recover to baseline levels after light cessation was independent of prior increases in firing rate (R2 = 0.016, p > 0.7; Figure 4C; n = 12 excited single units that decreased activity during the after light period). Thus, excited cells fired before, during, and after light exposure, in independent fashions. In contrast to excited units, for suppressed units decreases in firing rate during light exposure were highly correlated with baseline firing rate (R2 = 0.749, p < 0.0001; Figure 4D; n = 16 suppressed single units): for each additional Hz of baseline firing rate, light exposure decreased firing by an additional ∼0.6 Hz. In addition, the decrease in firing rate of suppressed neurons after light cessation was correlated with the decrease in firing rate during light exposure (R2 = 0.673, p < 0.0001; Figure 4E). However, as for excited cells, the time for the firing rate to recover to baseline level was independent of prior reductions in activity levels for suppressed cells (e.g., during the steady state period; R2 = 0.045, p > 0.4; Figure 4F; n = 13 suppressed single units that had significant decreases in activity during the after light period). Thus, for suppressed cells, but not for excited cells, light-induced changes in activity were correlated with baseline activity, as though the magnitudes of spontaneous and light-suppressed activity were both functions of a common neural network state. We further probed the response of excited cells with trains of light pulses at 10, 20, and 50 Hz (Figure S6), finding that while spike probability fell during long, high-frequency trains, spike timing remained reliable, and therefore ChR2 may subserve the ability to use light to control cortical synchrony.
Figure 4. Comparison of Neural Activity Levels within Excited and Suppressed Single Units, before, during, and after Light Exposure.

(A) Firing rate change during the beginning of light period (i.e., firing rate during beginning of light minus baseline firing rate) versus baseline firing rate, for excited cells (n = 15 excited single units). (B) Firing rate change during the after light period versus during the beginning of light period, for excited single units. (C) Time elapsed until activity recovery to baseline level after light cessation, versus firing rate change during the beginning of light period, for excited single units. (D) Firing rate change during the steady state period, versus baseline firing rate, for suppressed cells (n=16 suppressed single units). (E) Firing rate change during the after light period versus during the steady state period, for suppressed single units. (F) Time elapsed until activity recovery to baseline level after light cessation, versus firing rate change during the steady state period, for suppressed single units.
Discussion
We have demonstrated millisecond-timescale optical activation of excitatory neurons in the frontal cortex of nonhuman primates, using lentivirally delivered ChR2, and have characterized the impact of such optical control on cortical circuits. This work demonstrates the feasibility of applying optogenetic methods to primate neural circuits, and points the way toward the potential use of optical control in a new generation of therapies for the improvement of human health. Immediately, this technology makes it possible to activate a region containing a set of excitatory neurons while avoiding the modulation of fibers of passage, or of neurons projecting into the region. Light stimulation did result in a slow electrical artifact on the metal electrode, which was easily filtered out from our spike recordings. Single viral injections labeled on the order of 1 mm3 of brain tissue, comparable to the volume illuminated by single optical fibers, suggesting that arrays of viral injectors and optical fibers may enhance the ability to systematically modulate cell and circuit activities during behavior.
We found that whereas many neurons were excited during light activation of excitatory neurons, others were profoundly suppressed during light exposure. In addition, many excited and suppressed cells exhibited a period of reduced neural activity after cessation of light activation. These excited and suppressed effects were also observed in mouse cortical neurons under similar optical stimulation conditions. These effects may be due to biophysical properties of the neurons recorded, for example hyperpolarization after depolarization-induced opening of BK and SK potassium channels (Bekkers, 2000; Sah and Davies, 2000; Storm, 1987; Vogalis et al., 2003). But several independent lines of reasoning support the hypothesis that suppression emerges from recruitment of networks of inhibitory cells downstream of activated ChR2-positive excitatory cells. First, suppressed neurons underwent reductions in spike firing without having undergone prior increases in spike firing, which implies that cell-autonomous mechanisms such as postdepolarization hyperpolarization cannot be the sole mechanism mediating the observed suppression. Second, the latency to the decrease in firing after light onset was significantly longer for suppressed cells than the latency to the increase in firing was for excited cells, consistent with suppressed cells being downstream of light-activated cells. Third, suppressed neurons decreased activity during light exposure in proportion to their basal firing rate, whereas light-driven excitation was independent of basal firing rate, consistent with suppression being mediated through a mechanism related to the one that sustains baseline firing, i.e., network activity. Finally, postillumination reductions in neural activity were similar in duration across excited and suppressed neurons, again suggesting that these effects may be more due to emergent properties of the neural network that a given neuron is embedded in, rather than that cell's autonomous history of activity. Future studies will explore which inhibitory neurons are recruited by excitatory neurons to create activity patterns like those found here; one possibility is that somatostatin-positive interneurons that can be activated by stimulation of single excitatory neurons (Kapfer et al., 2007) could potentially mediate excitation-induced suppression. It will also be interesting to see if such dynamics can subserve oscillatory activity: in Figures 3H and 3J, the rebound depolarization after the end of activity suppression is suggestive of a possible natural timescale of activity fluctuation in the delta-theta range.
Electrical microstimulation is an important tool for both basic neuroscience and for therapeutic neuromodulation, but how it impacts neural circuit dynamics remains unclear. In microstimulation experiments where recording was also performed, some neurons dramatically decreased activity in response to electrical microstimulation (e.g., Butovas et al., 2006; Butovas and Schwarz, 2003; Seidemann et al., 2002). These electrical stimulation-induced decreases possessed some of the same attributes of the suppressions here observed; for example, the duration of the inhibition was largely independent of the amount of activation induced (Butovas and Schwarz, 2003). This inhibition was pharmacologically associated with GABAergic neuron recruitment (Butovas et al., 2006), but it remained unknown whether electrical microstimulation must directly recruit GABAergic neurons, or whether activation of excitatory neurons would be sufficient to create such inhibition. Here we prove that driving excitatory neurons alone is sufficient to result in periods of activity suppression in a significant population of cortical neurons. We found similar responses in mouse neocortex, which suggests that such neural dynamics might be a general property of neocortical circuits under neuromodulation. Thus, even when just one cell type is manipulated, its impact on the brain must be evaluated in the context of the neural network in which it is embedded. Principles must be derived for how to control a circuit, even given a delimited set of cell types to be controlled, in order to achieve a desired physiological, behavioral, or clinical outcome.
Optically activating excitatory neurons is just one step along the path of implementing cell-type-specific optical control in primates. Future viral, promoter, injection, and illumination innovations will need to be developed to match the manipulations possible in mice and other classical genetic model systems. Optical neural silencing strategies for primates will also be critical, perhaps involving light-activated chloride pumps such as halorhodopsins (Halo/NpHR) (Han and Boyden, 2007; Zhang et al., 2007). Especially for questions involving higher-order activity patterns such as neural synchrony, the ability to use ChR2 and Haloin concert to create “informational lesions,” in which precise neural patterns are disrupted, may prove especially useful (Han and Boyden, 2007).
Launching the verification of the safety and efficacy of ChR2 function in rhesus macaques is a critical step toward any potential clinical translational path for cell-type-specific optical neural control prosthetics. Given that in many disorders, the functions of specific cell types are compromised, it is possible that the ability to optically remedy aberrant activity in specific cell types will spur precise, side-effect-free treatments for neural disorders. As a first step toward this synthetic neurobiology goal, here we have shown that ChR2 performs efficaciously and without immune attack in the macaque brain, and appears to be safe over many months despite repeated viral injections and repeated illumination sessions.
Experimental Procedures
Detailed descriptions are provided in the Supplemental Data. All procedures were in accordance with the NIH Guide for Laboratory Animals and approved by the MIT Animal Care and Use and Biosafety Committees. Two rhesus monkeys, 7–11 years of age, weighing 8–15 kg, were equipped for awake headfixed physiology. High-titer replication-incompetent lentivirus encoding for ChR2-GFP was produced and injected into premotor cortex/frontal eye fields, via custom hardware. Optical stimulation proceeded via a 200 μm diameter optical fiber coupled to a 200 mW blue laser. Electrophysiological recording was performed using tungsten electrodes guided parallel to the optical fiber, using independent microdrives. Signal conditioning and acquisition were performed with a Plexon data acquisition system and analyzed with Matlab. The brain of one of the two monkeys was fixed and examined with immunostaining and confocal microscopy.
Supplementary Material
Supplemental Figure 1. Voltage deflections observed on tungsten electrodes immersed in saline (A) or brain (B), upon tip exposure to 200 ms blue light pulses (i) or trains of 10 ms blue light pulses delivered at 50 Hz (ii). Light pulses are indicated by blue dashes. Electrode data was hardware filtered using two data acquisition channels operating in parallel, yielding a low-frequency component (“field potential channel”) and a high-frequency component (“spike channel”). For the “spike channel” traces taken in brain (B), spikes were grouped into 100 ms bins, and then the binned spikes were displayed beneath corresponding parts of the simultaneously acquired “field potential channel” signal (59 and 53 repeated light exposures for Bi and Bii respectively). (Shown are the spikes in 8 such bins – the 2 bins before light onset, the 2 bins during the light delivery period, and the 4 bins after light cessation.) For all other signals shown, 10 overlaid traces are plotted.
Supplemental Figure 2. Light intensity dependence of neural activity driven by ChR2-mediated activation of excitatory neurons. A, neural activity during ‘Beginning of light’ (i), ‘Steady state’ (ii), and ‘After light’ (iii) time periods, upon illumination with different light power intensities (plotted is mean ± SE; n = 11 excited units). B, neural activity during ‘Beginning of light’ (i), ‘Steady state’ (ii), and ‘After light’ (iii) periods, when optical fibers were retracted, holding the electrode still (plotted is mean ± SE; n = 7 excited units).
Supplemental Figure 3. Increases and decreases in firing rates of neurons in mouse frontal cortex, during optical stimulation of excitatory neurons. A, increases in spiking activity in one neuron during blue light illumination (200 ms duration). Top, spike raster plot displaying each spike as a black dot; 25 trials are shown in horizontal rows, in this and subsequent raster plots; bottom, histogram of instantaneous firing rate (averaged across all trials; bin size, 5 ms, in this and subsequent histogram plots). B, decreases in spiking activity in one neuron during blue light illumination (200 ms duration). Top, spike raster plot; bottom, histogram of instantaneous firing rate.
Supplemental Figure 4. Fluorescence magnitudes measured along lines taken through the centers of spheres of virally-labeled neurons, each created by one injection of 1 microliter of ChR2-GFP lentivirus. Shown are the mean (black lines) and the mean ± SD (gray lines) for n = 6 well-isolated spheres of virally-labeled cells.
Supplemental Figure 5. Quantitation of the number of light-responsive and light-unresponsive units detected along five individual electrode tracks, each sampled every 300 microns. The farthest-separated depths where units responded to light were first identified, and each unit recorded between those two depths was then classified according to its activity profile – excitation, suppression, or nonresponsive – during light illumination.
Supplemental Figure 6. Driving oscillatory cortical activity by activating ChR2 expressing excitatory neurons with rhythmic blue light pulse trains, at 10 Hz (A), 20 Hz (B), and 50 Hz (C). For each of these three pulse rates, plotted in subpanel i is the histogram of instantaneous firing rate (bin size, 5 ms) averaged across all excited units tested (black lines, mean; gray lines, mean ± SE; n = 8 units for 10 Hz, n = 14 units for 20 Hz, and n = 9 units for 50 Hz). Plotted in subpanel ii is the firing rate divided by baseline firing rate, during the 20 ms following the onset of each light pulse, and averaged across all excited units tested (mean ± SE)). Plotted in subpanel iii is the jitter of spike times elicited by each pulse, calculated as the across-trial standard deviation of the time of the first spike elicited within 20 ms of each light pulse onset and averaged across all excited units tested (mean ± SE).
Acknowledgments
We thank the MIT Division of Comparative Medicine for help with serum collection, and H. Hall and P. Harlen for help with preparing cortical slices. We thank members of the Boyden and Desimone labs, and E. Tehovnik, for their suggestions on the manuscript. This work was supported by NIH-EY002621-31; a HHWF fellowship to X.H.; a McGovern Institute Neurotechnology Award to E.S.B., R.D., and X.H.; grant NIH-EY12848 to A.G.; and grants NIH-EY017292 and EY017921 to R.D. E.S.B. acknowledges support from the NIH Director's New Innovator Award (DP2 OD002002-01), NSF, Department of Defense, NARSAD, Alfred P. Sloan Foundation, Jerry Burnett Foundation, SFN Research Award for Innovation in Neuroscience, MIT Media Lab, MIT McGovern Institute, Benesse Foundation, MIT Neurotechnology Fund, and Wallace H. Coulter Foundation.
Footnotes
References
- Bekkers JM. Distribution of slow AHP channels on hippocampal CA1 pyramidal neurons. J Neurophysiol. 2000;83:1756–1759. doi: 10.1152/jn.2000.83.3.1756. [DOI] [PubMed] [Google Scholar]
- Bernstein JG, Han X, Henninger MA, Ko EY, Qian X, Franzesi GT, McConnell JP, Stern P, Desimone R, Boyden ES. Prosthetic systems for therapeutic optical activation and silencing of genetically-targeted neurons. Proc Soc Photo Opt Instrum Eng. 2008;6854:68540H. doi: 10.1117/12.768798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Butovas S, Schwarz C. Spatiotemporal effects of microstimulation in rat neocortex: a parametric study using multielectrode recordings. J Neurophysiol. 2003;90:3024–3039. doi: 10.1152/jn.00245.2003. [DOI] [PubMed] [Google Scholar]
- Butovas S, Hormuzdi SG, Monyer H, Schwarz C. Effects of electrically coupled inhibitory networks on local neuronal responses to intra-cortical microstimulation. J Neurophysiol. 2006;96:1227–1236. doi: 10.1152/jn.01170.2005. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Chong KY, Martinovich C, Simerly C, Schatten G. Transgenic monkeys produced by retroviral gene transfer into mature oocytes. Science. 2001;291:309–312. doi: 10.1126/science.291.5502.309. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lenti-virus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci USA. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SH, Jones EG, Emson PC, Lawson DE, Heizmann CW, Streit P. Two classes of cortical GABA neurons defined by differential calcium binding protein immunoreactivities. Exp Brain Res. 1989;76:467–472. doi: 10.1007/BF00247904. [DOI] [PubMed] [Google Scholar]
- Houser CR, Hendry SH, Jones EG, Vaughn JE. Morphological diversity of immunocytochemically identified GABA neurons in the monkey sensory-motor cortex. J Neurocytol. 1983;12:617–638. doi: 10.1007/BF01181527. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Kakuda M, Araki R, Yawo H. Kinetic evaluation of photosensitivity in genetically engineered neurons expressing green algae light-gated channels. Neurosci Res. 2006;54:85–94. doi: 10.1016/j.neures.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Jones EG, Huntley GW, Benson DL. Alphacalcium/calmodulin-dependent protein kinase II selectively expressed in a subpopulation of excitatory neurons in monkey sensory-motor cortex: comparison with GAD-67 expression. J Neurosci. 1994;14:611–629. doi: 10.1523/JNEUROSCI.14-02-00611.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat Neurosci. 2007;10:743–753. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- Li X, Gutierrez DV, Hanson MG, Han J, Mark MD, Chiel H, Hegemann P, Landmesser LT, Herlitze S. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci USA. 2005;102:17816–17821. doi: 10.1073/pnas.0509030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Richmond BJ, Murray EA, Saunders RC, Steenrod S, Stubble-field BK, Montague DM, Ginns EI. DNA targeting of rhinal cortex D2 receptor protein reversibly blocks learning of cues that predict reward. Proc Natl Acad Sci USA. 2004;101:12336–12341. doi: 10.1073/pnas.0403639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon RE, Bigner DD. Immunohistochemistry of the glial fibrillary acidic protein: basic and applied considerations. Brain Pathol. 1994;4:221–228. doi: 10.1111/j.1750-3639.1994.tb00837.x. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- Seidemann E, Arieli A, Grinvald A, Slovin H. Dynamics of depolarization and hyperpolarization in the frontal cortex and saccade goal. Science. 2002;295:862–865. doi: 10.1126/science.1066641. [DOI] [PubMed] [Google Scholar]
- Stettler DD, Yamahachi H, Li W, Denk W, Gilbert CD. Axons and synaptic boutons are highly dynamic in adult visual cortex. Neuron. 2006;49:877–887. doi: 10.1016/j.neuron.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EM, Yamaguchi Y, Horwitz GD, Gosgnach S, Lein ES, Goulding M, Albright TD, Callaway EM. Selective and quickly reversible inactivation of mammalian neurons in vivo using the Drosophila allatostatin receptor. Neuron. 2006;51:157–170. doi: 10.1016/j.neuron.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Tighilet B, Hashikawa T, Jones EG. Cell- and lamina-specific expression and activity-dependent regulation of type II calcium/calmodulin-dependent protein kinase isoforms in monkey visual cortex. J Neurosci. 1998;18:2129–2146. doi: 10.1523/JNEUROSCI.18-06-02129.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci. 2003;18:3155–3166. doi: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- Yang SH, Cheng PH, Banta H, Piotrowska-Nitsche K, Yang JJ, Cheng EC, Snyder B, Larkin K, Liu J, Orkin J, et al. Towards a transgenic model of Huntington's disease in a non-human primate. Nature. 2008;453:921–924. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Voltage deflections observed on tungsten electrodes immersed in saline (A) or brain (B), upon tip exposure to 200 ms blue light pulses (i) or trains of 10 ms blue light pulses delivered at 50 Hz (ii). Light pulses are indicated by blue dashes. Electrode data was hardware filtered using two data acquisition channels operating in parallel, yielding a low-frequency component (“field potential channel”) and a high-frequency component (“spike channel”). For the “spike channel” traces taken in brain (B), spikes were grouped into 100 ms bins, and then the binned spikes were displayed beneath corresponding parts of the simultaneously acquired “field potential channel” signal (59 and 53 repeated light exposures for Bi and Bii respectively). (Shown are the spikes in 8 such bins – the 2 bins before light onset, the 2 bins during the light delivery period, and the 4 bins after light cessation.) For all other signals shown, 10 overlaid traces are plotted.
Supplemental Figure 2. Light intensity dependence of neural activity driven by ChR2-mediated activation of excitatory neurons. A, neural activity during ‘Beginning of light’ (i), ‘Steady state’ (ii), and ‘After light’ (iii) time periods, upon illumination with different light power intensities (plotted is mean ± SE; n = 11 excited units). B, neural activity during ‘Beginning of light’ (i), ‘Steady state’ (ii), and ‘After light’ (iii) periods, when optical fibers were retracted, holding the electrode still (plotted is mean ± SE; n = 7 excited units).
Supplemental Figure 3. Increases and decreases in firing rates of neurons in mouse frontal cortex, during optical stimulation of excitatory neurons. A, increases in spiking activity in one neuron during blue light illumination (200 ms duration). Top, spike raster plot displaying each spike as a black dot; 25 trials are shown in horizontal rows, in this and subsequent raster plots; bottom, histogram of instantaneous firing rate (averaged across all trials; bin size, 5 ms, in this and subsequent histogram plots). B, decreases in spiking activity in one neuron during blue light illumination (200 ms duration). Top, spike raster plot; bottom, histogram of instantaneous firing rate.
Supplemental Figure 4. Fluorescence magnitudes measured along lines taken through the centers of spheres of virally-labeled neurons, each created by one injection of 1 microliter of ChR2-GFP lentivirus. Shown are the mean (black lines) and the mean ± SD (gray lines) for n = 6 well-isolated spheres of virally-labeled cells.
Supplemental Figure 5. Quantitation of the number of light-responsive and light-unresponsive units detected along five individual electrode tracks, each sampled every 300 microns. The farthest-separated depths where units responded to light were first identified, and each unit recorded between those two depths was then classified according to its activity profile – excitation, suppression, or nonresponsive – during light illumination.
Supplemental Figure 6. Driving oscillatory cortical activity by activating ChR2 expressing excitatory neurons with rhythmic blue light pulse trains, at 10 Hz (A), 20 Hz (B), and 50 Hz (C). For each of these three pulse rates, plotted in subpanel i is the histogram of instantaneous firing rate (bin size, 5 ms) averaged across all excited units tested (black lines, mean; gray lines, mean ± SE; n = 8 units for 10 Hz, n = 14 units for 20 Hz, and n = 9 units for 50 Hz). Plotted in subpanel ii is the firing rate divided by baseline firing rate, during the 20 ms following the onset of each light pulse, and averaged across all excited units tested (mean ± SE)). Plotted in subpanel iii is the jitter of spike times elicited by each pulse, calculated as the across-trial standard deviation of the time of the first spike elicited within 20 ms of each light pulse onset and averaged across all excited units tested (mean ± SE).