History, Molecular Mechanisms, and Endoscopic Treatment of Barrett’s Esophagus (original) (raw)
. Author manuscript; available in PMC: 2011 Mar 1.
Published in final edited form as: Gastroenterology. 2010 Jan 18;138(3):854–869. doi: 10.1053/j.gastro.2010.01.002
Abstract
This report is written as an adjunct to the American Gastroenterological Association Institute’s medical position statement and technical review on the management of Barrett’s esophagus, which will be published in the near future. Those documents will consider a number of broad questions on the diagnosis, clinical features, and management of patients with Barrett’s esophagus, and the reader is referred to the technical review for an in-depth discussion of those topics. In this report, we review historical, molecular, and endoscopic therapeutic aspects of Barrett’s esophagus that are of interest to clinicians and researchers.
History of Barrett’s Esophagus
Barrett’s esophagus is a condition named for the late Norman Rupert Barrett, an influential esophageal surgeon who was born in Adelaide, Australia in 1903.1 Barrett worked for most of his career as a consultant surgeon at St. Thomas’ Hospital in London. He was a pioneer in the field of thoracic surgery and a charismatic academic leader who served for more than 25 years as editor of Thorax. By all accounts, Norman Barrett was an outstanding surgeon, scholar, and teacher. However, Norman Barrett was not the first to describe the condition that now bears his name; in fact, his original contentions about the nature and pathogenesis of the condition were incorrect. The eponym “Barrett’s esophagus” continues to evoke confusion and controversy, and authorities still dispute the definition of the disorder.2 To appreciate these controversies, it is helpful to consider some key events in the relatively brief history of Barrett’s esophagus.
In 1950, Barrett published a treatise proposing that the esophagus should be defined as “that part of the foregut, distal to the cricopharyngeal sphincter, which is lined by squamous epithelium”.3 He commented on earlier reports describing patients with ulcerations in a tubular organ that grossly appeared to be the esophagus, but whose distal, ulcerated portion was lined by columnar epithelium. Since Barrett had defined the esophagus by its squamous lining, he argued that the ulcerated, columnar-lined viscus described in those reports was a tubular segment of stomach that had been tethered within the chest by a congenitally short esophagus. To support that contention, Barrett noted that the ulcerated columnar lining always was identified as “histologically gastric in type”.
Barrett himself claimed that peptic ulcer of the esophagus was first reported in 1839 by the German pathologist Albers.3 However, credit for describing the columnar-lined esophagus probably should go to Wilder Tileston, a pathologist who, while working in Boston in 1906, described 3 cases of “peptic ulcer of the oesophagus”, and noted “the close resemblance of the mucous membrane about the ulcer to that normally found in the stomach”.4 Tileston wrote that “the first requisite for the formation of the peptic ulcer of the oesophagus is an insufficiency of the cardia” (i.e. gastroesophageal reflux). Thus, almost a half-century before Barrett, Tileston described the columnar-lined esophagus and correctly attributed the pathogenesis of the associated ulceration to gastroesophageal reflux.
In 1953, Allison and Johnstone described 7 patients who had reflux esophagitis involving an “oesophagus lined with gastric mucous membrane”.5 In this report, they refuted Barrett’s contention that the tubular, intra-thoracic, columnar-lined viscus was stomach. They noted that, unlike the stomach, the columnar-lined organ lacked a peritoneal covering, harbored islands of squamous epithelium, and had submucosal glands and a muscularis propria typical of the esophagus. In deference to Barrett, the editor of the journal to which they had submitted their report, Allison and Johnstone suggested that ulcerations in the columnar-lined esophagus should be called “Barrett’s ulcers”. Barrett eventually accepted Allison and Johnstone’s arguments and, in a report published in 1957, suggested that the condition should be called “lower oesophagus lined by columnar epithelium”.6 Whether justified or not, the eponym Barrett’s esophagus has been retained.
In another influential report published in Thorax in 1961, an Australian surgeon named John Hayward elaborated his opinions on the histological features of the distal esophagus.7 He contended that the distal 1– 2 cm of the esophagus is normally lined by a mucus-secreting, junctional epithelium (also called gastric cardia-type epithelium). Hayward argued that “…if squamous epithelium joined gastric epithelium of fundal [acid-secreting] type directly, it would be liable to digestion at the junction. The buffer zone of junctional epithelium, which does not secrete acid or pepsin but is resistant to them, has to be interposed.” Hayward provided no data to support his contentions and Barrett remarked that Hayward’s report contained “a lot of nonsense”.1 Nevertheless, Barrett published the report essentially unaltered, and it has had substantial influence on the course of studies on Barrett’s esophagus.
The histology of the columnar-lined esophagus remains a controversial issue to this day. Barrett, and virtually all of the investigators who wrote about the condition before he did, described an acid-secreting, gastric type of columnar epithelium lining the esophagus.3 In 1951, Bosher and Taylor were the first to describe intestinal-type goblet cells in the columnar-lined esophagus.8 In 1952, Morson and Belcher reported a patient who had an adenocarcinoma in an esophageal mucosa that had “atrophic changes with a tendency towards intestinal type containing many goblet cells”.9 Still other investigators described cardia-type epithelium in Barrett’s esophagus.10
This confusing situation was clarified somewhat in 1976, when Paull et al. reported a systematic study of 11 patients with Barrett’s esophagus who had esophageal biopsy specimens taken above the lower esophageal sphincter using manometric guidance.11 Those patients were found to have as many as 3 types of columnar epithelia lining the distal esophagus: 1) a junctional (cardia-type) epithelium that comprised mucus-secreting cells, 2) a gastric fundic-type epithelium with parietal and chief cells, and 3) intestinal-type metaplasia, which the authors called specialized columnar epithelium, with prominent goblet cells. The 3 epithelial types occupied different zones in the columnar-lined esophagus, with intestinal-type metaplasia adjacent to squamous epithelium in the most proximal segment, followed by cardia-type epithelium, with gastric fundic-type epithelium lining the most distal esophageal segment.
By the 1970’s, it was well established that Barrett’s esophagus was associated with severe gastroesophageal reflux disease (GERD) and hiatal hernia,12-14 conditions that can obscure the endoscopic landmarks used to identify the gastroesophageal junction (GEJ). Endoscopists intent on collecting biopsy samples from the distal esophagus of patients with these disorders could mistakenly take those specimens from the proximal stomach, resulting in a spurious diagnosis of Barrett’s esophagus lined by gastric-type epithelium. Further complicating diagnostic issues, Hayward had contended that even the normal esophagus could be lined by up to 2 cm of cardia-type epithelium.7 Therefore, analysis of biopsy samples from this “normal” columnar mucosa also might result in a spurious diagnosis of Barrett’s esophagus. These factors created major problems for investigators, because accurate diagnostic criteria are a prerequisite for a meaningful study of a disease.
In the early 1980’s, investigators intent on avoiding the problem of spurious diagnoses established diagnostic criteria for Barrett’s esophagus based on an arbitrary extent of esophageal columnar lining above the GEJ. For example, Skinner et al. chose 3 cm as the extent of esophageal columnar lining required for patients to be enrolled in studies of Barrett’s esophagus.15 These arbitrary, investigative criteria were eventually used by clinicians as diagnostic criteria. As a result, endoscopists often dismissed columnar epithelium limited to the distal few centimeters of the esophagus as normal, and obtained biopsy specimens to confirm a diagnosis of Barrett’s esophagus only when columnar lining extended some arbitrary distance (e.g. >3 cm) above the GEJ. Furthermore, endoscopists sought to identify Barrett’s esophagus almost exclusively in patients who had symptoms and endoscopic signs of severe GERD. By adhering to such diagnostic guidelines, physicians minimized the problem of mis-diagnosis, but failed to identify short segments of metaplastic epithelium in the distal esophagus.
By the 1980s, it was well established that adenocarcinoma was associated with Barrett’s esophagus.16-18 When reports of esophageal adenocarcinomas in patients with Barrett’s esophagus provided a description of the associated Barrett’s epithelium, they almost invariably identified that epithelium as intestinal-type metaplasia, usually with dysplastic features.19 By the late 1980’s, intestinal metaplasia was widely regarded as both the most common type of Barrett’s epithelium and the epithelial type associated with cancer development.20,21 Barrett’s esophagus had little clinical importance outside of its malignant predisposition, and intestinal metaplasia was distinct in its histologic appearance (and unlike the gastric types of Barrett’s epithelia, clearly abnormal when located at the GEJ). Consequently, some researchers chose to define Barrett’s esophagus by the presence of intestinal metaplasia.22,23 This diagnostic criterion, arguably based more on convenience than on science, also was adopted into clinical practice.
An esophageal biopsy specimen showing intestinal-type metaplasia had become virtually a sine qua non for the diagnosis of Barrett’s esophagus by the early 1990s.22 Nevertheless, in the early 1990s, most endoscopists would not take esophageal biopsy specimens to seek a diagnosis of Barrett’s esophagus unless their patients had GERD with some minimal extent of esophageal columnar lining (e.g. at least 3 cm). That practice was challenged in 1994, when Spechler et al. showed that 18% of consecutive patients in a general endoscopy unit who had columnar epithelium that involved <3 cm of the distal esophagus also had intestinal metaplasia.24 Futhermore, they showed that symptoms and endoscopic signs of GERD were not reliable markers for intestinal metaplasia in the distal esophagus. Numerous subsequent studies confirmed that short segments of intestinal metaplasia frequently line the distal esophagus of individuals with no signs or symptoms of GERD.25-27 Since the late 1990s, therefore, Barrett’s esophagus has been categorized as long-segment (when the metaplastic epithelium extends at least 3 cm above the GEJ) or short-segment (when there is <3 cm of metaplastic epithelium lining the esophagus).28 Thus, 5 decades after the publication of Norman Barrett’s treatise, the diagnostic criteria for Barrett’s esophagus had evolved from an esophagus lined extensively by gastric epithelium in patients with severe GERD to an esophagus lined by any extent of intestinal epithelium in patients who might not have GERD.
A recent issue of contention is whether the presence of gastric cardia-type epithelium in the distal esophagus warrants a diagnosis of Barrett’s esophagus.29 The British Society of Gastroenterology’s recent guidelines specifically state that only “columnar lined oesophagus on histology” (i.e. cardia- or intestinal-type epithelium) is needed for the diagnosis of Barrett’s esophagus.30 Data indicate that cardia-type epithelium is not normal, as Hayward had contended, but rather a metaplastic lining that develops as a consequence of GERD.31 Histochemical and genetic studies of cardia-type epithelium have revealed molecular abnormalities, similar to those found in intestinal metaplasia, that could predispose patients to carcinogenesis,32,33 and recent clinical studies support the concept that cardia-type epithelium has malignant potential.34-36 This issue and others related to diagnostic criteria for Barrett’s esophagus are discussed in detail in the AGA Institute’s technical review on Barrett’s esophagus, which recommends that Barrett’s esophagus should now be defined as “the condition in which any extent of metaplastic columnar epithelium that predisposes to cancer development replaces the stratified squamous epithelium that normally lines the distal esophagus.”
Physicians should consider how and when the diagnostic criteria for Barrett’s esophagus changed when evaluating reports on the disorder. Short-segment Barrett’s esophagus was not widely recognized until 1994,24 and the vast majority of studies reported before that year included patients with only long-segment disease. More recent studies, however, include a substantial proportion of patients with short-segment Barrett’s esophagus. Patients with short-and long-segment Barrett’s esophagus can differ considerably in the severity of their associated GERD and risk for developing esophageal adenocarcinoma. It is therefore not appropriate to extrapolate the results of older studies on the epidemiology and natural history of Barrett’s esophagus to patients with short-segment disease.
Molecular Events that Underlie Metaplasia and Cancer in Barrett’s Esophagus
GERD and metaplasia
Long-segment Barrett’s esophagus is associated with GERD, and the epithelial metaplasia characteristic of Barrett’s esophagus is widely regarded as a consequence of GERD (Fig. 1, panels A,B). Early investigators proposed that Barrett’s esophagus developed from gastric columnar cells that migrated from the stomach into the esophagus to reconstitute GERD-damaged squamous epithelium.5 This hypothesis did not account for the intestinal metaplasia typical of Barrett’s esophagus, however. The prevailing hypothesis now is that Barrett’s esophagus forms when GERD damages the esophageal squamous epithelium, thereby exposing multipotential stem cells in the basal layers to refluxed gastric juice that stimulates abnormal differentiation.37-39 These progenitor cells may also be present in the ducts of esophageal mucosal glands.40 One recent study has suggested that circulating stem cells of bone marrow origin might contribute to the development of Barrett’s esophagus.41 Although the progenitor cell for Barrett’s esophagus remains unknown, metaplasias must arise from changes in cellular gene expression and, in Barrett’s esophagus, those changes appear to be induced by GERD.
Figure 1.
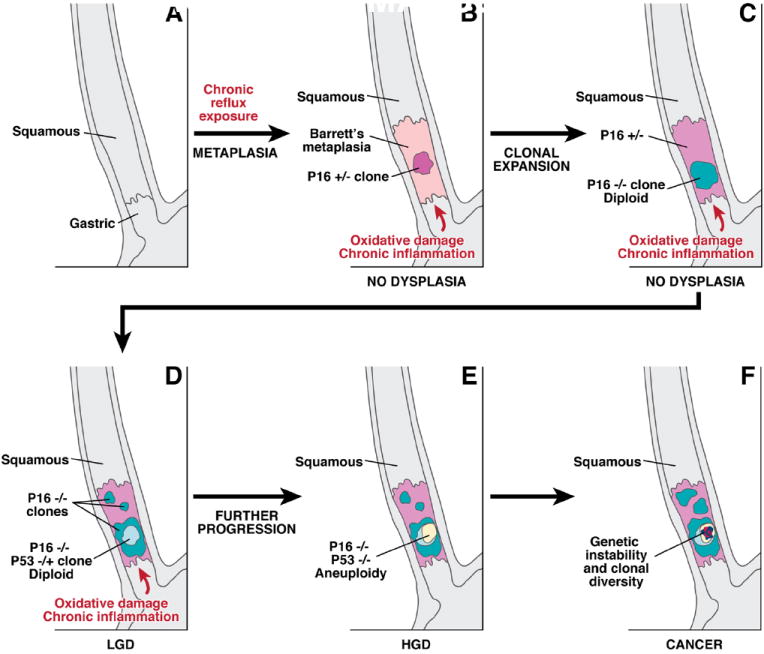
A schematic illustrating the sequential somatic genetic changes in the progression from the squamous esophagus to Barrett’s esophagus to adenocarcinoma. The normal squamous esophagus (A) undergoes a metaplastic transformation with the oxidative damage and chronic inflammation that accompanies chronic gastroesophageal reflux. The initial metaplastic change is followed early on by the loss of one p16 allele (B); this clone may then expand (pink area panel C), followed by loss of the 2nd p16 allele and the formation of some p16 null clones (blue area, C). The subsequent loss of p53 may be associated with morphological changes of low grade dysplasia (LGD), (D). Genetic instability may lead to aneuploidy, which is commonly seen with high grade dysplasia (HGD), (panel E). Numerous clones may develop, and there may be heterogeneity within clones especially as the degree of genetic instability increases and invasive adenocarcinoma develops (F).
Recent studies have reported molecular events in esophageal squamous epithelium that might be triggered by gastroesophageal reflux to cause Barrett’s metaplasia. In esophageal squamous cell lines, for example, acid and bile induce the expression of caudal homeobox genes such as CDX1 and CDX2.42-45 The word homeobox originates from the Greek homeosis, meaning a shift in structural development; homeobox genes encode transcription factors that regulate cell differentiation during embryogenesis. In adult cells, alterations in homeobox genes might alter cellular phenotypic features.46,47 Homeobox gene expression in adult cells can be regulated epigenetically, such as through alterations in gene promoter methylation, or via cell signaling pathways regulated by factors like bone morphogenetic proteins (BMPs) or fibroblast growth factors.45,48
Compared to normal esophageal squamous epithelium, BMP4 expression is increased in Barrett’s metaplasia; in a rat model of reflux-induced Barrett’s esophagus, BMP4 expression is increased in the stroma that underlies the Barrett’s metaplasia.49 Cultures of human esophageal squamous cells treated with BMP4 express cytokeratins that are characteristic of columnar cells.49 Bile acids, at neutral and acidic pH levels, cause a cancer cell line to express CDX2 through ligand-dependent transactivation of the epidermal growth factor receptor.50 It was therefore proposed that GERD induces the expression of Cdx genes through BMP4 and, perhaps, EGFR activation, and that GERD-induced Cdx expression might partially mediate the development of Barrett’s metaplasia.47
GERD and Carcinogenesis in Barrett’s esophagus
Tumor initiation is the process in which cells are changed so that they are able to form tumors. Although GERD clearly plays a role in the pathogenesis of Barrett’s metaplasia, it is not clear whether gastroesophageal reflux can initiate tumor formation in the metaplastic cells. Studies have shown that esophageal cells exposed to acid develop DNA double-strand breaks that might contribute to tumor initiation.51,52 In addition, deoxycholic acid (a bile acid) induces DNA damage in a dose-dependent, but nonlinear fashion.53,54 These DNA injuries appear to be mediated by reactive oxygen species, so antioxidants could have chemopreventive effects in patients with Barrett’s esophagus.
In addition to its potential role in initiating tumor development, GERD might also promote the growth of established neoplasms in Barrett’s esophagus (a process called tumor promotion). Acid and bile exposure alter Barrett’s cell kinetics55-58 so as to enable cells that have sustained DNA damage to resist apoptosis via activation of the nuclear factor (NF)-κB pathway.59 Other studies have shown that acid and bile salts increase NF-κB activity60 and have effects on molecules involved in inflammation and proliferation.61-63 For example, the farnesoid X receptor (FXR), a nuclear receptor that regulates inflammation, is up-regulated by deoxycholic acid in vitro.64 Tuberous sclerosis complex 1 (TSC1) is a tumor suppressor that regulates the mammalian target of rapamycin (mTOR) pathway; in neoplastic Barrett’s cells, bile acids deregulate the TSC/mTOR pathway.65 There are therefore several potential mechanisms by which gastroesophageal reflux of acid and bile might initiate and promote tumors in Barrett’s esophagus.
Obesity, metabolic syndrome, and Barrett’s esophagus
Obesity is a risk factor for Barrett’s esophagus and Barrett’s adenocarcinoma,66,67 especially an abdominal pattern of obesity, which has been associated both with GERD and Barrett’s esophagus.68 The mechanical effects of intra-abdominal fat might adversely affect the anatomical anti-reflux barrier.69 Metabolic syndrome is also frequently observed in patients with Barrett’s esophagus;70 the adipocytokines associated with the metabolic syndrome and central adiposity have been proposed to contribute to the development of GERD, Barrett’s esophagus and cancer.71 For example, adipose tissues secrete inflammatory cytokines like tumor necrosis factor-alpha and interleukin-6, as well as pro-proliferative hormones like leptin and insulin-like growth factor-1 that might promote carcinogenesis. Obesity is also associated with low levels of the anti-proliferative hormone adiponectin and low plasma levels of adiponectin have been described in patients with Barrett’s esophagus.72 Nevertheless, the precise nature of the relationships among the metabolic syndrome, GERD, Barrett’s esophagus, and cancer remains to be established.
Diet and Barrett’s esophagus
The role of specific dietary factors in the pathogenesis of Barrett’s esophagus is not clear. In patients with GERD, the distal esophagus can be exposed to high concentrations of nitric oxide and other nitrosating species, which are generated from the nitrate in green, leafy vegetables in the diet.73 Those nitrosating species are capable of causing DNA double-strand breaks, which are potentially carcinogenic.51 These types of oxidative injuries to DNA might be prevented by dietary antioxidant compounds such as polyphenols. Modest wine intake has been associated with a lower risk for Barrett’s esophagus and esophageal adenocarcinoma, possibly from the reduction in oxidative stress provided by polyphenols in wine.74
Iron also might have a role in the development of esophageal cancer. After the surgical induction of reflux in rats, the rate of development of esophageal adenocarcinoma is substantially higher in animals that receive iron supplementation.75 One study described overexpression of iron transport proteins in the progression of Barrett’s metaplasia to adenocarcinoma, and cultured cancer cells loaded with iron increased proliferation.76 Reduced iron levels in premenopausal women might underlie the delayed onset of adenocarcinoma in women with Barrett’s esophagus.77 However, no link has been found between hemochromatosis (excess iron storage) genes and Barrett’s esophagus, so the relationship between iron metabolism and this condition is complex.78
Genetics, Carcinogenesis, and Barrett’s esophagus
Unlike the well-characterized sequence of genetic alterations that lead to some forms of colon cancer, no well-defined pathway has been shown to mediate the pathogenesis of adenocarcinoma in Barrett’s esophagus. There is considerable genetic heterogeneity among the adenocarcinomas that develop in patients with Barrett’s esophagus, which frequently involve alterations in tumor suppressor genes.79,80 A number of the many molecular changes that have been described in Barrett’s-associated cancers are so-called “hitchhiker mutations” that do not directly contribute to carcinogenesis, but are retained in the cancer cell genome because they are linked to oncogenic mutations.81 It is not clear which of the many genetic changes described in Barrett’s esophagus are required for oncogenesis; when these are identified, they might be used in diagnosis and as therapeutic targets.
In general, both alleles of a tumor suppressor gene must be silenced to contribute to carcinogenesis. The first silencing event (allele 1) usually involves a DNA nucleotide sequence change mutation (e.g. a nucleotide base excision or deletion). The second silencing event (allele 2) can also be a sequence change mutation, but more commonly involves loss of a chromosome segment after a faulty cell division (so-called loss of heterozygosity or LOH). Methylation of DNA cytosine residues in the gene promoter region is another common gene silencing mechanism.
Alterations in genes that encode the tumor suppressors p16 (CDKN2A) and TP53 are frequently found in cells from Barrett’s esophagus samples. Loss of expression of a CDKN2A allele, usually through methylation of its promoter, is found in non-dysplastic Barrett’s metaplasia in 85% of cases (Figure, panel B).82-84 The CDKN2A mutations found in Barrett’s metaplasia appear to have been caused by oxidative damage in areas of chronic inflammation.85 LOH has been observed in CDKN2A in dysplastic regions of Barrett’s esophagus (Fig. 1, panel B,C).82,83,86,87 CDKN2A abnormalities do not always alter esophageal cell proliferation and the affected cells can remain diploid, so these defects might cause not histological abnormalities.85,88 However, there is little point in testing for loss of p16 expression to predict adenocarcinoma risk because it occurs with such high frequency that it is not a useful biomarker.81
In addition to being part of the coding sequence of p16, exon 2 of CDKN2A is also part of the region that encodes p14ARF (alternate reading frame). P14ARF is a tumor suppressor that prevents degradation of TP53. Because p16 expression is downregulated so frequently in Barrett’s metaplasia, there has been interest in whether p14ARF expression is also lost. p14ARF expression appears to be lost in approximately 30% of Barrett’s cancers. Data indicate that p14ARF loss is a relatively late event that is independent of the loss of p16 expression in Barrett’s metaplasia. The loss of p14ARF expression appears to occur primarily as the result of CpG or histone methylation, rather than LOH.89
The appearance of low-grade dysplasia in Barrett’s esophagus often coincides with the loss of TP53 expression, through methylation of the gene promoter, mutations, or LOH in cells that have already lost p16 expression (Fig. 1, panel D). LOH at TP53 is associated with a 16-fold increase in the rate of progression to cancer.90 Certain mutations in TP53 result in nuclear accumulation of the resulting non-functional p53 protein that can be detected by immunohistochemistry. A large, nested, case-control study found that TP53 staining was increased in biopsies from patients who developed esophageal adenocarcinoma compared with controls, with an odds ratio of 11.7 91,92
Cells that lose TP53 expression often acquire an abnormal content of DNA (tetraploidy or aneuploidy) that can be detected by flow cytometry. These DNA changes correlate with increased proliferation and expansion of the proliferative compartment towards the mucosal surface.93-95 The increased proliferation is associated with an increase in the proportion of cells in the S phase of the cell cycle and increased expression of cell cycle-related proteins such as cyclins.85,92,96 These molecular changes and the associated proliferative abnormalities are often accompanied by histological changes in tissues recognized as high-grade dysplasia (Fig. 1, panel E). Widespread LOH at TP53 and cytogenetic abnormalities are associated with increased cancer risk, perhaps because these changes increase sensitivity to mutagens.96-99
Aneuploidy has diverse effects on cell metabolism, proliferation, and immortalization.100 Chromosomal copy number changes (a manifestation of aneuploidy) do not appear to be random events in Barrett’s cells. Aneuploidy at chromosomes 4 and 7 usually occurs early in carcinogenesis, followed by aneuploidy at chromosomes 8 and 17 and then by the loss of the Y chromosome in cells from male patients.101,102 The presence of ploidy abnormalities in esophageal cells increases the risk for cancer among patients with Barrett’s esophagus (relative risks (RR)=4.4 and 11 for tetraploidy and aneuploidy, respectively; RR=20 when both are present).103
At advanced stages of neoplastic progression, the Barrett’s esophagus often harbors multiple distinct clones that exhibit variable degrees of expansion (Fig.1 panel F),104 and the level of clonal diversity correlates with the risk of adenocarcinoma development.97,105 One study found that samples of adenocarcinoma in Barrett’s esophagus had such high levels of genomic heterogeneity that even neighboring crypts had distinct genetic profiles.106 This genomic heterogeneity results from local variations in the microenvironment of different regions of the esophagus (so-called microenvironmental niches) or from genetic instability.105 However, microsatellite instability, which often underlies colorectal carcinomas, does not seem to contribute importantly to carcinogenesis in Barrett’s esophagus.107
The number of molecular abnormalities identified in Barrett’s esophagus has greatly increased with advances in high-throughput screening techniques for genomic and epigenetic alterations. Unfortunately, our understanding of the functional implications of these abnormalities and how they might be used to improve patient care lags behind. Application of concepts from other disciplines, such as evolutionary biology and bioinformatics, should facilitate progress in understanding causality. This information might be used to develop non-invasive tests for determining which patients will benefit most from the exciting new endoscopic techniques available for treatment of patients with Barrett’s esophagus.
Endoscopic Therapy for Barrett’s Esophagus
Endoscopic ablative therapy for Barrett’s esophagus is based on the principle that, when gastroesophageal acid reflux is controlled by medical or surgical means, squamous epithelium replaces ablated metaplastic epithelium. This principle was first validated more than 20 years ago in subjects whose Barrett’s epithelium was ablated by endoscopic laser treatment.108,109 Since then, endoscopic ablation has advanced from an experimental approach to a valid therapeutic option that is endorsed by professional societies.110,111 Large cohort studies with long durations of follow up have documented the durability of endoscopic therapies and the comparability of their outcomes to those of traditional surgical treatment for neoplasia in Barrett’s esophagus.112-114 The efficacy of endoscopic ablation has been confirmed in multicenter, randomized controlled trials.115-117 Nevertheless, many questions remain regarding which patients are appropriate candidates for endoscopic therapy and which endoscopic eradication technique is most effective in preventing cancer.118,119
Techniques of endoscopic therapy for Barrett’s esophagus can be categorized broadly into those that provide tissue for histological examination (endoscopic mucosal resection and endoscopic submucosal dissection) and those that do not (ablative therapies). The ablative therapies can be further classified as heat-generating thermal techniques (radiofrequency ablation, multipolar electrocoagulation, and argon plasma coagulation), photochemical techniques (photodynamic therapy), and cryotherapy. Multimodal endoscopic therapy, which uses a resection technique to remove visible abnormalities followed by an ablation technique to eradicate the remaining Barrett’s epithelium, is considered to be the most comprehensive endoscopic management of neoplasia in Barrett’s esophagus. Table 1 summarizes the evidence for efficacy, advantages, and limitations of each technique.
Table 1.
| Technique | Study | Study Design | Dysplasia grade included (sample size) | Outcomes | Advantages | Limitations |
|---|---|---|---|---|---|---|
| Endoscopic resection techniques | ||||||
| Focal Endoscopic resection | Pech et al$ 113 | Single center cohort | HGD (61)IMCA (288)Median follow up 63months | CR–D: 97%Recurrence of HGD/IMCa: 21.5%Overall 5 year survival : 84% | Allows precise determination of depth of invasion and assessment of marginsLess variability in pathological assessment | May need multiple sessions to achieve remissionFocal EMR alone may be associated with higher recurrence rates and positive marginsBleeding (0.6% to 6%)Perforation (0% to Stricture (4%) |
| Prasad et al # 114 | Single center cohort study (endoscopic : PDT/EMR and surgical cohorts) | IMCa (178)Median follow upSurg 64mEndo 43m | CR-D: 94%Recurrent Ca : 12%Overall 5 year survival: 83% in the Endo group and 95% in Surg group | |||
| Circumferential endoscopic resection | Gondrie et al 162 | Multicenter cohort study | HGD/IMCa (149)Median follow up 18months | CR-IM : 97%Recurrent neoplasia 3%2-3 sessions needed to achieve CR | Low rate of recurrence by removing all at-risk mucosaWith availability of EMRL easier to perform | Perforation 1%High stricture rate 52%Bleeding 1-4%%Ridges of tissue persist between EMR sites which may contribute to recurrenceBuried metaplasia (8%) |
| Larghi et al 123 | Multicenter cohort study | HGD/IMCa (26)Median follow up 28months | CR-IM : 88%Recurrent neoplasia 4% (IMCa) | |||
| Submucosal Dissection | Yoshinaga et al 163 | Single center cohort study | GE junction adenoCa (24)Median follow up 30m | CR : 72%No recurrence in those with CR | En bloc resection allows clear margins to be obtainedMay be more suitable in lesions > 2 cm in diameter | Long procedure timesStrictures |
| Endoscopic ablation techniques | ||||||
| Thermal | ||||||
| Multipolar electrocoagulation | Sampliner et al 135 | Multicenter cohort study | No dysplasia (58)Median follow up 6m | CR-IM : 78% | Technically easyRelatively inexpensiveWell tolerated (1/58 developed stricture, 1/58 hospitalized for chest pain)Persistent reversal of IM at 24 m in 68% | Difficult to treat longer segments (study used 10F probe via therapeutic endoscope)Not used for treatment of HGDShort follow up |
| Sharma at al 164 | Multicenter randomized controlled trial (comparing MEPC and APC) | No dysplasia and LGD (35)Median follow up 24 m | CR-IM : 83% (75% MPEC, 63% with APC)CR-IM at 24 m : 68% | |||
| Argon Plasma Coagulation | Attwood et al 165 | Single center cohort study | HGD (29)Median follow up 37m | CR-D :86%CR-IM: 76%4 patients progressed to EAC1 esophageal perforation | Technically easy to perform | Dosimetry variable across studiesSuperficial effect leads to high prevalence of buried metaplasiaPerforation reportedLimited evidence in HGD |
| Ferraris et al 166 | Multicenter cohort study | No dysplasia (96)Median follow up 36 m | CR-IM 96%Recurrence 18% | |||
| Radiofrequency Ablation | Shaheen et al 147 | Multicenter, sham controlled RCT | HGD (64)LGD (63) | CR-IM (12 m): 77% vs. 2.3%CR-D (12m)HGD : 81% vs. 19%LGD: 90% vs. 23%Progression to Ca: 19% vs. 2% | Well tolerated by most patientsLow stricture rate (6%)Low rate of subsquamous BE (5%) | Ablation requires multiple stepsLong term data on durability of ablation and recurrence not available |
| Fleischer et al 116 | Multicenter, cohort study | No dysplasia (100) | CR-IM (12m) : 70%CR-IM (30m): 96% | |||
| Photochemical | ||||||
| Porfimer PDT | Overholt et al 115 | Multicenter, partially blinded controlled RCT | HGD (208) | CR-HGD (24m): 77% Vs 38%CR-IM (24m) : 52% Vs 7%CR-D (24m): 59% Vs. 14% | Easy to administerResults of RCT durable at 5 years33 | Photosensitivity (60%)Strictures (27-39%)5Significant post procedure morbidity |
| Prasad et al 141 | Single center cohort study (endoscopic : PDT/EMR and surgical cohorts) | HGD (199) | Overall survival comparable at 5 years between endoscopic and surgical cohorts.No death from esophageal carcinoma in both groups | Compares favorably to esophagectomy36 | ||
| ALA PDT | Pech et al 167 | Single center cohort study | HGD (35)IMCA (31) | CR-D : 100%CR-D : 97%Median follow up 37m | Oral administration of 5-ALALimited photosensitivity | Minor adverse effects 40%No strictures reported29% recurrent carcinoma |
| Peters et al 144 | Single center cohort study | Residual HGD or IMCA after EMR (23) | CR-D : 75%CR-IM: 0%Median follow up 30mRecurrent HGD : 27% | Oral administration of 5-ALA | Major adverse effects : arrhythmia, hypotension, hematemesisBuried metaplasia in 33%High recurrence rateDeath has been reported | |
| Cryotherapy | ||||||
| Liquid N2 spray | Greenwald et al 150 | Multicenter cohort study | IMCA, HGD, Non dysplastic BE, severe squamous dysplasia (77) | CR-D : 88% (in HGD)CR-IM: 53%1 perforation (in patient with Marfan Syndrome)3 esophageal stricturesAdverse effects: chest pain (17%), dysphagia (13%) | No mucosal contact requiredWell tolerated by most patients | Dosimetry not well establishedNo controlled data availableTechnically challenging : need for accompanying decompression tube, visibility impaired due to freezing |
| CO2 spray | Canto et al 151 | Single center cohort study | HGD/IMCA (44)Median follow up 12m | CR-IM 86% (after median of 6 procedures) | Initial promising results in patients who failed other forms of ablation (n=25) including EMR | Additional decompression tube not needed. May be option for patients who fail RF ablation, PDT |
Endoscopic mucosal resection
Endoscopic mucosal resection (EMR) involves the removal of mucosal and submucosal tissue, usually after the targeted mucosal segment is elevated by the submucosal injection of fluid.120,121 The most commonly used EMR technique is the “suck and cut” method, wherein the mucosal lesion is sucked into a cap on the tip of the endoscope and then resected using a diathermic snare. There is also a “band and snare” method that uses a band ligating device, similar to that used for endoscopic variceal ligation, which deploys elastic bands around the suctioned mucosal segment. The banded segments are then removed with a snare. The band ligation method allows multiple resections to be completed in a single intubation and does not always require submucosal fluid injection before banding. The 2 EMR techniques have been compared; the suck and cut method provides larger tissue samples whereas the band ligation technique is faster and less expensive for multiple resections.122 EMR can be applied focally (to resect only a visible lesion) or circumferentially (to remove the entire segment of Barrett’s metaplasia).113,114,123
Compared to conventional endoscopic biopsy methods, EMR allows for more precise characterization of neoplasia, less inter-observer variation among pathologists, and more accurate assessments of dysplasia grades and the depths of invasion.124,125 In addition, the pathologist can provide information about neoplastic involvement in the margins of the resection specimens.126 All of this information is invaluable in determining whether endoscopic therapy is adequate or whether surgical resection is required to cure neoplasias in patients with Barrett’s esophagus. Results of the 2 approaches are summarized in table 1.
Accurate T staging of the depth of neoplastic involvement is required to determine whether endoscopic therapy can be considered definitive. Esophageal resections performed on patients with intramucosal carcinomas, which do not penetrate the muscularis mucosae, have revealed a low rate of metastases to lymph nodes (less than 5%). 127-130 Thus, endoscopic therapy might be curative for most patients with neoplasms confined to the mucosa. For tumors that involve the submucosa, however, the rate of metastasis to the lymph nodes exceeds 20%.126,131 Therefore, endoscopic therapy is generally not considered definitive for patients with submucosal neoplasia, despite reports documenting successful endoscopic therapy for some patients with tumors that invaded only the upper third of the submucosa.132 Non-invasive procedures like endoscopic ultrasonography have relatively poor accuracy for staging early esophageal neoplasia.133 Consequently, EMR has a major role in staging early esophageal neoplasms and determining whether endoscopic therapy is feasible.
Endoscopic submucosal dissection
Endoscopic submucosal dissection (ESD) has been used primarily in Japan, predominantly for the treatment of gastric neoplasia. ESD starts with injection of fluid into the submucosa, followed by incision around the mucosal segment to be removed using a cutting device (e.g. ceramic tip knife, triangle tip knife, flex knife, hook knife, standard needle knife) and, finally, submucosal dissection of the segment.121 This technique requires meticulous endoscopic control and the use of a cap to help with the submucosal dissection.
The major proposed advantage of ESD over EMR is the ability to remove a neoplastic lesion en-bloc, which provides more precise determination of its vertical and lateral margins and, perhaps, greater potential for the complete removal of all neoplastic cells. However, ESD is a technically challenging and lengthy procedure that sometimes requires hours to complete, and serious complications such as perforation are frequent. In the esophagus, furthermore, GERD-induced inflammation and fibrosis in the submucosa can make ESD difficult and even more hazardous. These concerns have limited the use of ESD in the treatment of Barrett’s esophagus.
Multipolar electrocoagulation
Multipolar electrocoagulation (MPEC) involves the application of thermal energy to the mucosa using an electrode-tipped, 7F or 10F catheter that is advanced through the working channel of the endoscope.134,135 The technique is time consuming and not practical for ablating large mucosal areas; there are no published data on the efficacy of MPEC for treating neoplasia in Barrett’s esophagus. Because MPEC is a bipolar device, however, it might be used to eradicate residual areas of intestinal metaplasia for patients who have implanted cardiac devices.
Argon Plasma Coagulation
Unlike MPEC, argon plasma coagulation (APC) is a non-contact technique in which monopolar energy is delivered to tissue using ionized argon gas. Energy settings from 40W to 90W have been used to ablate Barrett’s metaplasia and dysplasia, with short-term success rates ranging from 70% to 90%. However, some studies have reported recurrence rates as high as 66% and frequently found metaplastic glands underlying squamous epithelium (so-called “buried metaplasia”). This might reflect the relatively superficial injury inflicted by APC.136 In addition, reports of serious complications such as perforation, pneumomediastinum, and major bleeding have reduced endoscopists’ enthusiasm for APC ablation of Barrett’s esophagus.137,138 Newer ablation techniques (see below) have rendered both APC and MPEC largely obsolete for the eradication of Barrett’s esophagus, although APC and MPEC could still be used to eliminate small islands of Barrett’s metaplasia.
Photodynamic therapy
Photodynamic therapy (PDT) is an ablation technique that destroys Barrett’s epithelium using photochemical energy generated through the interaction between endoscopically delivered light and a photosensitizer that is concentrated in the tissue. This interaction leads to the generation of toxic, singlet oxygen molecules that damage tissue. The photosensitizers used for PDT include porfimer sodium, which is administered intravenously, and 5-aminolevulinic acid (ALA), which can be administered orally.139 The most data on efficacy, durability, and long-term outcome are available for the use of PDT with porfimer sodium.115,140,141 Common side effects of PDT using porfimer sodium include photosensitivity reactions in more than 60% of patients, a high rate of esophageal stricture formation (as much as 36%), and substantial short-term morbidity.115, 142 PDT with ALA is associated with a shorter duration of photosensitivity and less esophageal structuring than with porfimer sodium, but ALA appears to be less efficacious and has been associated with vascular instability and at least 1 death.143,144
Radiofrequency ablation
Radiofrequency ablation (RFA) uses radiofrequency energy, which is delivered by an endoscopic balloon catheter (Halo360 system) or a focal ablation device (Halo 90 system), to destroy Barrett’s epithelium. To perform RFA, the endoscopist first determines the esophageal diameter using a balloon measuring device. The mucosa is sprayed with 1% N-acetyl cysteine solution to remove mucus that might interfere with energy delivery to the mucosa, and a 3cm-long ablation balloon (whose diameter is determined by the aforementioned measurement) that is lined by circumferential electrodes is positioned in the esophageal segment to be ablated. If there is evidence of fibrosis in the wall of the esophagus, an ablation balloon with a diameter smaller than that determined by the measuring balloon is used to prevent esophageal tears; RFA is not recommended for patients with esophageal strictures because inflation of the treatment balloons (typical diameters of 28 to 31 mm) could cause perforations. Radiofrequency energy is delivered through the electrodes to produce heat that destroys the metaplastic tissue. Patients return 2–3 months after initial RFA treatment for endoscopic evaluation, and any residual metaplastic tissue is ablated using the focal ablation device (which measures 20 mm in its longest dimension).
Initial dose-finding and phase II studies showed the efficacy and safety of RFA for ablating non-dysplastic Barrett’s epithelium.145,146 By using a combination of the balloon-based and focal RFA devices, an uncontrolled study has described complete eradication of Barrett’s epithelium in 60 of 61 patients (98%) during a follow-up period of 30 months.116 A recent multicenter, randomized, sham-controlled study demonstrated the ability of RFA to eradicate dysplasia and intestinal metaplasia in patients with Barrett’s esophagus.147 Adverse events included esophageal stricture formation (6%), gastrointestinal bleeding (1%) and chest pain requiring hospitalization (2%); these rates of serious complications were substantially lower than those reported for PDT with porfimer sodium. Esophageal mucosal tears and perforations with RFA have been reported infrequently to the FDA MAUDE database (www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfMAUDE/search.CFM). Although these results are encouraging, the long-term durability of RFA remains to be determined. Furthermore, the randomized controlled trial enrolled patients who had high-grade dysplasia without associated nodularity, so only a small proportion had undergone EMR before RFA (7 of 84 patients in the ablation arm of the study). Moreover, the EMR was performed by endoscopists with substantial expertise, so it is not clear whether the results could be reproduced by community endoscopists.
Cryotherapy
Cryotherapy involves the use of an endoscopically delivered cryogen (liquid nitrogen or carbon dioxide) to inflict tissue injury. One of the 2 cryotherapy systems available (CryoSpray Ablation system) uses a 7F catheter passed through the working channel of the endoscope to spray liquid nitrogen onto the metaplastic epithelium. The other system (GI Supply) delivers cold carbon dioxide gas to the mucosa through a spray catheter. Tissue destruction from cryotherapy occurs in 2 phases: an immediate phase, caused by freezing of the cell and its organelles, followed by a delayed phase, in which cells undergo apoptosis.148 Cryotherapy is delivered by spraying without the need for mucosal contact with the catheter, which might be useful for application to uneven surfaces. Problems encountered with cryotherapy include over-distention with perforation (in a patient with Marfan’s syndrome), and fogging of the endoscope lens (with the liquid nitrogen device). Though initial results with cryotherapy are promising and its adverse-event profile seems reasonable, long-term data are lacking.149-151 Initial results are summarized in table 1.
Buried Metaplasia
A major concern for all of the endoscopic ablative procedures is that partially-ablated Barrett’s epithelium might heal with an overlying layer of squamous epithelium that “buries” metaplastic tissue (with neoplastic potential) and hides it from the endoscopist. Although some studies suggest that buried metaplasia has a low risk of malignant transformation,152 case reports of dysplasia and adenocarcinoma in buried metaplastic glands continue to cause concern.153,154
A recent study that explored the prevalence of buried metaplasia in a large, randomized trial of PDT for patients with high-grade dysplasia in Barrett’s esophagus has allayed some concerns about buried metaplasia.155 A systematic review of more than 33,000 esophageal biopsy specimens revealed that the frequency of buried metaplasia increased after PDT (from 5.8% to 30%). However, a similar increase in the frequency of buried metaplasia was noted for patients treated with PPIs alone (from 2.9% to 33%). In addition, the highest grade of dysplasia detected during any given endoscopic examination was not found exclusively in the buried metaplasia in any patient.
In the aforementioned sham-controlled trial of RFA for dysplasia in Barrett’s esophagus, the frequency of buried metaplasia decreased in the group treated with RFA and PPIs (from 25% to 5%) and increased in the control group who received sham therapy and PPIs (from 25% to 40%).147 Another study of 22 patients treated with RFA found no buried metaplasia 2 months after the procedure in biopsy specimens of neo-squamous epithelium taken using a “keyhole” technique designed to obtain deep specimens.156 Nevertheless, the importance of buried metaplasia remains controversial, and large, well done, long-term studies are needed to determine its risk for malignancy.
Factors that influence choice of endoscopic therapy
The choice among treatment options for patients with dysplasia in Barrett’s esophagus requires consideration of patient, esophageal, and institutional factors. Patient factors include age, fitness for therapy, and personal preferences.157 The risks of esophagectomy might be prohibitive for older patients with substantial co-morbidities. Before endoscopic therapy is chosen, patients should be informed of the failure rates of the procedure and of the need for careful endoscopic follow-up.158 Patients who choose endoscopic therapy should be willing to comply with follow-up requirements and be aware of the uncertainty regarding the long-term efficacy of endoscopy in preventing cancer. Esophageal factors include the length of the Barrett’s segment, the presence of visible lesions like nodules that can be targeted for EMR, and the presence of multifocal lesions.142,159,160 Extensive metaplasia involving long segments of the esophagus can be difficult to eradicate with ablative therapies. The absence of a visible lesion to target by EMR makes it difficult to stage the depth of the neoplasia or determine whether endoscopic therapy can be successful. Dysplasia that is focal might be easily resected or ablated, whereas multi-focal dysplasia can be much harder to eradicate with a reasonable degree of certainty. Institutional factors include the levels of expertise of the surgeons and endoscopists. Surgical volume has been repeatedly shown to be an important determinant of esophagectomy outcome.161 Undoubtedly, an endoscopist’s level of experience will affect results as well. An approach to choosing endoscopic therapy for neoplasia in patients with Barrett’s esophagus is illustrated in Figure 2.
Figure 2.
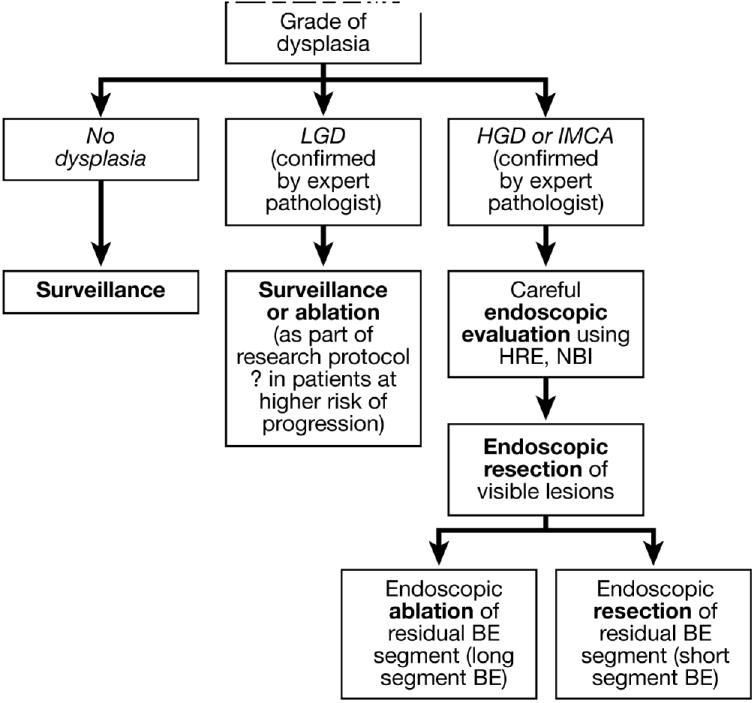
Algorithm for the endoscopic management of dysplasia in Barrett’s esophagus.
There are number of unresolved issues in the endoscopic treatment of Barrett’s esophagus. Dysplasia is an imperfect marker for cancer risk, and the identification of biomarkers to select patients who would benefit most from endoscopic or surgical treatments would represent a major advance. Although data from retrospective studies have shown that complete elimination of all metaplasia decreases the recurrence rate, prospective studies on this issue are needed. Predictors of recurrent dysplasia following ablation need to be defined to design interventions to limit that recurrence. New imaging techniques such as narrow band imaging, laser confocal endomicroscopy and endocytoscopy have the potential to improve the detection neoplasia in Barrett’s esophagus, and to direct endoscopic therapies. Finally, long-term data on the recurrence of metaplasia and dysplasia following ablation are required to determine the need for post-ablation surveillance and select appropriate surveillance intervals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lord RV. Norman Barrett, “doyen of esophageal surgery”. Ann Surg. 1999;229:428–39. doi: 10.1097/00000658-199903000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol. 2006;101:1900–20. doi: 10.1111/j.1572-0241.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 3.Barrett NR. Chronic peptic ulcer of the oesophagus and “oesophagitis”. Br J Surg. 1950;38:175–82. doi: 10.1002/bjs.18003815005. [DOI] [PubMed] [Google Scholar]
- 4.Tileston W. Peptic ulcer of the oesophagus. Am J Med Sci. 1906;132:240–65. [Google Scholar]
- 5.Allison PR, Johnstone AS. The oesophagus lined with gastric mucous membrane. Thorax. 1953;8:87–101. doi: 10.1136/thx.8.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett NR. The lower esophagus lined by columnar epithelium. Surgery. 1957;41:881–94. [PubMed] [Google Scholar]
- 7.Hayward J. The lower end of the oesophagus. Thorax. 1961;16:36–41. doi: 10.1136/thx.16.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosher LH, Taylor FH. Heterotopic gastric mucosa in the esophagus with ulceration and stricture formation. J Thorac Surg. 1951;21:306–12. [PubMed] [Google Scholar]
- 9.Morson BC, Belcher JR. Adenocarcinoma of the esophagus and ectopic gastric mucosa. Br J Cancer. 1952;6:127–30. doi: 10.1038/bjc.1952.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedersen SA, Hage E, Nielsen PA, Sorensen HR. Barrett’s syndrome: morphological and physiological characteristics. Scand J Thorac Cardiovasc Surg. 1971;6:191–205. doi: 10.3109/14017437209134800. [DOI] [PubMed] [Google Scholar]
- 11.Paull A, Trier JS, Dalton MD, Camp RC, Loeb P, Goyal RK. The histologic spectrum of Barrett’s esophagus. N Engl J Med. 1976;295:476–80. doi: 10.1056/NEJM197608262950904. [DOI] [PubMed] [Google Scholar]
- 12.Burgess JN, Payne WS, Andersen HA, Weiland LH, Carlson HC. Barrett esophagus: the columnar-epithelial-lined lower esophagus. Mayo Clin Proc. 1971;46:728–34. [PubMed] [Google Scholar]
- 13.Naef AP, Savary M, Ozzello L. Columnar-lined lower esophagus: an acquired lesion with maliganant predisposition: report on 140 cases of Barrett’s esophagus with 12 adenocarcinomas. J Thorac Cardiovasc Surg. 1975;70:826–34. [PubMed] [Google Scholar]
- 14.Borrie J, Goldwater L. Columnar cell-lined esophagus: assessment of etiology and treatment: a 22 year experience. J Thorac Cardiovasc Surg. 1976;71:825–34. [PubMed] [Google Scholar]
- 15.Skinner DB, Walther BC, Riddell RH, Schmidt H, Iascone C, DeMeester TR. Barrett’s esophagus: comparison of benign and malignant cases. Ann Surg. 1983;198:554–65. doi: 10.1097/00000658-198310000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler RH. The lower esophagus lined by columnar epithelium: its association with hiatal hernia, ulcer, stricture, and tumor. J Thorac Cardiovasc Surg. 1963;45:13–32. [PubMed] [Google Scholar]
- 17.Hawe A, Payne WS, Weiland LH. Adenocarcinoma in the columnar epithelial lined lower (Barrett) esophagus. Thorax. 1973;28:511–4. doi: 10.1136/thx.28.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haggitt RC, Tryzelaar J, Ellis FH, Colcher H. Adenocarcinoma complicating columnar epithelium-lined (Barrett’s) esophagus. Am J Clin Pathol. 1978;70:1–5. doi: 10.1093/ajcp/70.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Haggitt RC, Dean PJ. Adenocarcinoma in Barrett’s epithelium. In: Spechler SJ, Goyal RK, editors. Barrett’s esophagus: pathophysiology, diagnosis, and management. New York: Elsevier Science Publishing Co., Inc.; 1985. pp. 153–66. [Google Scholar]
- 20.Reid BJ, Weinstein WM. Barrett’s esophagus and adenocarcinoma. Annu Rev Med. 1987;38:477–92. doi: 10.1146/annurev.me.38.020187.002401. [DOI] [PubMed] [Google Scholar]
- 21.Reid BJ, Weinstein WM, Lewin KJ, Haggitt RC, VanDeventer G, DenBesten L, Rubin CE. Endoscopic biopsy can detect high-grade dysplasia or early adenocarcinoma in Barrett’s esophagus without grossly recognizable neoplastic lesions. Gastroenterology. 1988;94:81–90. doi: 10.1016/0016-5085(88)90613-0. [DOI] [PubMed] [Google Scholar]
- 22.Reid BJ. Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterol Clin N Am. 1991;20:817–834. [PubMed] [Google Scholar]
- 23.Weinstein WM, Ippoliti AF. The diagnosis of Barrett’s esophagus: goblets, goblets goblets. Gastrointest Endosc. 1996;44:91–95. doi: 10.1016/s0016-5107(96)70239-0. [DOI] [PubMed] [Google Scholar]
- 24.Spechler SJ, Zeroogian JM, Antonioli DA, Wang HH, Goyal RK. Prevalence of metaplasia at the gastro-oesophageal junction. Lancet. 1994;344:1533–1536. doi: 10.1016/s0140-6736(94)90349-2. [DOI] [PubMed] [Google Scholar]
- 25.Spechler SJ. The columnar lined oesophagus: a riddle wrapped in a mystery inside an enigma. Gut. 1997;41:710–711. doi: 10.1136/gut.41.5.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rex DK, Cummings OW, Shaw M, Cumings MD, Wong RK, Vasudeva RS, Dunne D, Rahmani EY, Helper DJ. Screening for Barrett’s esophagus in colonoscopy patients with and without heartburn. Gastroenterology. 2003;125:1670–7. doi: 10.1053/j.gastro.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 27.Ronkainen J, Aro P, Storskrubb T, Johansson SE, Lind T, Bolling-Sternevald E, Vieth M, Stolte M, Talley NJ, Agreus L. Prevalence of Barrett’s esophagus in the general population: an endoscopic study. Gastroenterology. 2005;129:1825–31. doi: 10.1053/j.gastro.2005.08.053. [DOI] [PubMed] [Google Scholar]
- 28.Sharma P, Morales TG, Sampliner RE. Short segment Barrett’s esophagus. The need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol. 1998;93:1033–6. doi: 10.1111/j.1572-0241.1998.00324.x. [DOI] [PubMed] [Google Scholar]
- 29.Riddell RH, Odze RD. Definition of Barrett’s esophagus: time for a rethink--is intestinal metaplasia dead? Am J Gastroenterol. 2009;104:2588–94. doi: 10.1038/ajg.2009.390. [DOI] [PubMed] [Google Scholar]
- 30.Playford RJ. New British Society of Gastroenterology (BSG) guidelines for the diagnosis and management of Barrett’s oesophagus. Gut. 2006;55:442. doi: 10.1136/gut.2005.083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandrasoma P. Pathophysiology of Barrett’s esophagus. Semin Thorac Cardiovasc Surg. 1997;9:270–278. [PubMed] [Google Scholar]
- 32.Liu W, Hahn H, Odze RD, Goyal RK. Metaplastic esophageal columnar epithelium without goblet cells shows DNA content abnormalities similar to goblet cell-containing epithelium. Am J Gastroenterol. 2009;104:816–24. doi: 10.1038/ajg.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahn HP, Blount PL, Ayub K, Das KM, Souza R, Spechler S, Odze RD. Intestinal differentiation in metaplastic, nongoblet columnar epithelium in the esophagus. Am J Surg Pathol. 2009 doi: 10.1097/PAS.0b013e31819f57e9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takubo K, Aida J, Naomoto Y, Sawabe M, Arai T, Shiraishi H, Matsuura M, Ell C, May A, Pech O, Stolte M, Vieth M. Cardiac rather than intestinal-type background in endoscopic resection specimens of minute Barrett adenocarcinoma. Hum Pathol. 2009;40:65–74. doi: 10.1016/j.humpath.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Kelty CJ, Gough MD, Van Wyk Q, Stephenson TJ, Ackroyd R. Barrett’s oesophagus: intestinal metaplasia is not essential for cancer risk. Scand J Gastroenterol. 2007;42:1271–4. doi: 10.1080/00365520701420735. [DOI] [PubMed] [Google Scholar]
- 36.Gatenby PA, Ramus JR, Caygill CP, Shepherd NA, Watson A. Relevance of the detection of intestinal metaplasia in non-dysplastic columnar-lined oesophagus. Scand J Gastroenterol. 2008;43:524–30. doi: 10.1080/00365520701879831. [DOI] [PubMed] [Google Scholar]
- 37.Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, Kerr D, Young LS. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol. 1999;154:965–973. doi: 10.1016/S0002-9440(10)65346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pera M, Pera M. Experimental Barrett’s esophagus and the origin of intestinal metaplasia. Chest Surg Clin N Am. 2002;12:25–37. doi: 10.1016/s1052-3359(03)00063-2. [DOI] [PubMed] [Google Scholar]
- 39.Seery JP. Stem cells of the oesophageal epithelium. J Cell Sci. 2002;115:1783–1789. doi: 10.1242/jcs.115.9.1783. [DOI] [PubMed] [Google Scholar]
- 40.Glickman JN, Chen YY, Wang HH, Antonioli DA, Odze RD. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett’s esophagus. Am J Surg Pathol. 2001;25:569–578. doi: 10.1097/00000478-200105000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, Souza RF, Zou YS, Shay JW, Spechler SJ. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus. 2008;21:43–50. doi: 10.1111/j.1442-2050.2007.00744.x. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y, Williams VA, Gellersen O, Jones C, Watson TJ, Peters JH. The pathogenesis of Barrett’s esophagus: secondary bile acids upregulate intestinal differentiation factor CDX2 expression in esophageal cells. J Gastrointest Surg. 2007;11:827–834. doi: 10.1007/s11605-007-0174-3. [DOI] [PubMed] [Google Scholar]
- 43.Kazumori H, Ishihara S, Kinoshita Y. Roles of caudal-related homeobox gene Cdx1 in oesophageal epithelial cells in Barrett’s epithelium development. Gut. 2009;58:620–628. doi: 10.1136/gut.2008.152975. [DOI] [PubMed] [Google Scholar]
- 44.Pera M, Pera M, de Bolos C, Brito MJ, Palacin A, Grande L, Cardesa A, Poulsom R. Duodenal-content reflux into the esophagus leads to expression of Cdx2 and Muc2 in areas of squamous epithelium in rats. J Gastrointest Surg. 2007;11:869–874. doi: 10.1007/s11605-007-0162-7. [DOI] [PubMed] [Google Scholar]
- 45.Wong NA, W J, Bartlett S, Liu Y, Warren BF, Piris J, Maynard N, Marshall R, Bodmer WF. CDX1 is an important molecular mediator of Barrett’s metaplasia. Proc Natl Acad Sci U S A. 2005;102:7565–7570. doi: 10.1073/pnas.0502031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beck F. The role of Cdx genes in the mammalian gut. Gut. 2004;53:1394–1396. doi: 10.1136/gut.2003.038240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Souza RF, Krishnan K, Spechler SJ. Acid, bile, and CDX: the ABCs of making Barrett’s metaplasia. Am J Physiol Gastrointest Liver Physiol. 2008;295:G211–218. doi: 10.1152/ajpgi.90250.2008. [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Zhang X, So CK, Wang S, Wang P, Yan L, Myers R, Chen Z, Patterson AP, Yang CS, Chen X. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis. 2007;28:488–496. doi: 10.1093/carcin/bgl176. [DOI] [PubMed] [Google Scholar]
- 49.Milano F, van Baal JW, Buttar NS, Rygiel AM, de Kort F, DeMars CJ, Rosmolen WD, Bergman JJ, VAM J, Wang KK, et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology. 2007;132:2412–2421. doi: 10.1053/j.gastro.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 50.Avissar NE, Toia L, Hu Y, Watson TJ, Jones C, Raymond DP, Matousek A, Peters JH. Bile acid alone, or in combination with acid, induces CDX2 expression through activation of the epidermal growth factor receptor (EGFR) J Gastrointest Surg. 2009;13:212–222. doi: 10.1007/s11605-008-0720-7. [DOI] [PubMed] [Google Scholar]
- 51.Clemons NJ, McColl KE, Fitzgerald RC. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology. 2007;133:1198–1209. doi: 10.1053/j.gastro.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 52.Zhang HY, Hormi-Carver K, Zhang X, Spechler SJ, Souza RF. In benign Barrett’s epithelial cells, acid exposure generates reactive oxygen species that cause DNA double strand breaks. Cancer Res. 2009;69:9083–9. doi: 10.1158/0008-5472.CAN-09-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jenkins GJ, D’Souza FR, Suzen SH, Eltahir ZS, James SA, Parry JM, Griffiths PA, Baxter JN. Deoxycholic acid at neutral and acid pH, is genotoxic to oesophageal cells through the induction of ROS: The potential role of anti-oxidants in Barrett’s oesophagus. Carcinogenesis. 2007;28:136–142. doi: 10.1093/carcin/bgl147. [DOI] [PubMed] [Google Scholar]
- 54.Jenkins GJ, Cronin J, Alhamdani A, Rawat N, D’Souza F, Thomas T, Eltahir Z, Griffiths AP, Baxter JN. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis. 2008;23:399–405. doi: 10.1093/mutage/gen029. [DOI] [PubMed] [Google Scholar]
- 55.Fitzgerald RC, Omary MB, Triadafilopoulos G. Dynamic effects of acid on Barrett’s esophagus. An ex vivo proliferation and differentiation model. J Clin Invest. 1996;98:2120–2128. doi: 10.1172/JCI119018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fitzgerald R, Omary M, Triadafilopoulos G. Altered sodium-hydrogen exchange activity is a mechanism for acid-induced hyperproliferation in Barrett’s Esophagus. American Journal of Physiology. 1998;275:G47–G55. doi: 10.1152/ajpgi.1998.275.1.G47. [DOI] [PubMed] [Google Scholar]
- 57.Kaur BS, Triadafilopoulos G. Acid- and bile-induced PGE(2) release and hyperproliferation in Barrett’s esophagus are COX-2 and PKC-epsilon dependent. Am J Physiol Gastrointest Liver Physiol. 2002;283:G327–334. doi: 10.1152/ajpgi.00543.2001. [DOI] [PubMed] [Google Scholar]
- 58.Souza RF, Shewmake K, Pearson S, Sarosi GA, Jr, Feagins LA, Ramirez RD, Terada LS, Spechler SJ. Acid increases proliferation via ERK and p38 MAPK-mediated increases in cyclooxygenase-2 in Barrett’s adenocarcinoma cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G743–748. doi: 10.1152/ajpgi.00144.2004. [DOI] [PubMed] [Google Scholar]
- 59.Hormi-Carver K, Zhang X, Zhang HY, Whitehead RH, Terada LS, Spechler SJ, Souza RF. Unlike esophageal squamous cells, Barrett’s epithelial cells resist apoptosis by activating the nuclear factor-kappaB pathway. Cancer Res. 2009;69:672–677. doi: 10.1158/0008-5472.CAN-08-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konturek PC, Nikiforuk A, Kania J, Raithel M, Hahn EG, Muhldorfer S. Activation of NFkappaB represents the central event in the neoplastic progression associated with Barrett’s esophagus: a possible link to the inflammation and overexpression of COX-2, PPARgamma and growth factors. Dig Dis Sci. 2004;49:1075–1083. doi: 10.1023/b:ddas.0000037790.11724.70. [DOI] [PubMed] [Google Scholar]
- 61.Abdalla SI, Lao-Sirieix P, Novelli MR, Lovat LB, Sanderson IR, Fitzgerald RC. Gastrin-induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin Cancer Res. 2004;10:4784–4792. doi: 10.1158/1078-0432.CCR-04-0015. [DOI] [PubMed] [Google Scholar]
- 62.Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, Saeed IT, Burnham WR, Farthing MJ. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut. 2002;50:451–459. doi: 10.1136/gut.50.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shirvani V, Ouatu-Luscar R, Kaur B, Omary M, Traidafilopoulos G. Cyclo-oxygenase 2 expression in Barrett’s esophagus and adenocarcinoma. ex vivo induction by bile salts and acid exposure. Gastroenterology. 2000;118:487–496. doi: 10.1016/s0016-5085(00)70254-x. [DOI] [PubMed] [Google Scholar]
- 64.Capello A, Moons LM, Van de Winkel A, Siersema PD, van Dekken H, Kuipers EJ, Kusters JG. Bile acid-stimulated expression of the farnesoid X receptor enhances the immune response in Barrett esophagus. Am J Gastroenterol. 2008;103:1510–1516. doi: 10.1111/j.1572-0241.2008.01908.x. [DOI] [PubMed] [Google Scholar]
- 65.Yen CJ, Izzo JG, Lee DF, Guha S, Wei Y, Wu TT, Chen CT, Kuo HP, Hsu JM, Sun HL, et al. Bile acid exposure up-regulates tuberous sclerosis complex 1/mammalian target of rapamycin pathway in Barrett’s-associated esophageal adenocarcinoma. Cancer Res. 2008;68:2632–2640. doi: 10.1158/0008-5472.CAN-07-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamat P, Wen S, Morris J, Anandasabapathy S. Exploring the association between elevated body mass index and Barrett’s esophagus: a systematic review and meta-analysis. Ann Thorac Surg. 2009;87:655–62. doi: 10.1016/j.athoracsur.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Lagergren J, Bergström R, Nyrén O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med. 1999;130:883–90. doi: 10.7326/0003-4819-130-11-199906010-00003. [DOI] [PubMed] [Google Scholar]
- 68.El-Serag H. The association between obesity and GERD: a review of the epidemiological evidence. Dig Dis Sci. 2008;53:2307–12. doi: 10.1007/s10620-008-0413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pandolfino JE, El-Serag HB, Zhang Q, Shah N, Ghosh SK, Kahrilas PJ. Obesity: a challenge to esophagogastric junction integrity. Gastroenterology. 2006;130:639–49. doi: 10.1053/j.gastro.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 70.Ryan AM, Healy LA, Power DG, Byrne M, Murphy S, Byrne PJ, Kelleher D, Reynolds JV. Barrett esophagus: prevalence of central adiposity, metabolic syndrome, and a proinflammatory state. Ann Surg. 2008;247:909–15. doi: 10.1097/SLA.0b013e3181612cac. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe S, Hojo M, Nagahara A. Metabolic syndrome and gastrointestinal diseases. J Gastroenterol. 2007;42:267–274. doi: 10.1007/s00535-007-2033-0. [DOI] [PubMed] [Google Scholar]
- 72.Rubenstein JH, Dahlkemper A, Kao JY, Zhang M, Morgenstern H, McMahon L, Inadomi JM. A pilot study of the association of low plasma adiponectin and Barrett’s esophagus. Am J Gastroenterol. 2008;103:1358–64. doi: 10.1111/j.1572-0241.2008.01823.x. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki H, Iijima K, Scobie G, Fyfe V, McColl KE. Nitrate and nitrosative chemistry within Barrett’s oesophagus during acid reflux. Gut. 2005;54:1527–35. doi: 10.1136/gut.2005.066043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Serag HB, Lagergren J. Alcohol drinking and the risk of Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 2009;136:1155–1157. doi: 10.1053/j.gastro.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen X, Yang G, Ding WY, Bondoc F, Curtis SK, Yang CS. An esophagogastroduodenal anastomosis model for esophageal adenocarcinogenesis in rats and enhancement by iron overload. Carcinogenesis. 1999;20:1801–8. doi: 10.1093/carcin/20.9.1801. [DOI] [PubMed] [Google Scholar]
- 76.Boult J, Roberts K, Brookes MJ, Hughes S, Bury JP, Cross SS, Anderson GJ, Spychal R, Iqbal T, Tselepis C. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin Cancer Res. 200814:379–387. doi: 10.1158/1078-0432.CCR-07-1054. [DOI] [PubMed] [Google Scholar]
- 77.Derakhshan MH, Liptrot S, Paul J, Brown IL, Morrison D, McColl KE. Oesophageal and gastric intestinal-type adenocarcinomas show the same male predominance due to a 17 year delayed development in females. Gut. 2009;58:16–23. doi: 10.1136/gut.2008.161331. [DOI] [PubMed] [Google Scholar]
- 78.Corley DA, Kubo A, Levin TR, Block G, Habel L, Rumore GJ, Quesenberry C, Buffler P. Hemochromatosis gene status as a risk factor for Barrett’s esophagus. Dig Dis Sci. 2008;53:3095–3102. doi: 10.1007/s10620-008-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fitzgerald RC. Molecular basis of Barrett’s oesophagus and oesophageal adenocarcinoma. Gut. 2006;55:1810–1820. doi: 10.1136/gut.2005.089144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wild CP, Hardie LJ. Reflux, Barrett’s oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer. 2003;3:676–684. doi: 10.1038/nrc1166. [DOI] [PubMed] [Google Scholar]
- 81.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 200464:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 82.Barrett MT, Sanchez CA, Neshat K, Galipeau PC, Reid BJ. Allelic loss of 9p21 and mutations of the CDKN2/p16 gene develop as early events in neoplastic progression in Barrett’s esophagus. Gastroenterology. 1996;110:A489. [PubMed] [Google Scholar]
- 83.Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, Rabinovitch PS, Reid BJ. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22:106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardie LJ, Darnton SJ, Wallis YL, Chauhan A, Hainaut P, Wild CP, Casson AG. p16 expression in Barrett’s esophagus and esophageal adenocarcinoma: association with genetic and epigenetic alterations. Cancer Lett. 2005;217:221–230. doi: 10.1016/j.canlet.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 85.Chao DL, Sanchez CA, Galipeau PC, Blount PL, Paulson TG, Cowan DS, Ayub K, Odze RD, Rabinovitch PS, Reid BJ. Cell proliferation, cell cycle abnormalities, and cancer outcome in patients with Barrett’s esophagus: a long-term prospective study. Clin Cancer Res. 2008;14:6988–6995. doi: 10.1158/1078-0432.CCR-07-5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galipeau P, et al. Gastroenterology. 2005;126:A822. [Google Scholar]
- 87.Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, Reid BJ. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res. 2001;61:8284–8289. [PubMed] [Google Scholar]
- 88.Lai LA, Paulson TG, Li X, Sanchez CA, Maley C, Odze RD, Reid BJ, Rabinovitch PS. Increasing genomic instability during premalignant neoplastic progression revealed through high resolution array-CGH. Genes Chromosomes Cancer. 2007;46:532–542. doi: 10.1002/gcc.20435. [DOI] [PubMed] [Google Scholar]
- 89.Huang Y, Peters CJ, Fitzgerald RC, Gjerset RA. Progressive silencing of p14ARF in oesophageal adenocarcinoma. J Cell Mol Med. 2009;13:398–409. doi: 10.1111/j.1582-4934.2008.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reid BJ, Prevo LJ, Galipeau PC, Sanchez CA, Longton G, Levine DS, Blount PL, Rabinovitch PS. Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol. 2001;96:2839–2848. doi: 10.1111/j.1572-0241.2001.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Murray L, Sedo A, Scott M, McManus D, Sloan JM, Hardie LJ, Forman D, Wild CP. TP53 and progression from Barrett’s metaplasia to oesophageal adenocarcinoma in a UK population cohort. Gut. 2006;55:1390–1397. doi: 10.1136/gut.2005.083295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, Wild CP. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92:1316–1321. doi: 10.1093/jnci/92.16.1316. [DOI] [PubMed] [Google Scholar]
- 93.Hong MK, Laskin WB, Herman BE, Johnston MH, Vargo JJ, Steinberg SM, Allegra CJ, Johnston PG. Expansion of the Ki-67 proliferative compartment correlates with degree of dysplasia in Barrett’s esophagus. Cancer. 1995;72:423–429. doi: 10.1002/1097-0142(19950115)75:2<423::aid-cncr2820750202>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 94.Sirieix P, O’Donovan M, Brown J, Save V, Coleman N, RC F. Surface expression of mini-chromosome maintenance proteins provides a novel method for detecting patients at risk for developing adeocarcinoma in Barrett’s oesophagus. Clin Cancer Res. 2003;9:2560–2566. [PubMed] [Google Scholar]
- 95.Williams GH, Swinn R, Prevost AT, De Clive-Lowe P, Halsall I, Going JJ, Hales CN, Stoeber K, Middleton SJ. Diagnosis of oesophageal cancer by detection of minichromosome maintenance 5 protein in gastric aspirates. Br J Cancer. 2004;91:714–719. doi: 10.1038/sj.bjc.6602028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lao-Sirieix P, B R, L L, C N, F RC. Cell cycle phase abnormalities do not account for disordered proliferation in Barrett’s carcinogenesis. Neoplasia. 2004;6:751–760. doi: 10.1593/neo.04280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–7633. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- 98.Vaughan TL, D L, Blount PL, Ayub K, Odze RD, Sanchez CA, Rabinovitch PS, Reid BJ. Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett’s oesophagus: a prospective study. Lancet Oncol. 2005;6:945–952. doi: 10.1016/S1470-2045(05)70431-9. [DOI] [PubMed] [Google Scholar]
- 99.Chao DL, Maley CC, Wu X, Farrow DC, Galipeau PC, Sanchez CA, Paulson TG, Rabinovitch PS, Reid BJ, Spitz MR, Vaughan TL. Mutagen sensitivity and neoplastic progression in patients with Barrett’s esophagus: a prospective analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1935–1940. doi: 10.1158/1055-9965.EPI-06-0492. [DOI] [PubMed] [Google Scholar]
- 100.Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cestari R, Villanacci V, Rossi E, Della Casa D, Missale G, Conio M, Grigolato P, Bassotti G. Fluorescence in situ hybridization to evaluate dysplasia in Barrett’s esophagus: a pilot study. Cancer Lett. 2007;251:278–287. doi: 10.1016/j.canlet.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 102.Chaves P, Crespo M, Ribeiro C, Laranjeira C, Pereira AD, Suspiro A, Cardoso P, Leitao CN, Soares J. Chromosomal analysis of Barrett’s cells: demonstration of instability and detection of the metaplastic lineage involved. Mod Pathol. 2007;20:788–796. doi: 10.1038/modpathol.3800787. [DOI] [PubMed] [Google Scholar]
- 103.Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett’s esophagus III: baseline flow cytometric variables. Am J Gastroenterol. 2001;96:3071–3083. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Galipeau PC, Prevo LJ, Sanchez CA, Longton GM, Reid BJ. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett’s) tissue. J Natl Cancer Inst. 1999;91:2087–2095. doi: 10.1093/jnci/91.24.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, Reid BJ. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 106.Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, Harrison R, Novelli MR, Jankowski JA, Wright NA. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008 doi: 10.1136/gut.2007.143339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muzeau F, Flejou JF, Belghiti J, Thomas G, Hamelin R. Infrequent microsatellite instability in oesophageal cancers. Br J Cancer. 1997;75:1336–1339. doi: 10.1038/bjc.1997.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brandt LJ, Kauvar DR. Laser-induced transient regression of Barrett’s epithelium. Gastrointest Endosc. 1992;38:619–22. doi: 10.1016/s0016-5107(92)70536-7. [DOI] [PubMed] [Google Scholar]
- 109.Berenson MM, Johnson TD, Markowitz NR, Buchi KN, Samowitz WS. Restoration of squamous mucosa after ablation of Barrett’s esophageal epithelium. Gastroenterology. 1993;104:1686–91. doi: 10.1016/0016-5085(93)90646-t. [DOI] [PubMed] [Google Scholar]
- 110.Curvers WL, Singh R, Song LM, Wolfsen HC, Ragunath K, Wang K, Wallace MB, Fockens P, Bergman JJ. Endoscopic tri-modal imaging for detection of early neoplasia in Barrett’s oesophagus: a multi-centre feasibility study using high-resolution endoscopy, autofluorescence imaging and narrow band imaging incorporated in one endoscopy system. Gut. 2008;57:167–72. doi: 10.1136/gut.2007.134213. [DOI] [PubMed] [Google Scholar]
- 111.Fernando HC, Murthy SC, Hofstetter W, Shrager JB, Bridges C, Mitchell JD, Landreneau RJ, Clough ER, Watson TJ. The Society of Thoracic Surgeons practice guideline series: guidelines for the management of Barrett’s esophagus with high-grade dysplasia. Ann Thorac Surg. 2009;87:1993–2002. doi: 10.1016/j.athoracsur.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 112.Das A, Singh V, Fleischer DE, Sharma VK. A comparison of endoscopic treatment and surgery in early esophageal cancer: an analysis of surveillance epidemiology and end results data. Am J Gastroenterol. 2008;103:1340–5. doi: 10.1111/j.1572-0241.2008.01889.x. [DOI] [PubMed] [Google Scholar]
- 113.Pech O, Behrens A, May A, Nachbar L, Gossner L, Rabenstein T, Manner H, Guenter E, Huijsmans J, Vieth M, Stolte M, Ell C. Long-term results and risk factor analysis for recurrence after curative endoscopic therapy in 349 patients with high-grade intraepithelial neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut. 2008;57:1200–6. doi: 10.1136/gut.2007.142539. [DOI] [PubMed] [Google Scholar]
- 114.Prasad GA, Wu TT, Wigle DA, Buttar NS, Wongkeesong LM, Dunagan KT, Lutzke LS, Borkenhagen LS, Wang KK. Endoscopic and Surgical Treatment of Mucosal (T1a) Esophageal Adenocarcinoma in Barrett’s Esophagus. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Overholt BF, Lightdale CJ, Wang KK, Canto MI, Burdick S, Haggitt RC, Bronner MP, Taylor SL, Grace MG, Depot M. Photodynamic therapy with porfimer sodium for ablation of high-grade dysplasia in Barrett’s esophagus: international, partially blinded, randomized phase III trial. Gastrointest Endosc. 2005;62:488–98. doi: 10.1016/j.gie.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 116.Fleischer DE, Overholt BF, Sharma VK, Reymunde A, Kimmey MB, Chuttani R, Chang KJ, Lightdale CJ, Santiago N, Pleskow DK, Dean PJ, Wang KK. Endoscopic ablation of Barrett’s esophagus: a multicenter study with 2.5-year follow-up. Gastrointest Endosc. 2008;68:867–76. doi: 10.1016/j.gie.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 117.Inadomi JM, Somsouk M, Madanick RD, Thomas JP, Shaheen NJ. A cost-utility analysis of ablative therapy for Barrett’s esophagus. Gastroenterology. 2009;136:2101–2114. e1–6. doi: 10.1053/j.gastro.2009.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sampliner RE. Endoscopic Therapy for Barrett’s Esophagus. Clin Gastroenterol Hepatol. 2009 doi: 10.1016/j.cgh.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 119.Falk GW. Radiofrequency Ablation of Barrett’s Esophagus: Should Everybody Get it? Gastroenterology. 2009;136:2399–401. doi: 10.1053/j.gastro.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 120.Prasad G, Wang KK. Endoscopic Mucosal Resection: Esophageal Applications. Curr Treat Options Gastroenterol. 2005;8:41–49. doi: 10.1007/s11938-005-0050-8. [DOI] [PubMed] [Google Scholar]
- 121.Seewald S, Ang TL, Gotoda T, Soehendra N. Total endoscopic resection of Barrett esophagus. Endoscopy. 2008;40:1016–20. doi: 10.1055/s-0028-1103401. [DOI] [PubMed] [Google Scholar]
- 122.Peters FP, Kara MA, Curvers WL, Rosmolen WD, Fockens P, Krishnadath KK, Ten Kate FJ, Bergman JJ. Multiband mucosectomy for endoscopic resection of Barrett’s esophagus: feasibility study with matched historical controls. Eur J Gastroenterol Hepatol. 2007;19:311–5. doi: 10.1097/MEG.0b013e328080ca90. [DOI] [PubMed] [Google Scholar]
- 123.Larghi A, Lightdale CJ, Ross AS, Fedi P, Hart J, Rotterdam H, Noffsinger A, Memeo L, Bhagat G, Waxman I. Long-term follow-up of complete Barrett’s eradication endoscopic mucosal resection (CBE-EMR) for the treatment of high grade dysplasia and intramucosal carcinoma. Endoscopy. 2007;39:1086–91. doi: 10.1055/s-2007-966788. [DOI] [PubMed] [Google Scholar]
- 124.Mino-Kenudson M, Hull MJ, Brown I, Muzikansky A, Srivastava A, Glickman J, Park DY, Zuckerberg L, Misdraji J, Odze RD, Lauwers GY. EMR for Barrett’s esophagus-related superficial neoplasms offers better diagnostic reproducibility than mucosal biopsy. Gastrointest Endosc. 2007;66:660–6. doi: 10.1016/j.gie.2007.02.063. quiz 767, 769. [DOI] [PubMed] [Google Scholar]
- 125.Peters FP, Brakenhoff KP, Curvers WL, Rosmolen WD, Fockens P, ten Kate FJ, Krishnadath KK, Bergman JJ. Histologic evaluation of resection specimens obtained at 293 endoscopic resections in Barrett’s esophagus. Gastrointest Endosc. 2008;67:604–9. doi: 10.1016/j.gie.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 126.Prasad GA, Buttar NS, Wongkeesong LM, Lewis JT, Sanderson SO, Lutzke LS, Borkenhagen LS, Wang KK. Significance of neoplastic involvement of margins obtained by endoscopic mucosal resection in Barrett’s esophagus. Am J Gastroenterol. 2007;102:2380–6. doi: 10.1111/j.1572-0241.2007.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Montgomery E, Bronner MP, Goldblum JR, Greenson JK, Haber MM, Hart J, Lamps LW, Lauwers GY, Lazenby AJ, Lewin DN, Robert ME, Toledano AY, Shyr Y, Washington K. Reproducibility of the diagnosis of dysplasia in Barrett esophagus: a reaffirmation. Hum Pathol. 2001;32:368–78. doi: 10.1053/hupa.2001.23510. [DOI] [PubMed] [Google Scholar]
- 128.Montgomery E, Goldblum JR, Greenson JK, Haber MM, Lamps LW, Lauwers GY, Lazenby AJ, Lewin DN, Robert ME, Washington K, Zahurak ML, Hart J. Dysplasia as a predictive marker for invasive carcinoma in Barrett esophagus: a follow-up study based on 138 cases from a diagnostic variability study. Hum Pathol. 2001;32:379–88. doi: 10.1053/hupa.2001.23511. [DOI] [PubMed] [Google Scholar]
- 129.Lewis JT, Wang KK, Abraham SC. Muscularis mucosae duplication and the musculo-fibrous anomaly in endoscopic mucosal resections for barrett esophagus: implications for staging of adenocarcinoma. Am J Surg Pathol. 2008;32:566–71. doi: 10.1097/PAS.0b013e31815bf8c7. [DOI] [PubMed] [Google Scholar]
- 130.Stein HJ, Feith M, Bruecher BL, Naehrig J, Sarbia M, Siewert JR. Early esophageal cancer: pattern of lymphatic spread and prognostic factors for long-term survival after surgical resection. Ann Surg. 2005;242:566–73. doi: 10.1097/01.sla.0000184211.75970.85. discussion 573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buskens CJ, Westerterp M, Lagarde SM, Bergman JJ, ten Kate FJ, van Lanschot JJ. Prediction of appropriateness of local endoscopic treatment for high-grade dysplasia and early adenocarcinoma by EUS and histopathologic features. Gastrointest Endosc. 2004;60:703–10. doi: 10.1016/s0016-5107(04)02017-6. [DOI] [PubMed] [Google Scholar]
- 132.Manner H, May A, Pech O, Gossner L, Rabenstein T, Gunter E, Vieth M, Stolte M, Ell C. Early Barrett’s carcinoma with “low-risk” submucosal invasion: long-term results of endoscopic resection with a curative intent. Am J Gastroenterol. 2008;103:2589–97. doi: 10.1111/j.1572-0241.2008.02083.x. [DOI] [PubMed] [Google Scholar]
- 133.Larghi A, Lightdale CJ, Memeo L, Bhagat G, Okpara N, Rotterdam H. EUS followed by EMR for staging of high-grade dysplasia and early cancer in Barrett’s esophagus. Gastrointest Endosc. 2005;62:16–23. doi: 10.1016/s0016-5107(05)00319-6. [DOI] [PubMed] [Google Scholar]
- 134.Dulai GS, Jensen DM, Cortina G, Fontana L, Ippoliti A. Randomized trial of argon plasma coagulation vs. multipolar electrocoagulation for ablation of Barrett’s esophagus. Gastrointest Endosc. 2005;61:232–40. doi: 10.1016/s0016-5107(04)02576-3. [DOI] [PubMed] [Google Scholar]
- 135.Sampliner RE, Faigel D, Fennerty MB, Lieberman D, Ippoliti A, Lewin K, Weinstein WM. Effective and safe endoscopic reversal of nondysplastic Barrett’s esophagus with thermal electrocoagulation combined with high-dose acid inhibition: a multicenter study. Gastrointest Endosc. 2001;53:554–8. doi: 10.1067/mge.2001.114418. [DOI] [PubMed] [Google Scholar]
- 136.Mork H, Al-Taie O, Berlin F, Kraus MR, Scheurlen M. High recurrence rate of Barrett’s epithelium during long-term follow-up after argon plasma coagulation. Scand J Gastroenterol. 2007;42:23–7. doi: 10.1080/00365520600825125. [DOI] [PubMed] [Google Scholar]
- 137.Ragunath K, Krasner N, Raman VS, Haqqani MT, Phillips CJ, Cheung I. Endoscopic ablation of dysplastic Barrett’s oesophagus comparing argon plasma coagulation and photodynamic therapy: a randomized prospective trial assessing efficacy and cost-effectiveness. Scand J Gastroenterol. 2005;40:750–8. doi: 10.1080/00365520510015737. [DOI] [PubMed] [Google Scholar]
- 138.Pereira-Lima JC, Busnello JV, Saul C, Toneloto EB, Lopes CV, Rynkowski CB, Blaya C. High power setting argon plasma coagulation for the eradication of Barrett’s esophagus. Am J Gastroenterol. 2000;95:1661–8. doi: 10.1111/j.1572-0241.2000.02197.x. [DOI] [PubMed] [Google Scholar]
- 139.Wang KK, Lutzke L, Borkenhagen L, Westra W, Song MW, Prasad G, Buttar NS. Photodynamic therapy for Barrett’s esophagus: does light still have a role? Endoscopy. 2008;40:1021–5. doi: 10.1055/s-0028-1103405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Overholt BF, Wang KK, Burdick JS, Lightdale CJ, Kimmey M, Nava HR, Sivak MV, Jr, Nishioka N, Barr H, Marcon N, Pedrosa M, Bronner MP, Grace M, Depot M. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest Endosc. 2007;66:460–8. doi: 10.1016/j.gie.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 141.Prasad GA, Wang KK, Buttar NS, Wongkeesong LM, Krishnadath KK, Nichols FC, 3rd, Lutzke LS, Borkenhagen LS. Long-term survival following endoscopic and surgical treatment of high-grade dysplasia in Barrett’s esophagus. Gastroenterology. 2007;132:1226–33. doi: 10.1053/j.gastro.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Prasad GA, Wang KK, Buttar NS, Wongkeesong LM, Lutzke LS, Borkenhagen LS. Predictors of stricture formation after photodynamic therapy for high-grade dysplasia in Barrett’s esophagus. Gastrointest Endosc. 2007;65:60–6. doi: 10.1016/j.gie.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 143.Hage M, Siersema PD, van Dekken H, Steyerberg EW, Haringsma J, van de Vrie W, Grool TE, van Veen RL, Sterenborg HJ, Kuipers EJ. 5-aminolevulinic acid photodynamic therapy versus argon plasma coagulation for ablation of Barrett’s oesophagus: a randomised trial. Gut. 2004;53:785–90. doi: 10.1136/gut.2003.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Peters F, Kara M, Rosmolen W, Aalders M, Ten Kate F, Krishnadath K, van Lanschot J, Fockens P, Bergman J. Poor results of 5-aminolevulinic acid-photodynamic therapy for residual high-grade dysplasia and early cancer in barrett esophagus after endoscopic resection. Endoscopy. 2005;37:418–24. doi: 10.1055/s-2005-861198. [DOI] [PubMed] [Google Scholar]
- 145.Sharma VK, Wang KK, Overholt BF, Lightdale CJ, Fennerty MB, Dean PJ, Pleskow DK, Chuttani R, Reymunde A, Santiago N, Chang KJ, Kimmey MB, Fleischer DE. Balloon-based, circumferential, endoscopic radiofrequency ablation of Barrett’s esophagus: 1-year follow-up of 100 patients. Gastrointest Endosc. 2007;65:185–95. doi: 10.1016/j.gie.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 146.Dunkin BJ, Martinez J, Bejarano PA, Smith CD, Chang K, Livingstone AS, Melvin WS. Thin-layer ablation of human esophageal epithelium using a bipolar radiofrequency balloon device. Surg Endosc. 2006;20:125–30. doi: 10.1007/s00464-005-8279-9. [DOI] [PubMed] [Google Scholar]
- 147.Shaheen NJ, Sharma P, Overholt BF, Wolfsen HC, Sampliner RE, Wang KK, Galanko JA, Bronner MP, Goldblum JR, Bennett AE, Jobe BA, Eisen GM, Fennerty MB, Hunter JG, Fleischer DE, Sharma VK, Hawes RH, Hoffman BJ, Rothstein RI, Gordon SR, Mashimo H, Chang KJ, Muthusamy VR, Edmundowicz SA, Spechler SJ, Siddiqui AA, Souza RF, Infantolino A, Falk GW, Kimmey MB, Madanick RD, Chak A, Lightdale CJ. Radiofrequency ablation in Barrett’s esophagus with dysplasia. N Engl J Med. 2009;360:2277–88. doi: 10.1056/NEJMoa0808145. [DOI] [PubMed] [Google Scholar]
- 148.Gage AA, Baust J. Mechanisms of tissue injury in cryosurgery. Cryobiology. 1998;37:171–86. doi: 10.1006/cryo.1998.2115. [DOI] [PubMed] [Google Scholar]
- 149.Dumot JA, Greenwald BD. Argon plasma coagulation, bipolar cautery, and cryotherapy: ABC’s of ablative techniques. Endoscopy. 2008;40:1026–32. doi: 10.1055/s-0028-1103414. [DOI] [PubMed] [Google Scholar]
- 150.Greenwald BD, Dumot JA, Horwhat JD, Lightdale CJ, Abrams JA. Safety, tolerability, and efficacy of endoscopic low-pressure liquid nitrogen spray cryotherapy in the esophagus. Dis Esophagus. 2009 doi: 10.1111/j.1442-2050.2009.00991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Canto MI, Gorospe EC, Shin EJ, Dunbar KB, Montogomery EA, Okolo P. Carbon Dioxide (CO2) Cryotherapy is a Safe and Effective Treatment of Barrett’s Esophagus (BE) with HGD/Intramucosal Carcinoma. Gastrointest Endosc. 2009;69:AB341. [Google Scholar]
- 152.Hornick JL, Mino-Kenudson M, Lauwers GY, Liu W, Goyal R, Odze RD. Buried Barrett’s epithelium following photodynamic therapy shows reduced crypt proliferation and absence of DNA content abnormalities. Am J Gastroenterol. 2008;103:38–47. doi: 10.1111/j.1572-0241.2007.01560.x. [DOI] [PubMed] [Google Scholar]
- 153.Mino-Kenudson M, Ban S, Ohana M, Puricelli W, Deshpande V, Shimizu M, Nishioka NS, Lauwers GY. Buried dysplasia and early adenocarcinoma arising in barrett esophagus after porfimer-photodynamic therapy. Am J Surg Pathol. 2007;31:403–9. doi: 10.1097/01.pas.0000213407.03064.37. [DOI] [PubMed] [Google Scholar]
- 154.Van Laethem JL, Peny MO, Salmon I, Cremer M, Devière J. Intramucosal adenocarcinoma arising under squamous re-epithelialisation of Barrett’s oesophagus. Gut. 2000;46:574–7. doi: 10.1136/gut.46.4.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bronner MP, Overholt BF, Taylor SL, Haggitt RC, Wang KK, Burdick JS, Lightdale CJ, Kimmey M, Nava HR, Sivak MV, Nishioka N, Barr H, Canto MI, Marcon N, Pedrosa M, Grace M, Depot M. International Photodynamic Therapy Group for High-Grade Dysplasia in Barrett’s Esophagus. Squamous overgrowth is not a safety concern for photodynamic therapy for Barrett’s esophagus with high-grade dysplasia. Gastroenterology. 2009;136:56–64. doi: 10.1053/j.gastro.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 156.Pouw RE, Gondrie JJ, Rygiel AM, Sondermeijer CM, ten Kate FJ, Odze RD, Vieth M, Krishnadath KK, Bergman JJ. Properties of the neosquamous epithelium after radiofrequency ablation of Barrett’s esophagus containing neoplasia. Am J Gastroenterol. 2009;104:1366–73. doi: 10.1038/ajg.2009.88. [DOI] [PubMed] [Google Scholar]
- 157.Wang KK. Selecting treatment for Barrett’s esophagus: no second chances to make a first impression. Clin Gastroenterol Hepatol. 2008;6:1181–2. doi: 10.1016/j.cgh.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 158.Yachimski P, Nishioka NS, Richards E, Hur C. Treatment of Barrett’s esophagus with high-grade dysplasia or cancer: predictors of surgical versus endoscopic therapy. Clin Gastroenterol Hepatol. 2008;6:1206–11. doi: 10.1016/j.cgh.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yachimski P, Puricelli WP, Nishioka NS. Patient predictors of histopathologic response after photodynamic therapy of Barrett’s esophagus with high-grade dysplasia or intramucosal carcinoma. Gastrointest Endosc. 2009;69:205–12. doi: 10.1016/j.gie.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 160.Yachimski P, Puricelli WP, Nishioka NS. Patient predictors of esophageal stricture development after photodynamic therapy. Clin Gastroenterol Hepatol. 2008;6:302–8. doi: 10.1016/j.cgh.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 161.Birkmeyer JD, Siewers AE, Finlayson EV, Stukel TA, Lucas FL, Batista I, Welch HG, Wennberg DE. Hospital volume and surgical mortality in the United States. N Engl J Med. 2002;346:1128–37. doi: 10.1056/NEJMsa012337. [DOI] [PubMed] [Google Scholar]
- 162.Gondrie JJ, Seewald S, Pouw RE, et al. Stepwise radical endoscopic resection for complete removal of Barrett’s esophagus with early neoplasia : an international multicenter study. Gastrointestinal Endoscopy. 2008;67:AB 175. [Google Scholar]
- 163.Yoshinaga S, Gotoda T, Kusano C, Oda I, Nakamura K, Takayanagi R. Clinical impact of endoscopic submucosal dissection for superficial adenocarcinoma located at the esophagogastric junction. Gastrointest Endosc. 2008;67:202–9. doi: 10.1016/j.gie.2007.09.054. [DOI] [PubMed] [Google Scholar]
- 164.Sharma P, Wani S, Weston AP, Bansal A, Hall M, Mathur S, Prasad A, Sampliner RE. A randomised controlled trial of ablation of Barrett’s oesophagus with multipolar electrocoagulation versus argon plasma coagulation in combination with acid suppression: long term results. Gut. 2006;55:1233–9. doi: 10.1136/gut.2005.086777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Attwood SE, Lewis CJ, Caplin S, Hemming K, Armstrong G. Argon beam plasma coagulation as therapy for high-grade dysplasia in Barrett’s esophagus. Clin Gastroenterol Hepatol. 2003;1:258–63. [PubMed] [Google Scholar]
- 166.Ferraris R, Fracchia M, Foti M, Sidoli L, Taraglio S, Vigano L, Giaccone C, Rebecchi F, Meineri G, Senore C, Pera A. Barrett’s oesophagus: long-term follow-up after complete ablation with argon plasma coagulation and the factors that determine its recurrence. Aliment Pharmacol Ther. 2007;25:835–40. doi: 10.1111/j.1365-2036.2007.03251.x. [DOI] [PubMed] [Google Scholar]
- 167.Pech O, Gossner L, May A, Rabenstein T, Vieth M, Stolte M, Berres M, Ell C. Long-term results of photodynamic therapy with 5-aminolevulinic acid for superficial Barrett’s cancer and high-grade intraepithelial neoplasia. Gastrointest Endosc. 2005;62:24–30. doi: 10.1016/s0016-5107(05)00333-0. [DOI] [PubMed] [Google Scholar]