Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing (original) (raw)
. Author manuscript; available in PMC: 2010 Aug 24.
Abstract
Clinical management of human cancer is dependent on the accurate monitoring of residual and recurrent tumors. The evaluation of patient-specific translocations in leukemias and lymphomas has revolutionized diagnostics for these diseases. We have developed a method, called personalized analysis of rearranged ends (PARE), which can identify translocations in solid tumors. Analysis of four colorectal and two breast cancers with massively parallel sequencing revealed an average of nine rearranged sequences (range, 4 to 15) per tumor. Polymerase chain reaction with primers spanning the breakpoints was able to detect mutant DNA molecules present at levels lower than 0.001% and readily identified mutated circulating DNA in patient plasma samples. This approach provides an exquisitely sensitive and broadly applicable approach for the development of personalized biomarkers to enhance the clinical management of cancer patients.
INTRODUCTION
A nearly universal feature of human cancer is the widespread rearrangement of chromosomes as a result of chromosomal instability (1). Such structural alterations begin to occur at the earliest stages of tumorigenesis and persist throughout tumor development. The consequences of chromosomal instability can include copy number alterations (duplications, amplifications, and deletions), inversions, insertions, and translocations (2). Historically, the ability to detect such alterations has been limited by the resolution of genetic analyses. However, a number of more recent approaches, including high density oligonucleotide arrays and high-throughput sequencing, have allowed detection of changes at much higher resolution (3–15).
Tumor-specific (somatic) chromosomal rearrangements have the potential to serve as highly sensitive biomarkers for tumor detection. Such alterations are not present in normal cells and should be exquisitely specific. Rearrangement-associated biomarkers therefore offer a reliable measure that would be useful for monitoring tumor response to specific therapies, detecting residual disease after surgery, and long-term clinical management. Recurrent somatic structural alterations, such as those involving the BCR-ABL oncogene (the target of the Philadelphia chromosome translocation), immunoglobulin genes, T cell receptor genes, and the retinoic acid receptor a gene, have been shown to be useful as diagnostic markers in certain hematopoietic malignancies (16–20). However, recurrent structural alterations do not generally occur in most solid tumors. We reasoned that any structural alteration identified in an individual's tumor could be used for analogous purposes, whether it was found in tumors of the same type in other individuals and whether it was a “driver”—causing a selective growth advantage—or a “passenger.” We describe herein our efforts to implement this concept in representative examples of common solid tumors.
RESULTS
Description of the approach
The PARE (personalized analysis of rearranged ends) approach, shown schematically in Fig. 1, requires the identification of patient specific rearrangements in tumor samples. To determine the feasibility of identifying such alterations using next-generation sequencing approaches, we initially analyzed four tumor samples (two colon and two breast tumors) and their matched normal tissue samples using the Applied Biosystems SOLiD System (Table 1). Genomic DNA from each sample was purified, sheared, and used to generate libraries with mate-paired tags ~1.4 kb apart. Libraries were digitally amplified by emulsion polymerase chain reaction (PCR) on magnetic beads (21), and 25-bp mate-paired tags were sequenced using the sequencing-by-ligation approach (15, 22). An average of 198.1 million 25-bp reads was obtained for each sample, where each read aligned perfectly and was uniquely localized in the reference human genome(hg18), resulting in 4.95 Gb of mappable sequence per sample. An average of 40 million mate-paired reads, where both tags were perfectly mapped to the reference human genome, was obtained for each sample. The total amount of genome base pairs covered by the mate-paired analysis (that is, distance between mate-paired tags × number of mate-paired tags) was 53.6 Gb per sample, or an 18-fold physical coverage of the human genome.
Fig. 1.

Schematic of PARE approach. The method is based on next-generation mate-paired analysis of resected tumor DNA to identify individualized tumor-specific rearrangements. Such alterations are used to develop PCR-based quantitative analyses for personalized tumor monitoring of plasma samples or other bodily fluids.
Table 1.
Summary of mate-paired tag libraries.
| Single tag analyses | Mate-paired tag analyses | |||||||
|---|---|---|---|---|---|---|---|---|
| Samples | Number of beads* | Number of tags matching human genome | Total bases sequenced (bp) | Expected coverage per 3-kb bin | Number of mate-paired tags matching human genome | Distance between mate-paired tags (bp) | Total physical coverage by mate-paired tags (bp) | Expected genome coverage |
| Colon cancer | ||||||||
| Col08 tumor | 526,209,780 | 121,527,707 | 3,038,192,675 | 122 | 21,899,809 | 1371 | 30,024,693,714 | 10.0 |
| Co108 normal | 328,599,033 | 86,032,253 | 2,150,806,325 | 86 | 11,694,361 | 1254 | 14,665,530,804 | 4.9 |
| Co84 tumor | 677,137,128 | 256,065,437 | 6,401,635,925 | 256 | 58,678,410 | 1488 | 87,292,060,006 | 29.1 |
| Co84 normal | 486,663,520 | 218,280,146 | 5,457,003,650 | 218 | 59,019,031 | 1384 | 81,690,396,379 | 27.2 |
| Hx402 tumor | 523,745,015 | 198,342,749 | 4,958,568,725 | 198 | 43,457,431 | 1629 | 70,789,547,653 | 23.6 |
| Hx403 tumor | 475,658,760 | 164,061,938 | 4,101,548,450 | 164 | 37,123,395 | 1705 | 63,295,388,475 | 21.1 |
| Breast cancer | ||||||||
| B7 tumor | 840,979,999 | 281,027,274 | 7,025,681,850 | 281 | 27,548,989 | 1220 | 33,604,662,404 | 11.2 |
| B7 normal | 705,704,265 | 253,482,262 | 6,337,056,550 | 253 | 57,878,644 | 1404 | 81,271,654,770 | 27.1 |
| B5 tumor | 444,249,217 | 147,612,941 | 3,690,323,525 | 148 | 29,961,045 | 1193 | 35,730,144,651 | 11.9 |
| B5 normal | 549,237,156 | 220,669,795 | 5,516,744,875 | 221 | 53,611,974 | 1205 | 64,591,276,025 | 21.5 |
Identification of somatic rearrangements
Two methods were used to identify somatic rearrangements from these data (fig. S1). The first approach involved searching for tags whose mate pairs were derived from different chromosomes (interchromosomal rearrangements). The high physical coverage of breakpoints provided by the ~40 million mate-paired sequences per sample (Table 1) suggested that a large fraction of such translocations could be identified. End sequences from such mate-paired tags were grouped into 1-kb bins, and those bin pairs that were observed at least five times were analyzed further. The requirement for five or more occurrences minimized the chance that the presumptive fusion sequences represent incorrect mapping to the reference genome or artifacts of library construction. Comparison with SOLiD libraries made from the matched normal samples reduced the possibility that the fusion sequences represented rare germline variants rather than somatic events.
The second approach combined mate-paired tag data with copy number alterations identified by analyses of individual 25-bp tags. Tumor-specific copy number alterations are often associated with de novo rearrangements (23), and the boundaries of such alterations would be expected to contain novel junctions not present in the human genome. To identify somatic copy number gains, losses, high-amplitude amplifications, and homozygous deletions, we grouped tags into nonoverlapping 3-kb bins. Normalized tag densities, defined as the number of tags per bin divided by average number of tags per bin, were determined for all 3-kb bins in each sample. Bins that displayed tag density ratios of >1.75 or <0.25 in two or more normal tissue samples (corresponding to <6% of all bins) were discarded from the analysis. This eliminated confounding regions of common germline copy number variation and resulted in 892,567 bins that were analyzed in each tumor sample. Comparison of 256 million reads from colorectal tumor sample Co84 with Illumina arrays containing ~1 million single-nucleotide polymorphism (SNP) probes and with a ~1 million Digital Karyotyping tag library obtained with Sanger sequencing showed high concordance for copy number alterations among the three platforms (fig. S2 and table S1). With the higher resolution afforded by the SOLiD data, we were able to identify additional copy number changes not detected with the other methods (table S2). Boundary regions of copy number alteration were analyzed to identify mate-paired tags corresponding to rearranged DNA sequences. These included fusion of DNA sequences that have inappropriate spacing, order, or orientation on the same chromosome (intrachromosomal rearrangements), or inappropriate joining of sequences from different chromosomes (interchromosomal rearrangements).
Through these two approaches, we identified 57 regions containing putative somatic rearrangements, with an average of 14 rearrangements per sample (Table 2). Of these, an average of seven represented interchromosomal rearrangements and seven represented intrachromosomal rearrangements. For confirmation, we designed primers to 42 of the paired-end regions and used them for PCR spanning the putative breakpoints. Thirty-five of these (83%) yielded PCR products of the expected size in the tumor samples but not in the normal samples (Fig. 2 and table S3). Sanger sequencing of seven PCR products confirmed the rearrangements in all cases tested. Although there was variation in the number of detected alterations per sample (range, 7 to 21), all four tumor samples were found to have at least four bona fide somatic rearrangements through this approach.
Table 2.
Summary of rearrangements identified in tumor samples.
| Sample | Rearrangement type | Total rearrangements | Tested rearrangements | Confirmed somatic rearrangements | |
|---|---|---|---|---|---|
| Intrachromosomal | Interchromosomal | ||||
| Tumor and normal libraries | |||||
| B5 | 7 | 4 | 11 | 7 | 5 (71%) |
| B7 | 17 | 4 | 21 | 16 | 15 (94%) |
| Co84 | 0 | 7 | 7 | 6 | 4 (67%) |
| Co108 | 6 | 12 | 18 | 13 | 11 (85%) |
| Tumor libraries | |||||
| Hx402 | 7 | 2 | 9 | 9 | 4 (44%) |
| Hx403 | 17 | 0 | 17 | 12 | 7 (58%) |
Fig. 2.

Detection of tumor-specific rearrangements in breast and colorectal cancers. Two representative rearrangements are shown for each tumor sample. (A) PCR amplification across breakpoint regions. MW, molecular weight; T, tumor; N, normal. (B) Genomic coordinates for a representative mate pair of each rearrangement.
Further examination revealed that rearrangements could be readily identified with high confidence even in the absence of data from matched normal DNA by using the copy number and mate-pair coupled approach. Elimination of analysis of the matched normal would reduce the cost and simplify the identification of rearrangements. To test this strategy, we analyzed two additional tumor samples (H×402 and H×403) through the SOLiD approach but without generation of matching normal DNA libraries. We found that it was possible to identify putative rearrangements resulting in interchromosomal and intrachromosomal rearrangements at the border of copy number variations with high specificity even in the absence of a matched normal library. We were able to identify 11 confirmed somatic alterations (4 in H×402 and 7 in H×403) out of 21 candidate changes tested (table S3).
Development of PARE biomarkers from rearranged sequences
Each of the rearranged sequences identified through PARE was unique, as no identical rearrangement was found in any of the other five tumor samples. To determine the utility of these rearranged sequences to serve as potential biomarkers, we designed PCR assays to detect them in the presence of increasing amounts of normal DNA. These conditions simulate detection of tumor DNA from patient blood or other bodily fluids, where tumor DNA constitutes a minority of total DNA. PCR products representing a rearranged region from each of the six dilutions of tumor DNA could be identified, even in mixtures of DNA containing 1 cancer genome equivalent among ~390,000 normal genome equivalents (Fig. 3). Furthermore, no background PCR products were discernible when DNA from normal tissues was used as control.
Fig. 3.
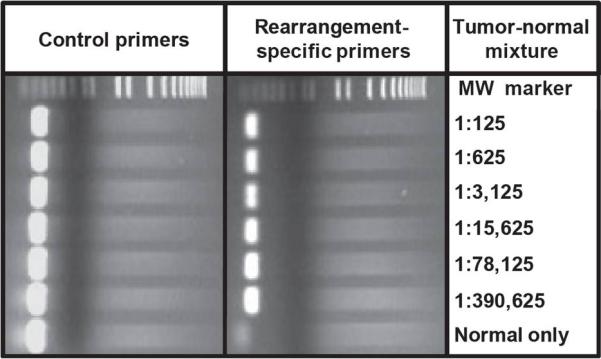
Detection of tumor-specific rearrangements in mixtures of tumor and normal DNA. Decreasing amounts of tumor DNA were mixed with increasing amounts of normal tissue DNA (300 ng total) and were used as template molecules for PCR using chromosome 4:8 translocation-specific primers or chromosome 3 control primers (see Materials and Methods for additional information).
To determine whether the rearranged sequences could actually be detected in clinical samples, we evaluated circulating DNA from plasma samples of patients H×402 and H×403. The sample from patient H×403 was obtained before surgery, whereas the samples from patient H×402 were obtained before and after surgery. A chromosome 4:8 translocation associated with an amplification was used in tumor H×402, and an intrachromosomal rearrangement associated with a homozygous deletion of chromosome 16 was used in tumor H×403. PCR amplification of plasma DNA using primers spanning the breakpoints produced products of the expected sizes only in the plasma samples from patients with disease and not in plasma from healthy controls (Fig. 4A). Sequencing of the PCR products from plasma DNA identified the identical breakpoints observed in the tumor DNA samples.
Fig. 4.
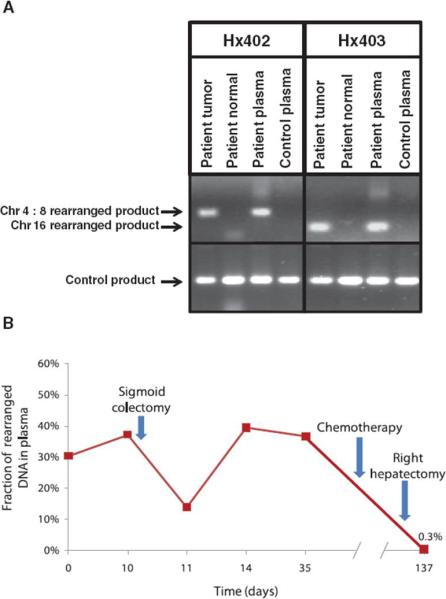
Detection of tumor-specific rearrangements in plasma of cancer patients. (A) The identified chromosome 4:8 and 16 rearrangements were used to design PCR primers spanning breakpoints and to amplify rearranged DNA from tumor tissue and plasma from patients H×402 and H×403, respectively. A plasma sample from an unrelated healthy individual was used as a control for both rearrangements. (B) Plasma samples from patient H×402 were analyzed at different time points using digital PCR to determine the fraction of genomic equivalents of plasma DNA containing the chromosome 4:8 rearrangement. The fraction of rearranged DNA at day 137 was 0.3%, consistent with residual metastatic lesions present in the remaining lobe of the liver.
We next determined whether circulating tumor-derived rearranged DNA could be quantitatively measured in patient plasma. Digital PCR analyses of plasma samples from patient H×402 indicated that rearranged tumor DNA was detectable at all six time points analyzed throughout the course of treatment (Fig. 4B). The fraction of mutant DNA present in plasma samples was 37% just before surgery but decreased to 14% 1 day after resection of the primary tumor. The mutant DNA fraction decreased further after chemotherapy and subsequent removal of metastatic lesions from the right lobe of the liver. However, the fraction of mutant tumor DNA did not reach zero (remaining at 0.3% at day 137), consistent with the fact that this patient had residual metastatic lesions in the remaining left lobe of the liver.
DISCUSSION
These results demonstrate that massively parallel sequencing can be used to develop personalized biomarkers based on somatic rearrangements. We were able to identify tumor-specific markers in each of the six breast and colorectal cancer cases analyzed. Moreover, we demonstrated that the identified breakpoints can be used to detect tumor DNA in the presence of large quantities of normal DNA and in patient plasma. These results highlight the sensitivity and specificity of the approach and suggest broad clinical utility of the PARE method.
Virtually all tumors of clinical consequence are thought to have rearranged DNA sequences resulting from translocations and copy number alterations, and these sequences are not present in normal human plasma or nontumor tissues. A recent genome-wide analysis of 24 breast cancers showed that all analyzed samples contained at least one genomic rearrangement that could be detected by next generation sequencing (24). From a technical perspective, PARE derived clinical assays should have no false positives: The PCR amplification of aberrant fusions of DNA sequences that are normally thousands of base pairs apart or on different chromosomes should not occur in nontumor DNA. In contrast, approaches that rely on monitoring of residual disease by analysis of somatic single-base alterations in specific genes are limited by polymerase error rates at the bases of interest (25). The PCR process generates background single-base mutations that are identical to bona fide mutations but does not generate false-positive rearrangements with carefully chosen primers. Because of the higher signal-to-noise ratio thereby obtained, PARE theoretically permits more sensitive monitoring of tumor burden.
The PARE approach, however, is not without limitations. Although somatic alterations in oncogenes and tumor suppressor genes persist throughout the clonal evolution of a tumor, it is conceivable that some rearranged sequences could be lost during tumor progression. The identification of several PARE biomarkers, each specific for different chromosomal regions, would mitigate this concern, as it is unlikely that all such markers would be lost in any particular patient. Another limitation is the cost of identifying a patient-specific alteration. In this prototype study, we obtained an average of 194.7 million reads per patient, resulting in ~200 tags in each 3-kb bin. The current cost for such an assay is ~$5000, which is expensive for general clinical use. This cost is a consequence of the high physical coverage and the inefficiencies associated with stringent mapping of 25-bp sequence data to the human genome. As read quality and length continue to improve, less stringent mapping criteria and lower physical coverage will permit analyses similar to those in this study but with substantially less sequencing effort. Moreover, the cost of massively parallel sequencing, which has decreased substantially over the last 2 years, continues to spiral downwards. Finally, there are clinical settings where the fraction of any DNA from tumors, including rearranged sequences, in the patient plasma is exceedingly small and undetectable. To be detectable by PARE, there must be at least one rearrangement template molecule in the plasma sample analyzed. When disease burden is this light, PARE may yield false-negative results. Larger studies will be needed to determine the clinical utility of PARE and its prognostic capabilities.
Despite these caveats, there are numerous potential applications of PARE. These include the more accurate identification of surgical margins free of tumor and the analysis of regional lymph nodes as well as the measurement of circulating tumor DNA after surgery, radiation, or chemotherapy. Short-term monitoring of circulating tumor DNA may be particularly useful in the testing of new drugs, as it could provide an earlier indication of efficacy than is possible through conventional diagnostic methods such as computed tomography (CT) scanning. Given current enthusiasm for the personalized management of cancer patients, PARE affords a timely method for uniquely sensitive and specific tumor monitoring.
MATERIALS AND METHODS
Clinical samples and cell lines
DNA samples were obtained from early-passage xenografts and cell lines of breast and colorectal cancers as described (26). Normal DNA samples were obtained from matched normal tissue. Plasma samples were collected from colorectal cancer patients H×402 and H×403 and from an unrelated normal control. All samples were obtained in accordance with the Health Insurance Portability and Accountability Act.
Digital Karyotyping and Illumina BeadChip arrays
A Digital Karyotyping library for colorectal cancer cell line Co84C was constructed as previously described (6). In summary, 17-bp genomic DNA tags were generated using the NlaIII and SacI restriction enzymes. The experimental tags obtained were concatenated, cloned, and sequenced. Previously described software was used to extract the experimental tags from the sequencing data. The sequences of the experimental tags were compared to the predicted virtual tags extracted from the human genome reference sequence. Amplifications were identified using sliding windows of variable sizes, and windows with tag density ratios of ≥6 were considered to represent amplified regions.
The Illumina Infinium II Whole Genome Genotyping Assay using the BeadChip platform was used to analyze the colorectal cancer cell line Co84C at 317K SNP loci from the Human HapMap collection. This assay is a two-step procedure: First the sample is hybridized to a 50-nucleotide oligo, and then the SNP position is interrogated by a two-color fluorescent single-base extension. Image files and data normalization were processed as previously described (10). Amplifications were defined as regions having at least 1 SNP with a LogR ratio of ≥1.4, at least 1 in 10 SNPs with a LogR ratio of ≥1, and an average LogR ratio of the entire region of ≥0.9.
SOLiD library preparation and sequencing
Mate-pair libraries were generated for the SOLiD platform as described (15). In brief, genomic DNA was sheared into ~1.4-kb fragments and used as template in emulsion PCR. Fragments were coupled to beads via an adapter sequence and clonally amplified. A 3′ modification of the DNA fragments allowed for covalent attachment to a slide. Sequencing primers hybridized to the adapter sequence and four fluorescently labeled di-base probes were used in ligation-based sequencing. Each nucleotide is sequenced twice in two different ligation reactions, resulting in two-base encoding, which has been shown to reduce sequencing artifacts.
Sequence data were mapped to the human genome reference sequence (hg18) using the Corona SOLiD software pipeline. All 25-bp tags (for both individual tag and mate-paired tag analyses) were required to match the reference genome uniquely and without mismatches.
Analysis of single tags for copy number alterations
The SOLiD tags were filtered and the remaining tags were grouped by genomic position in nonoverlapping 3-kb bins. A tag density ratio was calculated for each bin by dividing the number of tags observed in the bin by the average number of tags expected to be in each bin (on the basis of the total number of tags obtained for chromosomes 1 to 22 for each library divided by 849,434 total bins). The tag density ratio thereby allowed a normalized comparison between libraries containing different numbers of total tags. A control group of SOLiD libraries made from the four matched normal samples from Table 1 and two additional normal samples [CEPH (Centre d'Etude du Polymorphisme Humain) samples NA07357 and NA18507] was used to define areas of germline copy number variation or that contained a large fraction of repeated or low-complexity sequences. Any bin where at least two of the normal libraries had a tag density ratio of <0.25 or >1.75 was removed from further analysis.
Homozygous deletions were identified as three or more consecutive bins with tag ratios of <0.25 and at least one bin with a tag ratio of <0.005. Amplifications were identified as three or more consecutive bins with tag ratios of >2.5 and at least one bin with a tag ratio of >6. Single-copy gains and losses were identified through visual inspection of tag density data for each sample.
Analysis of mate-paired tags
Mate-paired tags mapping the reference genome uniquely and without mismatches were analyzed for aberrant mate-pair spacing, orientation, and ordering and categorized in 13 three-letter data formats (27). Mate pairs from the same chromosome that map at appropriate distances (~1.4 kb) and in the appropriate orientation and ordering are categorized as AAA. Mate pairs mapping to different chromosomes are categorized as C**. For the analysis of translocations of the PARE approach, we focused on C** mate pairs, whereas for analysis of rearrangements adjacent to copy number alterations, we chose all non-AAA (including C**) mate pairs for further analysis.
PARE identification and confirmation of candidate rearrangements
To identify candidate translocations, we grouped C** mate-pair tags in 1-kb bins and looked for bin pairs that were observed five or more times in the tumor sample but that were not observed in the matched normal sample. For identification of candidate rearrangements associated with copy number alterations, we analyzed the 10-kb boundary regions of amplifications, homozygous deletions, or lower copy gains and losses for neighboring non-AAA tags observed more than two times in the tumor but not in the matched normal sample. In the case of H×402 and H×403, the analysis of rearrangements adjacent to copy number alterations was performed in the absence of SOLiD libraries from normal tissue.
Mate-pair tag sequences associated with a candidate rearrangement were used as target sequences for primer design using Primer3 (28). When primers could not be designed from tag sequences alone, adjacent genomic sequence up to 100 bp was used for primer design. The observed rearranged tag ordering and orientation was used for Primer3 queries. Primers were used for PCR on tumor and matched normal samples as previously described (26). The candidate rearrangement was confirmed if a PCR product of the expected size was seen in the tumor but not in the matched normal sample. Sanger sequencing of PCR products was used to identify sequence breakpoint in a subset of cases.
Detection of PARE biomarker in human plasma
To determine the sensitivity of rearranged biomarkers in the presence of normal DNA, we used serial dilutions of tumor-normal DNA mixtures as templates for PCR using primers for the chromosome 4:8 translocation in H×402. The tumor DNA dilution began at 1:125 tumor-normal and continued as a one-in-five serial dilution until reaching 1:390,625 tumor-normal mixture. PCR was performed for each of the six tumor-normal DNA mixtures and for the normal DNA control using translocation-specific primers as well as control primers from chromosome 3.
Human plasma samples (1 ml) were obtained from patients H×402 and H×403 and from a control individual, and DNA was purified as described (29). Whole-genome amplification of plasma DNA was performed by ligation of adaptor sequences and PCR amplification with universal primers from the Illumina Genomic DNA Sample Prep kit.
Primers designed to amplify <200-bp fragments spanning each PARE rearrangement were used in PCR from total plasma DNA using patient or control samples. Digital PCR of plasma DNA dilutions from patient H×402 using rearrangement-specific and control primers was used to quantitate the fraction-mutated DNA molecules.
Supplementary Material
S/D
Acknowledgments
We thank H. Peckham for genome mapping analyses of SOLiD data.
Funding: The project was supported by NIH grants CA121113, CA057345, CA62924 and CA043460; Lustgarten Foundation; National Colorectal Cancer Research Alliance; The Virginia and D. K. Ludwig Fund for Cancer Research; and United Negro College Fund–Merck Fellowship.
Footnotes
SUPPLEMENTARY MATERIAL
www.sciencetranslationalmedicine.org/cgi/content/full/2/20/20ra14/DC1
Table S1. Comparison of SOLiD sequencing, Illumina SNP arrays, and Digital Karyotyping for analysis of copy number alterations.
Table S2. Putative copy number alterations identified by SOLiD sequencing in Co84 that were not identified by Illumina SNP arrays or Digital Karyotyping.
Table S3. Confirmed somatic rearrangements in breast and colorectal cancer samples. Fig. S1. Flow chart of approach used to identify rearranged sequences.
Fig. S2. Comparison of Digital Karyotyping, Illumina SNP array, and SOLiD sequencing results on chromosome 8.
Competing interests: Under a licensing agreement between Johns Hopkins University and Genzyme, B.V., V.E.V., and K.W.K. are entitled to a share of royalties received by the University on sales of products related to research described in this paper. The terms of these arrangements are managed by Johns Hopkins University in accordance with its conflict-of-interest policies.
REFERENCES AND NOTES
- 1.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 2.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 3.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 4.Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, Rodgers L, Brady A, Sebat J, Troge J, West JA, Rostan S, Nguyen KC, Powers S, Ye KQ, Olshen A, Venkatraman E, Norton L, Wigler M. Representational oligonucleotide microarray analysis: A high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, Cheung SW, Shen RM, Barker DL, Gunderson KL. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang TL, Maierhofer C, Speicher MR, Lengauer C, Vogelstein B, Kinzler KW, Velculescu VE. Digital karyotyping. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16156–16161. doi: 10.1073/pnas.202610899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang TL, Diaz LA, Jr., Romans K, Bardelli A, Saha S, Galizia G, Choti M, Donehower R, Parmigiani G, Shih I-M, Iacobuzio-Donahue C, Kinzler KW, Vogelstein B, Lengauer C, Velculescu VE. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3089–3094. doi: 10.1073/pnas.0308716101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di C, Liao S, Adamson DC, Parrett TJ, Broderick DK, Shi Q, Lengauer C, Cummins JM, Velculescu VE, Fults DW, McLendon RE, Bigner DD, Yan H. Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all-trans retinoic acid. Cancer Res. 2005;65:919–924. [PubMed] [Google Scholar]
- 9.Nakayama K, Nakayama N, Davidson B, Katabuchi H, Kurman RJ, Velculescu VE, Shih I-M, Wang TL. Homozygous deletion of MKK4 in ovarian serous carcinoma. Cancer Biol. Ther. 2006;5:630–634. doi: 10.4161/cbt.5.6.2675. [DOI] [PubMed] [Google Scholar]
- 10.Leary RJ, Lin JC, Cummins J, Boca S, Wood LD, Parsons DW, Jones S, Sjöblom T, Park BH, Parsons R, Willis J, Dawson D, Willson JK, Nikolskaya T, Nikolsky Y, Kopelovich L, Papadopoulos N, Pennacchio LA, Wang TL, Markowitz SD, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16224–16229. doi: 10.1073/pnas.0808041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders AC, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, Teague JW, Menzies A, Goodhead I, Turner DJ, Clee CM, Quail MA, Cox A, Brown C, Durbin R, Hurles ME, Edwards PA, Bignell GR, Stratton MR, Futreal PA. Identification of somatically acquired rearrangements in cancer using genomewide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, Zhang Z, Ranade SS, Dimalanta ET, Hyland FC, Sokolsky TD, Zhang L, Sheridan A, Fu H, Hendrickson CL, Li B, Kotler L, Stuart JR, Malek JA, Manning JM, Antipova AA, Perez DS, Moore MP, Hayashibara KC, Lyons MR, Beaudoin RE, Coleman BE, Laptewicz MW, Sannicandro AE, Rhodes MD, Gottimukkala RK, Yang S, Bafna V, Bashir A, MacBride A, Alkan C, Kidd JM, Eichler EE, Reese MG, De La Vega FM, Blanchard AP. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimwade D, Jovanovic JV, Hills RK, Nugent EA, Patel Y, Flora R, Diverio D, Jones K, Aslett H, Batson E, Rennie K, Angell R, Clark RE, Solomon E, Lo-Coco F, Wheatley K, Burnett AK. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J. Clin. Oncol. 2009;27:3650–3658. doi: 10.1200/JCO.2008.20.1533. [DOI] [PubMed] [Google Scholar]
- 17.Bregni M, Siena S, Neri A, Bassan R, Barbui T, Delia D, Bonadonna G, Dalla Favera R, Gianni AM. Minimal residual disease in acute lymphoblastic leukemia detected by immune selection and gene rearrangement analysis. J. Clin. Oncol. 1989;7:338–343. doi: 10.1200/JCO.1989.7.3.338. [DOI] [PubMed] [Google Scholar]
- 18.van der Velden VH, Panzer-Grümayer ER, Cazzaniga G, Flohr T, Sutton R, Schrauder A, Basso G, Schrappe M, Wijkhuijs JM, Konrad M, Bartram CR, Masera G, Biondi A, van Dongen JJ. Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multi-center setting. Leukemia. 2007;21:706–713. doi: 10.1038/sj.leu.2404535. [DOI] [PubMed] [Google Scholar]
- 19.Lion T. Minimal residual disease. Curr. Opin. Hematol. 1999;6:406–411. doi: 10.1097/00062752-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, Baccarani M, Cortes J, Cross NC, Druker BJ, Gabert J, Grimwade D, Hehlmann R, Kamel-Reid S, Lipton JH, Longtine J, Martinelli G, Saglio G, Soverini S, Stock W, Goldman JM. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: Review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 23.Bignell GR, Santarius T, Pole JC, Butler AP, Perry J, Pleasance E, Greenman C, Menzies A, Taylor S, Edkins S, Campbell P, Quail M, Plumb B, Matthews L, McLay K, Edwards PA, Rogers J, Wooster R, Futreal PA, Stratton MR. Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome Res. 2007;17:1296–1303. doi: 10.1101/gr.6522707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, Greenman CD, Jia M, Latimer C, Teague JW, Lau KW, Burton J, Quail MA, Swerdlow H, Churcher C, Natrajan R, Sieuwerts AM, Martens JW, Silver DP, Langerød A, Russnes HE, Foekens JA, Reis-Filho JS, van 't Veer L, Richardson AL, Børresen-Dale AL, Campbell PJ, Futreal PA, Stratton MR. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M, Diehl F, Dressman D, Vogelstein B, Kinzler KW. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods. 2006;3:95–97. doi: 10.1038/nmeth850. [DOI] [PubMed] [Google Scholar]
- 26.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 27.SOLiD data format and file definitions guide. http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_058717.pdf.
- 28.Primer3 version 0.4.0. http://frodo.wi.mit.edu/primer3/
- 29.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA., Jr. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S/D