TagRecon: High-Throughput Mutation Identification through Sequence Tagging (original) (raw)
. Author manuscript; available in PMC: 2011 Apr 5.
Published in final edited form as: J Proteome Res. 2010 Apr 5;9(4):1716–1726. doi: 10.1021/pr900850m
Abstract
Shotgun proteomics produces collections of tandem mass spectra that contain all the data needed to identify mutated peptides from clinical samples. Identifying these sequence variations, however, has not been feasible with conventional database search strategies, which require exact matches between observed and expected sequences. Searching for mutations as mass shifts on specified residues through database search can incur significant performance penalties and generate substantial false positive rates. Here we describe TagRecon, an algorithm that leverages inferred sequence tags to identify unanticipated mutations in clinical proteomic data sets. TagRecon identifies unmodified peptides as sensitively as the related MyriMatch database search engine. In both LTQ and Orbitrap data sets, TagRecon outperformed state of the art software in recognizing sequence mismatches from data sets with known variants. We developed guidelines for filtering putative mutations from clinical samples, and we applied them in an analysis of cancer cell lines and an examination of colon tissue. Mutations were found in up to 6% of identified peptides, and only a small fraction corresponded to dbSNP entries. The RKO cell line, which is DNA mismatch repair deficient, yielded more mutant peptides than the mismatch repair proficient SW480 line. Analysis of colon cancer tumor and adjacent tissue revealed hydroxyproline modifications associated with extracellular matrix degradation. These results demonstrate the value of using sequence tagging algorithms to fully interrogate clinical proteomic data sets.
Keywords: mutation, bioinformatics, hydroxyproline, sequence tagging
Introduction
Shotgun proteomics identifies proteins in complex samples by generating large collections of tandem mass spectra from peptides produced through enzymatic digestion. Many of these MS/MS scans can be identified through standard database search algorithms, but in most cases, identification fails for a considerable fraction of the spectra.1 Some of these are unidentifiable because they represent nonpeptide contaminants or very low-signal peptides, but in clinical proteomics, many spectra fail identification due to post-translational modifications (PTMs) or amino acid mutations.1,2 Amino acid mutations are of significant interest in cancer,3 pharmacogenomics,4 hereditary diseases,4 and population proteomics.5 As a result, developing algorithms to match mutated peptide sequences to spectra has become a priority for the field of proteome bioinformatics.
Many different approaches have been employed for this task. A special version of Sequest was employed by Gatlin et al. in 20006 to introduce all possible SNPs in a DNA sequence database to generate singly mutant forms of hemoglobin. This strategy was recently refined by Bunger et al.7 to incorporate only noncoding SNPs from dbSNP8 in generating the list of sequences to be compared to spectra from a cell line. In a departure from this strategy, Edwards demonstrated in 2007 that starting from EST databases rather than whole proteome FASTA databases enables the identification of novel peptides.9 These approaches, along with refinement search strategies,10 demonstrate that database search algorithms can be pressed into service for mutated peptides with some effect. Each of these techniques, however, greatly increases the number of candidate peptides compared to each spectrum with costs in processing time, sensitivity, and specificity.
This issue can be partially addressed with “_de novo_” algorithms. These algorithms infer full-length sequences from tandem mass spectra. Inferred sequences are automatically reconciled against database sequences while making allowances for mutations and post-translational modifications.11–13 Great advances have been made by de novo algorithms during recent years,14 but they still fail to sequence a large portion of identifiable spectra. Meanwhile, more sensitive sequence tagging algorithms that use inferred sequence tags15 to recognize database sequences are gaining ground. The GutenTag algorithm from the Yates Laboratory automated the inference of partial sequences from tandem mass spectra, enabling the identification of variant ocular lens sequences.16 The Pevzner Laboratory soon thereafter introduced the InsPecT sequence tagging algorithm, enabling the identification of peptides with unknown post-translational modifications.17 Numerous other algorithms have also extended the sequence tagging technique to find post-translational modifications in peptides.18,19 The MultiTag algorithm adapted the technique for sequence homology searches across organisms.20 In recent work, we have introduced the DirecTag algorithm for highly accurate sequence tag inference.21 All these tools hold the potential to substantially alter the way in which proteomic information is derived from shotgun data sets, but they have been slow to emerge from their originating laboratories.
We perceive several challenges to the broader use of de novo/sequence tagging algorithms. The first is that database search tools are generally encapsulated in software pipelines with easy-to-use graphical or web user interfaces; this infrastructure does not exist for sequence tagging tools. Next is the widespread perception that these algorithms produce large numbers of false identifications. It must be acknowledged that most laboratories are unlikely to change their instrument configurations in order to facilitate identification (for example, by collecting tandem mass spectra within the slower but higher resolution Orbitrap rather than the faster linear trap of the same instrument). Likewise, most laboratories are unlikely to adopt protocols in which samples are routinely digested with multiple enzymes. Enabling the broad use of sequence tagging will depend upon adapting the algorithms to the data sets rather than vice versa.
In this study, we describe TagRecon, a novel sequence tagging algorithm designed to identify mutant peptides present in clinical proteomics LC-MS/MS experiments. TagRecon is part of an integrated bioinformatics pipeline that produces HTML reports of protein and peptide identifications. In this report, we compare its performance to a high-performance database search tool and to the InsPecT sequence tagging software with carefully controlled false discovery rates (FDRs). We also compared its mutation identification accuracy to that of Paragon and X! Tandem. We demonstrate that the software can be used successfully in both Orbitrap and LTQ data sets. We extend this analysis into complex clinical data sets, demonstrating the identification of widespread mutations in cell lines. We use the tool to profile important changes in colon tumor cells versus neighboring tissue.
Materials and Methods
Figure 1 illustrates the computational pipeline used to identify peptides from LC-MS/MS experiments. The standard database search paradigm employs a database search engine (MyriMatch, in this case) to compare candidate peptides to tandem mass spectra.22 A protein assembler (IDPicker) then filters the identifications and assembles a protein list from the confident peptide identifications.23,24 In this work, we have substituted sequence tag inference and reconciliation for the database search engine component of this pathway. DirecTag infers sequence tags from MS/MS scans.21 TagRecon matches these partial sequences and spectra to protein sequences, reconciling mass differences as it runs. Because the identifications are reported in pepXML format from both database search and sequence tagging pathways, IDPicker works equally well with either technique. The source code and binaries of all the software used in the workflow are available for download from our Web site: http://fenchurch.mc.vanderbilt.edu/.
Figure 1.
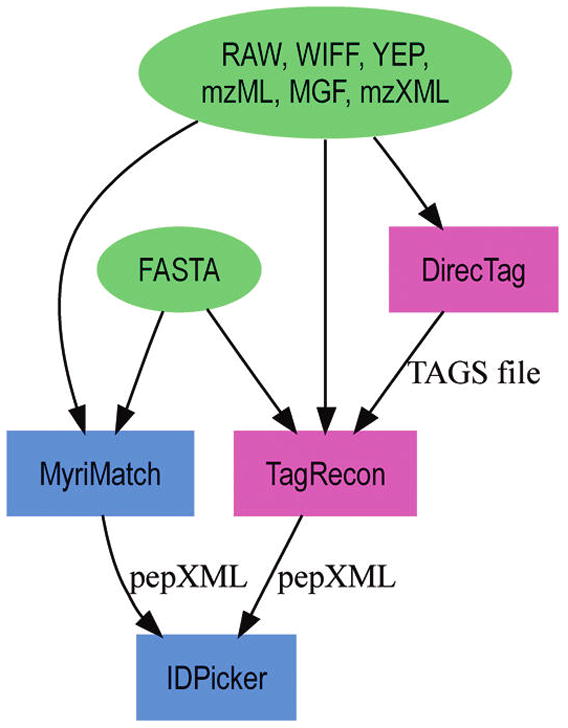
This flowchart illustrates the computational pipeline used to identify peptides from LC-MS/MS experiments. Myri-Match is a database search engine. TagRecon is a mutation-tolerant search engine, which reconciles partial sequence tags generated by DirecTag against a protein database. IDPicker is a parsimonious protein assembler, which filters peptide identifications using a target FDR.
Overview of TagRecon
TagRecon accepts three types of inputs. It accepts MS/MS in a variety of instrument-native and derived formats (see Figure 1 for a partial list) via the Proteo-Wizard library.25 It reads protein sequences from a user-specified FASTA database. It reads inferred tags from the output files of DirecTag. The output of TagRecon is a pepXML file for each input MS/MS file. As the HUPO-PSI mzIdentML format evolves support for sequence tagging,26 output will shift to this format to facilitate comparison to other tools. TagRecon was written in C++ and its multithreaded and message passing architecture can take advantage of multiple CPUs or multicore CPUs.
When TagRecon detects that a sequence tag matches a protein sequence, the software compares the flanking regions of both spectrum and protein sequence to determine whether or not the masses match within a user-defined mass error (see Figure 2). If either of the flanking masses is a match, the database peptide sequence may be used to explain the remainder of the spectrum. The software allows for only one mass mismatch to occur during the mass matching; this prevents identification when mutations or modifications can be found on both sides of the tag sequence but greatly improves search speed and accuracy. The software can apply a BLOSUM6227 matrix to determine which amino acid substitutions are permitted in reconciling the mass mismatches. Finally, peptide match scores for complete spectrum interpretations are computed for all candidate matches using the scoring algorithm embodied in MyriMatch.22
Figure 2.

The flowchart illustrates how TagRecon reconciles mass differences between database peptides and spectrum sequence tags as single amino acid mutations.
When a mismatch occurs during mass matching, the software computes the delta mass (ΔM) between the corresponding database sequence and spectral flanking masses (see Figure 2). The ΔM and the amino acids in the mass mismatch region are used to identify potential substitutions from a BLOSUM62 log-odds substitution matrix. The amino acid substitutions in the matrix are indexed based on their mass differences, the affected amino acids, and the log-odds score of the substitution. The potential substitutions are filtered using a user-defined log-odds score threshold, and permissible mutations are identified. The database sequence in the mass mismatch region is modified using the permissible mutations and several variants are generated (one per permissible mutation). For each full-length sequence, the list of expected fragment ions is compared to the observed spectrum using the peptide scoring system. The highest scoring mutant peptide is stored as TagRecon’s interpretation for the spectrum.
For each peptide–spectrum match (PSM), TagRecon generates a list of m/z values expected to be intense in the MS/MS. TagRecon examines these positions in the MS/MS and computes two probabilistic subscores: an intensity-based MVH22 score and a mass error-based mzFidelity score. The MVH score assesses the intensity classes of fragments found at the m/z values expected to be peaks for the candidate peptide. The mzFidelity score (Supplemental File 1) measures how well the predicted fragment ions match the experimental peaks in m/z space. The software computes and reports both score probabilities in the logarithmic domain. TagRecon employs the MVH score as the primary sort order for sequences, with mzFidelity serving as a tie-breaker.
Data Sets
Four different shotgun proteomics data sets were used to demonstrate the utility of TagRecon. Detailed sample processing protocols are presented in Supplemental File 2.
1. Yeast LTQ
Yeast whole cell lysates were obtained from NCI CPTAC (Clinical Proteomic Technology Assessment for Cancer). Proteins were reduced and alkylated with iodoacetamide prior to trypsin digestion. Digests were analyzed in reversed-phase liquid chromatography using an LTQ mass spectrometer (Thermo, San Jose, CA) at Vanderbilt University. A total of 10 replicate LC-MS/MS experiments were performed and 262 568 MS/MS spectra were collected. Binary spectral data present in the centroided raw files were converted to mzXML and MGF formats using the msConvert tool of the ProteoWizard library.25
2. Yeast LTQ-Orbi
Another aliquot of the yeast whole cell lysate sample described above was also analyzed on a LTQ Orbitrap mass spectrometer at Vanderbilt University. A total of eight LC-MS/MS replicate analyses were performed, and 105 496 MS/MS spectra were collected; the slower rate of MS/MS collection relative to the LTQ reflects the use of charge state exclusion on the Orbitrap. Binary spectral data present in the raw files were converted to either mzXML or MGF formats using msConvert,25 configured to compute and report accurate masses for the MS/MS precursors whenever possible and to centroid the MS scans.
3. Mismatch Repair Cell Lines
RKO (mismatch repair deficient) and SW480 (mismatch repair proficient) human adenocarcinoma cell line proteins were reduced, alkylated with iodoacetamide, and digested with trypsin. The resulting peptide mixtures were separated into 10 fractions using isoelectric focusing (IEF). Each fraction from the two cell lines was analyzed in triplicate on a LTQ-Orbitrap mass spectrometer using LC-MS/MS, and a total of 486 252 MS/MS spectra were collected. Binary spectral data present in the raw files were processed using msConvert as above.
4. Colon Tissue
Colorectal adenocarcinoma and adjacent normal tissue specimens were collected from two human subjects and fixed in polyvinyl alcohol. Two 60 _μ_m slices of each tissue sample were homogenized, reduced, alkylated with iodoacetamide, and digested with trypsin. Peptide mixtures were separated into 20 fractions using IEF. Each fraction was analyzed on an LTQ-Orbitrap mass spectrometer using LC-MS/MS, and a total of 967 938 MS/MS spectra were collected. Binary spectral data present in the raw files were processed using msConvert as above.
Bioinformatics Methods. Peptide Identification
The MS/MS scans present in the four data sets were identified using five different algorithms: MyriMatch,22 X! Tandem,28 Paragon,29 InsPecT,17 and TagRecon. Table 1 summarizes the data sets, protein sequence databases, and mass tolerances used in all searches. Detailed configuration parameters for all search engines are listed in Supplemental File 3. All search engines were configured to use a static mass shift of 57.0125 Da for alkylated cysteines. Oxidation of methionine (+15.996 Da), formation of N-terminal pyroglutamate (−17.0265 Da), and N-terminal acetylation (+42.013 Da) were allowed as variable modifications. MyriMatch was configured to derive semitryptic peptides from the protein database. X! Tandem (version 2008.12.01.1) was configured to derive fully tryptic peptides from the protein database, whereas Paragon employed its “Thorough ID” mode. InsPecT (version 20090202) was configured to derive 50 valid tags per MS/MS and reconcile them against the sequence database. DirecTag21 generated partial sequences for MS/MS scans from each mzXML file. The software was configured to generate the top 50 tags of three amino acids from each spectrum (see Supplemental File 3 for complete details). TagRecon was configured to derive either fully tryptic (for comparison to InsPecT) or semitryptic peptides from the sequence database while reconciling the sequence tags generated from the DirecTag software. All identifications were processed in pepXML format. Peptide identifications from InsPecT were converted into pepXML format using the InsPecTToPepXML.py script (part of the InsPecT package). The peptide–protein associations in the InsPecT’s pepXML files were corrected using RefreshParser tool (Trans-Proteomics Pipe Line, Institute of Systems Biology, Seattle, WA). X! Tandem search results were transcoded into pepXML format using Tandem2XML tool (Trans-Proteomics Pipe Line, Institute of Systems Biology, Seattle, WA), which was slightly altered to improve reporting of mutant peptides. Paragon (version 3.0) search results were exported to pepXML format using a newly developed group2PepXML tool, which can be obtained by contacting Applied Biosystems (Foster City, CA).
Table 1.
Summary of the Data Sets, Search Engines, And Protein Sequence Databases Used in This Study
| parent/fragment mass tolerancesa | |||||||
|---|---|---|---|---|---|---|---|
| data setb | replicates | total no. of MS/MS scans | sequence databasesc | MyriMatch and TagRecon | InsPecT | X! Tandem | Paragon |
| Yeast LTQ | 10 | 262568 | 20090124_sgd-orf-trans, Simulated Mutation Databasesd | 1.25/0.5 | 2.5/0.5 | 3.0/0.5 | 0.7/0.6 |
| Yeast LTQ-Orbi | 8 | 105496 | 20090124_sgd-orf-trans, Simulated Mutation Databases | 0.1/0.5 | 1.0/0.5 | 0.05/0.5* | 0.05/0.5* |
| MMR Cell Lines | 6 | 486252 | 20090205_IPI_Human, 20090205_IPI_Human_subset | 0.1/0.5 | |||
| Colon Tissue | 4 | 967938 | 20090205_IPI_Human, 20090205_IPI_Human_subset | 0.1/0.5 | 1.0/0.5 | 0.05/0.5 |
Conducting mutation-tolerant searches required changes in configuration. TagRecon was configured to interpret mass mismatches as single amino acid variations, where any amino acid was allowed to replace any other (BLOSUM62 filtering of potential mutations was not employed). InsPecT’s configurations were changed to search for unrestrictive modifications in “blind” mode following the recommendations in its manual. Ideally, this software would have employed a more restrictive mutation search mode (“freemods”), but this option was not implemented. X! Tandem was configured to search for point mutations in refinement mode. Paragon was configured to search for “Amino acid substitutions” in “Thorough ID” mode. The relevant configuration parameters for all search programs are shown in Supplemental File 3.
Assisted Attestation of Mutations Using IDPicker
IDPicker23,24 filtered peptide identifications from all search engines at a false discovery rate (FDR) of 2%. For MyriMatch and TagRecon search results, IDPicker was configured to automatically combine the MVH and mzFidelity scores for FDR filtering.24 For InsPecT searches, IDPicker combined the MQScore (“Match Quality”) and the DeltaScore for FDR filtering. IDPicker filtered X! Tandem search results using a static “hyperscore-expect” score. Paragon results were filtered using the “peptide confidence” score. Peptides passing the FDR thresholds were assembled into protein identifications using parsimony rules.23 Protein identifications with at least two distinct peptide identifications were considered for further analysis.
IDPicker produces a spectra-per-peptide text report containing all filtered peptide identifications. This report relates every peptide to the total number of spectra matched per replicate and the proteins containing its sequence. The spectra-per-peptide report is very useful for postprocessing but provides very little contextual information about the mutant peptides. Hence, we added the following new modules to IDPicker24 software for providing optional contextual information about each mutant peptide identification present in the spectra-per-peptide text report.
(1) UniMod Annotation
All identified mutations are annotated using the UniMod30 protein modifications database. IDPicker reads the annotations of all known post-translational and chemical modifications from an XML version of UniMod. Each mass shift and amino acid links to references for previous observations of that modification.
(2) CanProVar Annotation
All identified mutations are compared to known amino acid variations present in the Human Cancer Proteome Variation Database (CanProVar).31 The CanProVar database maps all known nonsynonymous coding SNPs (nsSNPs) to their corresponding amino acid variations in protein sequences. The database contains both population-wide nsSNPs from the dbSNP8 database and cancer-related nsSNPs extracted from various public and published sources. When mutations are identified in UniProtKB or IPI database searches, CanProVar enables users to check whether the mutation has been published previously.
(3) Best Nonmutant Peptide Annotation
MyriMatch identified peptides against the full IPI or UniProt databases, while TagRecon employed a smaller “subset” database of proteins observed to match two distinct sequences in MyriMatch. IDPicker reports the best score from the initial MyriMatch search alongside the best score from the TagRecon search to flag identifications that do not increase substantially in score when mutations are permitted.
High-throughput attestation of mutant peptides is still an open problem. In this study, we applied proven PTM attestation principles32 to validating mutant peptides present in complex mixtures. We created a series of Microsoft Excel 2007 macros to enforce the following filtering guidelines:
- Peptides must match to at least four different spectra (potentially of different precursor charges),
- Peptides should not match to contaminant proteins (like keratin, trypsin, etc.),
- Mutations cannot be explained as sample processing artifacts or known PTMs (for example, Met → Phe substitution can be explained away as oxidation of methionine, which is a common sample processing artifact),
- Mutations of lysine or arginine residues cannot be trypsin cutting sites,
- Mutations located at the peptide termini were rejected if there was any potential for misassignment, and
- Mutant peptides must improve upon the score of the nonmutant identification by 10%. Any smaller margin is counted as a misidentification.
Simulated Mutations for Performance Testing
We wanted to estimate the fraction of potential mutations that were detected by TagRecon. We also wanted to estimate the number of false detections produced by the software. Rather than work with samples that have known sequence variations, we chose to introduce sequence changes in FASTA databases for samples that correspond well to the original FASTA files. We first identified sequence coverage zones in the yeast proteome using the MyriMatch search engine. MyriMatch identified 5158 distinct peptides in the “Yeast LTQ” data set and 6603 distinct peptides in the “Yeast LTQ-Orbi” data set. Next, we screened the peptide identifications from the “Yeast LTQ” data set to remove subset peptides (for example, the peptide “CMEDK” is a subset of peptide “CMEDKEIGR”) and overlapping peptides (for example, peptides “ITISKGELK” and “GELKSILR” overlap each other). From the remaining results, we randomly selected 1050 peptides for sequence distortion in a test of mutation identification. The random selection process sampled peptides with a broad range of abundance (15% of peptides have one spectrum match, 70% of peptides have ≥2–10 spectral matches, and the rest have >10 spectral matches). For each of these selected peptides, a script in the AWK language randomly changed one amino acid residue in each peptide to a new residue. The altered sequences were changed in special versions of the full proteome and subset FASTA sequence databases. The same process was repeated for the “Yeast LTQ-Orbi” data set.
TagRecon, InsPecT, Paragon, and X! Tandem matched the MS/MS scans present in the “Yeast LTQ” and “Yeast LTQ-Orbi” data sets to the respective modified FASTA databases, configured to find mutations (see above). In this scenario, all search engines were forced to match the changed sequences to the corresponding spectra by introducing mutations that reconstruct the original sequences. IDPicker filtered peptides identified by all search engines at a 2% FDR threshold. Only the fully tryptic mutant peptides matching to at least two different spectra across all replicates were considered for further analysis. Any mutant peptide identification that could not be explained as a sample processing artifact was considered as a positive mutation; these artifacts included oxidation of M (+16), pyroglutamate formation of Q (−17), N-succinimide (−17), deamidation (+1) of N or Q, dehydration (−18) of D or E, Na adduct (+22) on D or E, and formylation (+28) of S or T. A positive mutation was considered as a true positive (TP) if it was in the simulated database (i.e., the mutation accurately reconstructed the original sequence) or a false positive (FP) otherwise. The overall performance of all search engines on simulated mutation databases was compared using the metrics developed for the evaluation of information retrieval systems: recall, precision, and _F_1-measure. Recall (R) is defined as the proportion of identified true positives to the number of positives in the simulated mutation database. Precision (P) is defined as the proportion of identified true positives to the identified positives. The F_1-measure is defined as the harmonic mean of recall and precision: 2_RP/(R + P).
Mutation-Tolerant Searches for Cell Lines and Tissue Data Sets
A subset database search strategy was employed to identify mutant peptides in the mismatch repair and colon tissue data sets. In the first pass, MyriMatch identified peptides from the data sets using an IPI Human protein database (version 3.47). IDPicker filtered the resulting peptide identifications at 2% FDR, and protein identifications with at least two distinct peptide matches were included in a subset database. Reversed sequences (decoys) were added to the subset database for estimation of FDRs. In the second pass, TagRecon, InsPecT, and Paragon used the subset databases to search for mutant peptides present in the data sets. TagRecon derived semitryptic peptides from the databases for improved sequence coverage. Paragon was configured in “Thorough ID” mode. InsPecT automatically looks for semitryptic peptides whenever possible. IDPicker filtered the peptide identifications from mutation-tolerant searches at 2% FDR.
Differential Abundance Testing Using Spectral Counting
The spectral counting method is a semiquantitative measure for comparing the abundance of proteins between two different samples.33 In this study, we used Fisher’s exact test to compare spectral counts between samples. In essence, the test separates the identified spectra for each sample into those that match the protein and those that do not match the protein. This produces a 2 × 2 contingency table for two samples, where the first column gives these two values for the first sample and the second column gives these two values for the second sample. Fisher’s exact test then produces a _p_-value reflecting the probability that these or more extreme results would appear if equal proportions of spectra matched this protein from each of the two samples. These tables can also be used to estimate the fold change for a protein between samples. These tests were conducted by means of the R Statistical Environment.34
Results and Discussion
Because shotgun proteomics experiments routinely incorporate database searching algorithms, we designed experiments that compared TagRecon performance to MyriMatch, a database search algorithm that employs an identical scoring system. We compared to the InsPecT algorithm since it represents an alternative implementation for leveraging sequence tags for MS/MS identification. We have also compared to X! Tandem and Paragon because they represent important methods of recognizing mutant peptides. We initially characterized performance in yeast lysate data and then shifted to cancer data to demonstrate the value of sequence tag-based identification in a biological context.
Standard Identification Performance
Identifying peptides is the most basic task of shotgun proteome informatics. In this study, we compared the identification performance of Myri-Match,22 InsPecT,17 and TagRecon using multiple replicates of yeast analyzed on LTQ and LTQ-Orbitrap mass spectrometers. All search engines were configured to use standard search parameters while deriving either fully tryptic or semitryptic peptides from the SGD ORF protein database. Peptide identifications from all searches were filtered at 2% FDR using IDPicker23,24 (see Materials and Methods). Figure 3 compares the number of confident peptides across replicates identified by a TagRecon search to that of a comparable MyriMatch or InsPecT search. In both instruments, TagRecon identified a larger number of peptides than did InsPecT. TagRecon also outperformed the MyriMatch database search engine when using LTQ data but fell behind by a smaller margin when using Orbitrap data (Figure 3). This difference by instrument probably results from the very large number of candidates MyriMatch compares to spectra in the LTQ compared to a relatively small number in Orbitrap data. In general, Figure 3 establishes TagRecon as a high-performance proteomic identification tool.
Figure 3.

These images compare the identification performance of MyriMatch, InsPecT, and TagRecon on multiple replicates of yeast from LTQ and Orbitrap instruments. Each row in the graph indicates a specific algorithm and configuration for the search of multiple replicates. “FT” reports the use of a fully tryptic search as opposed to an “ST” or semitryptic search. “PM” indicates that precursor mass filtering was used to select candidate peptides for comparison to the spectrum rather than “FM” or flanking mass filtering. TagRecon outperformed MyriMatch on the LTQ replicates but fell behind by a small margin on Orbitrap data. In both instruments, however, TagRecon achieved a larger number of identifications than did InsPecT at the same 2% FDR.
In both data sets, a majority of the peptides identified by TagRecon were also identified by MyriMatch (on average, 74% in LTQ data and 79% in Orbitrap data). This is because both search engines use the same peptide scoring system. The peptide overlap between TagRecon and InsPecT, however, was lower (on average, 61% in LTQ data and 69% in Orbitrap data). In both data sets, TagRecon identified more distinct peptides than InsPecT (on average 23% more in LTQ data and 21% more in Orbitrap data).
We tested the speed of TagRecon using a computer equipped with dual quad-core 2.4-GHz Intel processors, 4GB of RAM, and a RedHat Linux OS (kernel version 2.6). All timing statistics were gathered by confining TagRecon to a single core of the computer to match usage for InsPecT. When configured to disallow mutations, TagRecon required 7.9 s to sequence tag and match 1K spectra to 1K protein sequences, whereas a similar InsPecT search took 9.7 s. When configured to allow mutations, TagRecon search time increased to 19.4 s due to the increase in number of comparisons. In the future, we plan to improve the computational time of TagRecon by indexing the protein database.
Performance Comparisons with Simulated Mutations
We tested the mutation identification accuracy of TagRecon, InsPecT,17 Paragon,29 and X! Tandem.28 These tools were carefully chosen to represent three different ways of identifying mutant peptides: TagRecon and InsPecT use sequence-tag reconciliation, Paragon employs fraglet-taglet29 searching, and X! Tandem relies on database searching with refinement. For this test, we employed simulated mutation databases created for multiple replicates of yeast samples analyzed on LTQ and LTQ-Orbi instruments. To begin, MyriMatch identified peptides in the “Yeast LTQ” and “Yeast LTQ-Orbi” data sets. Next, 1050 peptides were randomly selected from each data set and mutated stochastically. These mutations were introduced in the corresponding FASTA entries of a large SGD ORF protein database and a smaller subset of the SGD database. The MS/MS present in the data sets were matched to the modified sequence databases using TagRecon, InsPecT, Paragon, and X! Tandem, configured to find mutations. In this scenario, all search engines were forced to match the changed sequences to the corresponding spectra by introducing mutations that reconstruct the original sequences. Mutation identifications that accurately reconstructed the original sequences were considered as true positives (TP) or false positives (FP) otherwise. IDPicker23,24 filtered the resulting peptide identifications at 2% FDR (see Materials and Methods for details). Compensating for errors in the sequence database poses the same bioinformatic challenge as recognizing observed peptides that differ by mutation from true protein sequences.
Figure 4 shows the number of true positive and false positive mutants identified by TagRecon, InsPecT, Paragon, and X! Tandem when searching the yeast replicates against simulated mutation databases. In all replicates, TagRecon identified more true positive mutations than did the rest of the search engines (Figure 4). All search engines identified more true positive mutations from the replicates when the mutations are present in a smaller subset database instead of a larger SGD ORF protein database (Figure 4). One reason is that the small subset databases offer fewer distracting sequences to accidentally outscore correct sequences.
Figure 4.

These images compare the mutation identification performance of TagRecon, InsPecT, Paragon, and X! Tandem when using multiple replicates of yeast samples analyzed on LTQ and LTQ-Orbi instruments. MS/MS were matched to sequence databases containing simulated mutations. All databases contained the same numbers of simulated mutations. “SGD” indicates that mutations were contained in a larger SGD ORF database. “SGD-SS” indicates that same mutations were restricted to a smaller subset of SGD database. “TP” indicates that software reported a mutation present in the simulated mutation database. “FP” indicates otherwise. In both samples, TagRecon identified more true positive mutations than any other software at 2% FDR. All search engines improved performance when looking for mutations present in a smaller subset database. Accurate precursor masses from LTQ-Orbi improved true positive mutation identification.
As described in Materials and Methods, we measured the ability of TagRecon, InsPecT, Paragon, and X! Tandem to recover simulated mutations from yeast data sets using metrics developed to gauge the performance of information retrieval systems: recall, precision, and _F_1-measure (Table 2). In all samples, TagRecon recovered more mutations from the simulated mutation databases at a higher precision than did InsPecT and Paragon. X! Tandem enjoyed higher precision at the expense of the lowest recall because of the following reasons: (a) the software ignores mutations that are disallowed by the PAM35 substitution matrix, (b) proteins have to pass an initial database search before they can be targeted for a mutation search (“refinement”). InsPecT performance was hampered by conducting an unrestrictive modification (“blind”) search to find mutations, rather than targeting only mutations (see Materials and Methods). As expected, all search engines also recovered more mutations when using a smaller subset database rather than a larger database (Table 2). Even though all mutation databases contained the same numbers of mutants, the software recovered more mutants from the “Yeast LTQ-Orbi” data set than “Yeast LTQ” data set because of highly accurate precursor masses (Table 2).
Table 2.
Comparison of the Ability of TagRecon, InsPecT, Paragon, and X! Tandem To Recover Simulated Mutations Using Multiple Replicates of Yeast Samples Analyzed on LTQ and LTQ-Orbi Mass Spectrometersa
| SGDb | SGD-SSc | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| sample | search engine | recovered mutantsd | recall | precision | _F_1-measure | recovered mutants | recall | precision | _F_1-measure |
| Yeast LTQ-Orbi | TagRecon | 445 | 0.42 | 0.72 | 0.53 | 477 | 0.45 | 0.67 | 0.54 |
| Paragon | 383 | 0.36 | 0.70 | 0.48 | 455 | 0.43 | 0.63 | 0.51 | |
| Tandem | 296 | 0.28 | 0.84 | 0.42 | 309 | 0.29 | 0.86 | 0.44 | |
| InsPecT | 342 | 0.33 | 0.46 | 0.38 | 380 | 0.36 | 0.39 | 0.42 | |
| Yeast LTQ | TagRecon | 298 | 0.30 | 0.67 | 0.41 | 347 | 0.35 | 0.67 | 0.46 |
| Paragon | 249 | 0.25 | 0.62 | 0.36 | 307 | 0.31 | 0.61 | 0.41 | |
| Tandem | 185 | 0.19 | 0.76 | 0.30 | 192 | 0.19 | 0.71 | 0.30 | |
| InsPecT | 151 | 0.15 | 0.49 | 0.23 | 174 | 0.17 | 0.46 | 0.25 |
The highest recall value achieved was only 45% because we required two spectra per peptide to accept mutant peptides (Table 2). This aggressive filter discriminates against low-abundance peptides but safeguards against false discoveries. When this filter was relaxed, TagRecon achieved the highest recall value of 60%, at the expense of precision (Supplemental File 4). The fact that many sequence changes were left undetected underscores the challenge of retrieving peptide identifications when the sequence database is not a perfect match.
Mutant Peptide Identification Comparison with Real Life Data Sets
TagRecon, InsPecT, and Paragon were compared in the context of the Colon Tissue data set to examine their performance in a real-world setting. All tools analyzed 20 fractions for a tumor and 20 fractions for adjacent normal tissue for an individual colon sample to produce identification lists that achieved a 2% FDR (see Materials and Methods). We inspected the search results of TagRecon and Paragon and retained fully tryptic peptides with at most two modifications to their sequences (the programs were allowed to incorporate common modifications as in standard database search). This step was essential in order to remove peptides that cannot be identified by InsPecT because of its limited mutation-tolerant search configuration (see search engine configurations in Supplemental File 3). The resulting mutant peptide identifications were attested following the guidelines outlined in Materials and Methods. Panels A and B of Figure 5 show the attested and unattested mutant peptide overlap, respectively, between TagRecon, InsPecT, and Paragon. Three quarters of the attested mutant peptides were identified by at least two search engines (Figure 5A). However, TagRecon also identified an additional 12 attested mutant peptides from the data set that were missed by InsPecT and Paragon (Figure 5A). A sizable pool of other sequences fell short of the stringent attestation criteria (Figure 5B). No single search engine was able to identify all mutant peptides present in the sample.
Figure 5.
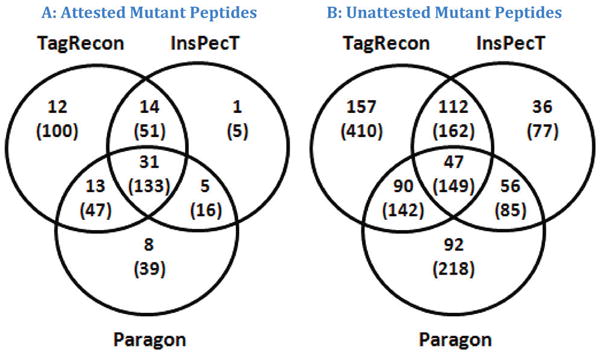
These images compare the mutant peptide overlap between TagRecon, InsPecT, and Paragon when analyzing data for an individual sample of the colon tissue data set. Mutant peptides were attested following the stringent attestation guidelines outlined in Materials and Methods. The numbers in parentheses represent the spectral counts. TagRecon recognized larger numbers of mutations that passed attestation criteria and provided hints for many other possible mutations. No single search engine can identify all mutant peptides present in a sample.
Frequency of Mutations in Biological Samples
Because standard database search engines silently fail to identify sequence changes in peptides, the prevalence of proteomic mutations in biological samples has been unknown. We used TagRecon to identify the mutant peptides present in the MMR cell lines and complex colon tissue samples. IDPicker then filtered identifications at 2% FDR. We subjected the resulting reports to the strict attestation guidelines outlined in Materials and Methods. The amino acid substitutions present in the mutant peptides were reconciled against known nsSNP annotations from dbSNP.8 Table 3 presents the percentage of peptide identifications with attested mutations (mutation frequency), percentage of the MS/MS identifications with attested mutations (mutation abundance), and the percentage of the attested mutants with matching dbSNP annotations for each replicate of these samples.
Table 3.
Extent of Mutations in Each Replicate Analysis of Simple MMR Cell Lines and Complex Colon Tissue Samples
| samplea | replicate ID | mutant peptides | mutant spectra | mutation frequencyb | mutation abundancec | known mutantsd |
|---|---|---|---|---|---|---|
| RKO | Rep1 | 117 | 166 | 4.6% | 4.9% | 3% |
| Rep2 | 106 | 147 | 4.3% | 4.6% | 2% | |
| Rep3 | 112 | 165 | 4.5% | 5.0% | 3% | |
| SW480 | Rep1 | 88 | 115 | 3.8% | 3.9% | 3% |
| Rep2 | 81 | 111 | 3.7% | 3.8% | 1% | |
| Rep3 | 67 | 89 | 3.1% | 3.2% | 3% | |
| CTS#747 | Rep1 | 536 | 2150 | 4.2% | 7.1% | 3% |
| Rep2 | 528 | 2604 | 3.9% | 6.7% | 3% | |
| CTS#823 | Rep1 | 519 | 1669 | 5.8% | 5.9% | 3% |
| Rep2 | 536 | 2238 | 5.9% | 6.5% | 3% |
The percentage of spectra that contribute evidence of mutations ranged from 3.2% to 7.1%, depending upon sample type. It is worth noting that a single mutation in a spectrum will typically prevent its identification. As a result, mutation-tolerant identification increases sequence coverage for proteins. The mutations revealed by this analysis did not overlap significantly with dbSNP; 3% or fewer of the mutations for each replicate matched to dbSNP. Tools to discover protein sequence variation from biological samples are likely to increase these collections significantly.
Differentiating between Mismatch Repair (MMR) Proficient and Deficient Cell Lines
The MMR cell lines data set contains the MMR proficient SW480 cell line and the MMR deficient RKO cell line. Because of the MMR deficiency, the genetic material of the RKO cell line accumulates mutations at a higher rate than SW480 cell line.36,37 For the first time, TagRecon enables us to differentiate between these two cell lines based on protein sequence variation rather than DNA variation. Table 3 shows that an average of 4.5% of the peptide sequences in RKO contain attested mutations, while SW480 cells produce mutations in an average of 3.5% of their peptide sequences. A t test comparing the sets of triplicates yields a _p_-value of 0.0228 (assuming unequal variances and one tail). Examining the percentage of mutated spectra produced an even more extreme _p_-value of 0.0063. While both of these colon cancer cell lines include mutations, the MMR deficient one has accumulated more of them.
The identified mutations conform to simple genetic changes. Twenty-three spectra in the RKO cell line (versus three spectra in the SW480 line) matched to a gain of 16 Da at proline residues in the sequence database, producing the mass of leucine or isoleucine. This mutation can be associated with a 2-fold elevation in cytosine (C) to thymine (T) transition due to MMR deficiency36–38 because all four codons of proline can be changed to codons for leucine by changing the C to T at the second position. Threonine was replaced by proline in 18 spectra for the RKO cell line and 7 in the SW480 line. This sequence change would result from an adenine to cytosine transversion. Some changes were not the result of single base changes. Arginine replaced glutamic acid in 10 spectra for RKO and 14 spectra for SW480. The mutation required transposition of GA to AG in the DNA. Because these cell lines were derived from cancer cells, their sequence diversity may not be typical of clinical samples from diseases that do not enhance mutation rates.
Identification of Mutations in Clinical Samples
The “Colon Tissue” data set contains adjacent normal and cancerous tissue excised from colons of two human subjects (Subject #747 and Subject #823). All tissue samples were prepared separately for MS analysis. We attested the mutations found by TagRecon. Table 4 scrutinizes the mutations observed in overlapping peptides that match at least 10 MS/MS. A relatively small fraction of the identified mutations were listed in dbSNP. While some peptides were observed only in mutated form, others were found in mixture with wild-type forms.
Table 4.
Mutations Identified in the “Colon Tissue” Samplea

Table 4 demonstrates that sequence tagging can identify real mutations and support them with credible evidence. Each tandem mass spectrum is identified independently by TagRecon. Assembling these identifications in IDPicker makes the discovery of overlapping peptides straightforward. Given the heterogeneity of cancer samples (mixing tumor and adjacent tissue and potentially reflecting diverse sequences), it seems only reasonable that evidence for mutated forms should always be accompanied by the evidence for wild-type forms. The framework we demonstrate in this work addresses these challenges, making complex biological reporting feasible.
Extracellular Matrix Degradation in Human Colon Cancerous Tissue
Table 4 excludes many apparent substitutions of isoleucine/leucine for proline (a shift of 16 Da). As mentioned in the cell line data above, this could be a simple transition from cytosine to thymine at the second position of the codon. This mass shift, however, can also be explained by the enzymatic hydroxylation of proline. Most of the observed hydroxyprolines were located in the cross-linking domains of α-1(III), α-1(I), and α-2(I) collagens. The collagen hydroxyprolines are critical for stabilizing the extracellular matrix (ECM) of tissues via covalent cross-linking.39 We used the number of collagen hydroxyproline modifications as a proxy for the structural integrity of ECM between cancerous and surrounding normal tissues in Table 5.
Table 5.
Comparison of the Amount of Collagen Hydroxyproline Modification between Adjacent Normal and Cancerous Tissue Samples Excised from the Colon of Two Human Subjects (Subject #747 and Subject #823)a
| spectral countsc | cancer vs normal | ||||||
|---|---|---|---|---|---|---|---|
| subject no. | IPI accession | protein name | Pro + 16 peptidesb | normal | cancer | fold changed | _p_-value |
| 747 | IPI00021033 | isoform 1 of collagen alpha-1(III) chain | 13 | 138 | 45 | −1.5 | 1.8 × 10−13 |
| IPI00297646 | collagen alpha-1(I) chain | 12 | 296 | 118 | −1.4 | 2.3 × 10−16 | |
| IPI00304962 | collagen alpha-2(I) chain | 7 | 94 | 54 | −0.7 | 5.2 × 10−4 | |
| Overall | 32 | 528 | 217 | −1.4 | 2.2 × 10−16 | ||
| 823 | IPI00021033 | isoform 1 of collagen alpha-1(III) chain | 8 | 64 | 23 | −1.5 | 7.9 × 10−6 |
| IPI00297646 | collagen alpha-1(I) chain | 4 | 120 | 52 | −1.2 | 1.5 × 10−7 | |
| IPI00304962 | collagen alpha-2(I) chain | 5 | 45 | 18 | −1.3 | 5.9 × 10−4 | |
| Overall | 17 | 229 | 93 | −1.3 | 2.2 × 10−16 |
Confirming previous reports,40,41 the collagens in the cancerous colon tissue have abnormally low levels of hydroxyproline content compared to the surrounding normal colon tissue. However, the unmodified content of the same collagens is not significantly different between these two tissues (Supplemental Table 1). This supports the hypothesis that the structural integrity of ECM in cancerous tissue is significantly compromised.40 Degradation of the ECM is often associated with proteolysis followed by tumor invasion.41
Our data sets revealed that only some sites in the collagens showed lower levels of hydroxyproline modification in the cancerous tissue compared to that of the normal tissue (data not shown). A majority of the collagen hydroxyproline (Hyp) sites that were under-hydroxylated in cancerous tissue have a Gly-Pro-Hyp-Gly motif. Recent discoveries suggest that the hydroxylases specific to this motif are epigenetically inactivated in cancerous tissue.42 We are conducting verification studies confirming the role of particular collagen hydroxyproline modification sites in the progression of colorectal cancer.
Conclusion
Here we show that TagRecon can identify unexpected mutant peptides from complex mixtures. When configured to disallow mutations, TagRecon identifies as many peptides from a sample as does the MyriMatch database search engine and more unique peptides than the InsPecT sequence tag search engine. When configured to allow mutations, TagRecon improves on the existing mutation-tolerant search algorithms in several ways. TagRecon does not require prior knowledge of mutations present in the sample. The software employs a peptide scoring system that improves the sensitivity of mutation-tolerant searches. TagRecon identifies mutant peptides with higher sensitivity and accuracy than existing algorithms. We incorporated TagRecon into an open-source computational pipeline that can be used for routine, large-scale identification of mutant peptides.
Sequence tagging and database search were introduced in the same year: 1994. Sequence tagging, however, has languished while database search identification has flourished. We believe that recent advances in automated tag inference and scoring have paved the way for sequence tagging to take its role as a powerful complement to database searching. In this work, we have shown its potential in the identification of mutations. We next intend to demonstrate its value for automated identification of post-translational modifications. Although plenty of work remains to make sequence tagging as easy to use as database search tools, we believe that the software infrastructure we have made available is a solid step in the right direction.
Supplementary Material
Collagen modifications
Mutation recovery rate
Sample production
Search configurations
mzFidelity scoring system
Acknowledgments
D. L. Tabb and M. C. Chambers were supported by NIH grants R01 CA126218 and U24 CA126479. S. Dasari was supported by NIH grant R01 CA126218. The “Yeast LTQ” and “Yeast LTQ-Orbi” data sets were collected under NIH grant U24 CA126479 at Vanderbilt University. The “Colon Tissue” data set was collected by Corbin Whitwell and Misti Martinez at Jim Ayers Institute of Precancer Detection and Diagnosis at Vanderbilt University (Nashville, TN). The “MMR Cell Lines” data set was collected at Jim Ayers Institute of Precancer Detection and Diagnosis under the NIH grant U24 CA126479. We are greatly indebted to Daniel C. Liebler for giving us access to the invaluable “Colon Tissue” and “MMR Cell Lines” data sets. We also appreciate Patrick J. Halvey for allowing us to use “MMR Cell Lines” data set generated as a part of his postdoctoral training. We are grateful to Bing Zhang and Jing Li at Vanderbilt University Department of Biomedical Informatics for providing us access to the CanProVar database.
Footnotes
Supporting Information Available: mzFidelity peptide scoring system; materials and methods used for acquiring shotgun proteomics data; detailed configurations of all software used in the data analysis; a figure comparing the recovery rate of mutations under various mutant peptide attestation criteria; and a table comparing the non-hydroxyproline modification content of collagens between the cancerous and normal colon tissues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bern M, Goldberg D, McDonald WH, Yates JR. Bioinformatics. 2004;20(Suppl 1):49–54. doi: 10.1093/bioinformatics/bth947. [DOI] [PubMed] [Google Scholar]
- 2.Nesvizhskii AI, Roos FF, Grossmann J, Vogelzang M, Eddes JS, Gruissem W, Baginsky S, Aebersold R. Mol Cell Proteomics. 2006;5:652–670. doi: 10.1074/mcp.M500319-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Bacolod MD, Schemmann GS, Giardina SF, Paty P, Notterman DA, Barany F. Cancer Res. 2009;69:723–727. doi: 10.1158/0008-5472.CAN-08-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao G, Yang F, Yuan Y, Gao X, Zhang J. Yichuan. 2005;27:123–129. [PubMed] [Google Scholar]
- 5.Nedelkov D. Expert Rev Proteomics. 2005;2:315–324. doi: 10.1586/14789450.2.3.315. [DOI] [PubMed] [Google Scholar]
- 6.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates JR. Anal Chem. 2000;72:757–763. doi: 10.1021/ac991025n. [DOI] [PubMed] [Google Scholar]
- 7.Bunger MK, Cargile BJ, Sevinsky JR, Deyanova E, Yates NA, Hendrickson RC, Stephenson JL. J Proteome Res. 2007;6:2331–2340. doi: 10.1021/pr0700908. [DOI] [PubMed] [Google Scholar]
- 8.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards NJ. Mol Syst Biol. 2007;3:102. doi: 10.1038/msb4100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig R, Beavis RC. Rapid Commun Mass Spectrom. 2003;17:2310–2316. doi: 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JA, Johnson RS. Anal Chem. 2001;73:2594–2604. doi: 10.1021/ac001196o. [DOI] [PubMed] [Google Scholar]
- 12.Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, Nagalla SR. J Proteome Res. 2005;4:546–554. doi: 10.1021/pr049781j. [DOI] [PubMed] [Google Scholar]
- 13.Han Y, Ma B, Zhang K. J Bioinform Comput Biol. 2005;3:697–716. doi: 10.1142/s0219720005001247. [DOI] [PubMed] [Google Scholar]
- 14.Frank A, Pevzner P. Anal Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- 15.Mann M, Wilm M. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 16.Tabb DL, Saraf A, Yates JR. Anal Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanner S, Shu H, Frank A, Wang L, Zandi E, Mumby M, Pevzner PA, Bafna V. Anal Chem. 2005;77:4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 18.Na S, Jeong J, Park H, Lee K, Paek E. Mol Cell Proteomics. 2008;7:2452–2463. doi: 10.1074/mcp.M800101-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Liu C, Yan B, Song Y, Xu Y, Cai L. Bioinformatics. 2006;22:e307–313. doi: 10.1093/bioinformatics/btl226. [DOI] [PubMed] [Google Scholar]
- 20.Sunyaev S, Liska AJ, Golod A, Shevchenko A, Shevchenko A. Anal Chem. 2003;75:1307–1315. doi: 10.1021/ac026199a. [DOI] [PubMed] [Google Scholar]
- 21.Tabb DL, Ma Z, Martin DB, Ham AL, Chambers MC. J Proteome Res. 2008;7:3838–3846. doi: 10.1021/pr800154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabb DL, Fernando CG, Chambers MC. J Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Chambers MC, Tabb DL. J Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ze-Qiang M, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. J Proteome Res. 2009;8(8):3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kessner D, Chambers M, Burke R, Agus D, Mallick P. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.mzIdentML: exchange format for peptides and proteins identified from mass spectra. home page, http://www.psidev.info/index.php?q=node/403.
- 27.Henikoff S, Henikoff JG. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig R, Beavis RC. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 29.Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA. Mol Cell Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Creasy DM, Cottrell JS. Proteomics. 2004;4:1534–1536. doi: 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Duncan DT, Zhang B. Hum Mutat. 2010;31(3):219–228. doi: 10.1002/humu.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilmarth PA, Tanner S, Dasari S, Nagalla SR, Riviere MA, Bafna V, Pevzner PA, David LL. J Proteome Res. 2006;5:2554–2566. doi: 10.1021/pr050473a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Sadygov RG, Yates JR. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 34.Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]
- 35.Pearson WR. Methods Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- 36.Baross-Francis A, Makhani N, Liskay RM, Jirik FR. Oncogene. 2001;20:619–625. doi: 10.1038/sj.onc.1204138. [DOI] [PubMed] [Google Scholar]
- 37.Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AMC, Lipkin M, Edelmann W. Proc Natl Acad Sci USA. 2002;99:14937–14942. doi: 10.1073/pnas.232579299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mark SC, Sandercock LE, Luchman HA, Baross A, Edelmann W, Jirik FR. Oncogene. 2002;21:7126–7130. doi: 10.1038/sj.onc.1205861. [DOI] [PubMed] [Google Scholar]
- 39.Uitto J. J Invest Dermatol. 1979;72:1–10. doi: 10.1111/1523-1747.ep12530093. [DOI] [PubMed] [Google Scholar]
- 40.Wobbes T, Hendriks T, de Boer HH. Dis Colon Rectum. 1988;31:778–780. doi: 10.1007/BF02560106. [DOI] [PubMed] [Google Scholar]
- 41.Bode MK, Karttunen TJ, Mäkelä J, Risteli L, Risteli J. Scand J Gastroenterol. 2000;35:747–752. doi: 10.1080/003655200750023435. [DOI] [PubMed] [Google Scholar]
- 42.Shah R, Smith P, Purdie C, Quinlan P, Baker L, Aman P, Thompson AM, Crook T. Br J Cancer. 2009;100:1687–1696. doi: 10.1038/sj.bjc.6605042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Collagen modifications
Mutation recovery rate
Sample production
Search configurations
mzFidelity scoring system