Apoptosis Protection by Mcl-1 and Bcl-2 Modulation of Inositol 1,4,5-Trisphosphate Receptor-dependent Ca2+ Signaling (original) (raw)
Abstract
Members of the Bcl-2 protein family play a central role in the regulation of apoptosis. An interaction between anti-apoptotic Bcl-xL and the endoplasmic reticulum (ER)-localized inositol trisphosphate receptor Ca2+ release channel (InsP3R) enables Bcl-xL to be fully efficacious as an anti-apoptotic mediator (White, C., Li, C., Yang, J., Petrenko, N. B., Madesh, M., Thompson, C. B., and Foskett, J. K. (2005) Nat. Cell Biol. 7, 1021–1028). Physiologically, Bcl-xL binds to the InsP3R to enhance its gating and Ca2+ signaling. Here we have discovered that structurally related proteins Bcl-2 and Mcl-1 function similarly. Bcl-2, Mcl-1 and Bcl-xL bind with comparable affinity to the carboxyl termini of all three mammalian InsP3R isoforms with important functional consequences. Stable expression of Bcl-2 or Mcl-1 lowered ER Ca2+ content and enhanced the rate of InsP3-mediated Ca2+ release in response to submaximal InsP3 stimulation in permeabilized wild-type DT40 cells but not in cells lacking InsP3R. In addition, expression of either Bcl-2 or Mcl-1 enhanced spontaneous InsP3R-dependent Ca2+ oscillations and spiking in intact cells in the absence of agonist stimulation. Bcl-2- and Mcl-1-mediated protection from apoptosis induced by staurosporine or etoposide was enhanced in cells expressing InsP3R, demonstrating that their interactions with InsP3R enable Bcl-2 and Mcl-1 to be fully efficacious anti-apoptotic mediators. Our data suggest a molecular mechanism that is shared by several anti-apoptotic Bcl-2 proteins that provides apoptosis resistance by direct interactions at the ER with the InsP3R that impinges on cellular Ca2+ homeostasis.
Keywords: Calcium, Calcium Intracellular Release, Cell Death, Endoplasmic Reticulum (ER), Protein-Protein Interactions, Bcl-XL, DT40, IP3
Introduction
The Bcl-2 family of proteins are important regulators of programmed cell death with both pro- and anti-apoptotic members. In response to apoptotic stimuli, activation of the principle pro-apoptotic Bax and Bak causes increased mitochondrial membrane permeability and release of cytochrome c, a critical mediator in the commitment to cell death (1, 2). The ability of pro-apoptotic Bcl-2 proteins to disrupt mitochondrial membrane integrity is held in check through heterodimeric interactions with anti-apoptotic members, including Bcl-2, Bcl-xL, and Mcl-1 (3, 4).
In addition to mitochondria, both pro-and anti-apoptotic Bcl-2 proteins localize to the endoplasmic reticulum (ER),2 where they regulate Ca2+ fluxes across the ER membrane (5, 6). The inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R), a family of Ca2+ release channels localized predominately in the ER, has been identified as a target for anti-apoptotic Bcl-2 and Bcl-xL (reviewed in Ref. 6). A direct interaction of Bcl-xL with all three isoforms of the InsP3R sensitizes InsP3R channel gating to extremely low [InsP3] that exists in cells under resting conditions, and can result in reduced ER [Ca2+] (7, 8). The interaction modifies Ca2+- and InsP3-dependent regulation of channel activity in vitro and enhances Ca2+ signaling in vivo. Enhanced Ca2+ signaling in the form of spontaneous cytoplasmic Ca2+ concentration ([Ca2+]i) oscillations or spiking, is correlated with increased apoptosis resistance (7, 8). Similar effects have been observed in cells lacking both Bak and Bax, which had Bcl-xL or other anti-apoptotic Bcl-2 proteins more available to interact with the InsP3R (9). These data provide a molecular basis for the involvement of the ER as a major effector organelle in apoptosis, and they support a paradigm in which Bcl-xL is a direct effector of the InsP3R, increasing its sensitivity to InsP3 and enabling ER Ca2+ release to be more sensitively coupled to extracellular signals that enhances resistance to apoptotic stimuli.
Different pro-survival Bcl-2 proteins are differentially expressed in various cell types and engaged in responses to specific cellular stimuli (10, 11). However, it is unknown whether the interaction of InsP3R with Bcl-xL, and the functional consequences for Ca2+ signaling, ER Ca2+ homeostasis, and apoptosis protection are more generally applicable to other anti-apoptotic proteins. Bcl-2 family members can have distinct binding specificities (12, 13). Furthermore, Bcl-2 has been reported to interact with the InsP3R (14) at different regions of the channel and with opposite functional effects on channel gating (14, 15) compared with Bcl-xL (7, 8). Thus, there is no a priori reason to assume that different anti-apoptotic Bcl-2 family members will interact with and affect InsP3R Ca2+ release activity similarly. To address this, in the present study we explored the role of Bcl-2 and Mcl-1, anti-apoptotic Bcl-2 family members with structural homology to Bcl-xL. We have assessed the binding of Bcl-2, Bcl-xL and Mcl-1 to the carboxyl termini of all three mammalian InsP3R isoforms and examined the relationship between ER Ca2+ store content, [Ca2+]i oscillations and apoptosis resistance conferred by the InsP3R-Bcl-2 protein interaction.
EXPERIMENTAL PROCEDURES
Cell Culture and Generation of Stable Cell Lines
COS-7 cells were grown in high glucose Dulbecco's modified Eagle's medium (GIBCO) containing 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin mixture at 37 °C, 95% air/5% CO2 atmosphere. DT40 cells were maintained in suspension culture at 37 °C (95/5% air/CO2) in RPMI 1640 medium (GIBCO) supplemented with 10% (v/v) fetal bovine serum, 1% chicken serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Full-length human Bcl-2 and Mcl-1 cDNAs were cloned into the pIRES2-DsRed2 vector (Clontech). DT40-WT and InsP3R-KO cells stably expressing either Bcl-2, Mcl-1, or empty vector were generated as described (7).
Biochemistry
Human Bcl-xL, Bcl-2, and Mcl-1 cDNAs were cloned into pFlag-C1 (Clontech). Sequences containing transmembrane helix 6 to the carboxyl terminus of rat InsP3R-1, InsP3R-2, and InsP3R-3 were subcloned into pEGX-6P-1 to create InsP3R-TM6+C constructs. Recombinant protein expression in E. coli and purification using glutathione beads were performed as described (8). The amounts of GST-InsP3R-TM6+C on beads were normalized by Western blotting using anti-GST antibody (GE Healthcare). Lipofectamine 2000 (Invitrogen) was used for Bcl-2 family member plasmid transfection into COS-7 cells. Concentrations of expressed Flag-tagged Bcl-2 family member fusion proteins in COS-7 cell lysates were normalized using anti-Flag antibody M2 (Sigma). Pull-down assays were performed as described (8). Transfected COS-7 cells were washed twice with phosphate-buffered saline and harvested into 1 ml of phosphate-buffered saline containing 0.5% glycerol, 1% Triton X-100, 1 mm dithiothreitol, and protease inhibitor mixture (Sigma). After brief sonication (10 s) and centrifugation, the total protein concentration in the lysate was adjusted to 5 mg/ml and incubated with GST fusion protein (1 h, 4 °C). Beads were centrifuged, washed three times, and prepared for Western blot. Western blot analyses were performed according to standard protocols. Flag-tagged Bcl-xL, Bcl-2, and Mcl-1 bound to GST-TM6+C beads were detected by SDS-PAGE and Western blotting using anti-Flag M2 antibody. We have determined that these protocols, with appropriate numbers of replicates and controls for amounts of proteins, exposure time, and gel loading, can discriminate affinities that differ by greater than a factor of 5.3
Cytoplasmic [Ca2+] Measurements
DT40 cells were plated onto a glass-bottomed perfusion chamber mounted on the stage of an inverted microscope (Olympus IX71) and incubated with Fura-2 AM (2 μm; Invitrogen) for 30 min at room temperature in normal culture media. Cells were then continuously perfused with Hanks Balanced Salt solution (Sigma) containing (mm) 1.8 CaCl2 and 0.8 MgCl2, pH 7.4. Fura-2 was alternately excited at 340 and 380 nm and the emitted fluorescence filtered at 510 nm was collected and recorded using a CCD-based imaging system running SimplePCI software (Hamamatsu Corp.). Dye calibration was achieved by applying experimentally determined constants in Equation 1.
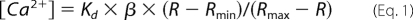
ER [Ca2+] Measurement
Cells were loaded with mag-Fura-2AM (5 μm) in normal culture medium for 45 min at room temperature and then perfused with Hanks Balanced Salt solution for 15 min prior to recording. Cells were washed briefly with intracellular-like medium (ICM) containing: 125 mm KCl, 19 mm NaCl, 10 mm Hepes (pH 7.3 with KOH), and 1 mm EGTA and permeabilized by 2–3 min exposure to ICM containing β-escin (Sigma; 25 μm). Store loading was achieved by switching to ICM containing Ca2+ (free concentration 100 nm) and MgATP (1.5 mm). To induce Ca2+ release, various concentrations of InsP3 were applied in ICM without MgATP to prevent reuptake and containing a free [Ca2+] of 1 μm to produce optimal InsP3R activation (16). Data were acquired and calibrated as described for Fura-2.
Apoptosis Assays
Cells were dual-labeled with AlexaFluor488-conjugated Annexin V and TOTO-3 (Invitrogen) following manufacturer's instructions and analyzed using flow cytometry (FACs Calibur; Beckton Dickinson) with CELLquest software. All experiments were performed at the RFUMS Flow Cytometry/Cell Sorting Research Support Laboratory.
Analyses and Statistics
In all experiments data from two independent clones expressing vector, Bcl-2 or Mcl-1 in WT or InsP3R-KO background were pooled and summarized as mean ± S.E. For multiple comparisons, one-way ANOVA with Fisher's LSD (Least Significant Difference) posthoc comparisons were used to assess statistical significance of differences between means. Unpaired comparisons for normal and non-normally distributed data employed the Student's t test and Mann-Whitney test, respectively. Differences between independent proportions were determined using a z-test. For all tests the differences between means were accepted as statistically significant at the 95% level (p < 0.05).
RESULTS
Bcl-xL, Bcl-2, and Mcl-1 All Bind with Similar Affinities to the Carboxyl Terminus of All Three InsP3R Isoforms
It was previously demonstrated that Bcl-xL binds to all three mammalian InsP3R isoforms in a region localized to the carboxyl terminus extending from the luminal loop between transmembrane helices 5 and 6 to the carboxyl terminus (TM6+C) (8). This region contains the pore, including the putative pore helix, selectivity filter and TM6, and a distal 160 amino acids located in the cytoplasm. Therefore, we assessed whether or not Bcl-2 and Mcl-1 could also bind to this same channel region. GST-TM6+C fusion proteins of the rat types 1, 2, and 3 were immobilized on glutathione beads. The quantities of beads were titrated, and the amounts of GST-InsP3R fragments used for pull-down experiments, as well as the concentrations of Flag-tagged Bcl-2 family member proteins expressed in COS-7 cell lysates, were adjusted to equivalent levels. The carboxyl-terminal fragments of each of the three InsP3R isoforms effectively pulled-down Mcl-1, Bcl-xL, and Bcl-2 with quantitatively similar (within a factor of 5) apparent affinities (Fig. 1A).
FIGURE 1.

Bcl-xL, Bcl-2, and Mcl-1 bind to the carboxyl terminus of each InsP3R isoform. A, left, anti-Flag Western blots of Flag-tagged Mcl-1, Bcl-xL, and Bcl-2 pulled down by InsP3R carboxyl terminus GST fusion proteins (GST-InsP3R-TM6+C; residues 2570–2749 in rat type 1 numbering) from each rat channel isoform. Right, anti-Flag and anti-GST Western blots demonstrating equal amounts of Bcl-2 family proteins and equal amounts of InsP3R isoform carboxyl termini fusion proteins used in pull-downs. B, expression of recombinant Bcl-2 and Mcl-1 in DT40-WT and InsP3R-KO cells. The empty vector pIRES2-DsRed2, Bcl-2-IRES2-DsRed2, or Mcl-1-IRES2-DsRed2 were stably expressed in DT40-WT and DT40-InsP3R-KO cells. Expression levels of Mcl-1 (left) and Bcl-2 (right) in two independent clones by Western blot. InsP3R types 1 and 3 shown. β-actin used as loading control. Depicted blots representative of three independent gels.
Bcl-2 and Mcl-1 Expression Alter Steady-State ER [Ca2+]
The functional effects of Bcl-2 and Mcl-1 on steady-state ER luminal [Ca2+] ([Ca2+]ER) and ER Ca2+ permeability were examined in stably transfected wild-type DT40 cells (DT40-WT) and in DT40 cells with all three InsP3R isoforms genetically deleted (DT40-InsP3R-KO) (17). The cell lines were characterized by Western blot and all subsequent experiments were carried out on two independent clones (shown in Fig. 1B). To monitor the ER store capacity directly, cells were loaded with the low affinity Ca2+ indicator mag-Fura-2, which compartmentalizes in the ER lumen and cytoplasm. The plasma membrane was then permeabilized to remove cytoplasmic dye and the cells were bathed in a Ca2+-free intracellular solution (18). After equilibration, cells were exposed to MgATP and 100 nm free Ca2+ to stimulate ER Ca2+ uptake until a new steady-state filling level was achieved (Fig. 2, A–D). Of note, steady-state [Ca2+]ER was markedly lower in DT40-WT cells expressing Bcl-2 and Mcl-1 compared with control cells expressing the vector only (Fig. 2A). In contrast, steady-state [Ca2+]ER was normalized in Bcl-2- and Mcl-1-expressing cells when ER Ca2+ uptake was activated in the presence of the InsP3R inhibitor heparin (Fig. 2B). Because steady-state [Ca2+]ER is determined by the balance between active uptake and passive release, these data suggest that reduced [Ca2+]ER in the presence of Bcl-2 or Mcl-1 is due to increased Ca2+ release mediated by InsP3R. To test this, similar experiments were performed in Bcl-2- or Mcl-1-expressing DT40-InsP3R-KO cells. In contrast to the effects observed in WT cells, Bcl-2 or Mcl-1 expression had no effect on [Ca2+]ER in the InsP3-KO cells (Fig. 2, C and D). These results indicate that both Bcl-2 and Mcl-1 reduce steady-state [Ca2+]ER by a mechanism that requires InsP3R.
FIGURE 2.

Bcl-2 and Mcl-1 reduce steady state [ Ca2+] ER by enhancing InsP3R-mediated Ca2+ release. A–D, effects of Bcl-2 and Mcl-1 on [Ca2+]ER. Representative recordings in permeabilized cells of [Ca2+]ER during store filling initiated by addition of 100 nm Ca2+ and 1.5 mm MgATP in DT40-WT cells expressing Bcl-2 (red), Mcl-1 (green), or vector (black) in the absence (A) or presence (B) of 100 μg ml−1 heparin, and in InsP3R-KO cells expressing Bcl-2, Mcl-1, or vector-only (C). Each trace represents mean of 15–20 cells in a single image field. D, summary data showing the steady-state [Ca2+]ER after store filling (mean ± S.E.) for 40–100 cells pooled from at least three independent trials. In the DT40-WT cells, mean [Ca2+]ER was lower in Bcl-2- and Mcl-1-expressing cells, 30.3 ± 1 μm and 28.8 ± 1 μm, respectively, compared with vector-only control cells (53.9 ± 3 μm; *, p < 0.001; ANOVA). Presence of heparin increased steady-state [Ca2+]_ER_ in both Bcl-2- and Mcl-1-expressing cells to 45.1 ± 2 and 39.5 ± 2 μm, respectively, but was without effect on vector-only control cells (49.6 ± 4.3 μm; *, _p_ < 0.001; ANOVA). [Ca2+]_ER_ inBcl-2- (55.6 ± 2 μm) and Mcl-1- (52.7 ± 2 μm) expressing DT40-InsP3R-KO cells were not different from DT40-InsP3R-KO controls (59 ± 2 μm, _p_ > 0.05; ANOVA). E–H, effects of Bcl-2 and Mcl-1 on InsP3 sensitivity of ER Ca2+ release. Representative recordings of [Ca2+]ER in response to 10 μm (E) or 100 nm (F) InsP3 in permeabilized DT40-WT cells expressing Bcl-2 (red), Mcl-1 (green), or empty vector (black). Stores filled by preincubation with Ca2+ and MgATP and then challenged with InsP3. Each trace represents mean response of 15–20 cells within single image field. Bar graphs (insets) summarize InsP3-induced change of [Ca2+]ER (mean ± S.E.) for 100–200 cells pooled from at least five independent trials. In cells exposed to 10 μm InsP3, Δ[Ca2+]ER = 47.1 ± 2.1 μm, 31.3 ± 1.1 μm and 31.9 ± 1.5 μm in control and Bcl-2 and Mcl-1 expressing cells, respectively (*, p < 0.001; ANOVA). In response to 100 nm InsP3, Δ[Ca2+]_ER_ = 26.7 ± 1.6, 24.2 ± 0.8, and 23.0 ± 0.9 μm in control, Bcl-2- and Mcl-1-expressing cells, respectively (_p_ > 0.05; ANOVA). G and H, rates of InsP3-evoked ER Ca2+ release, calculated as the first order derivative of average Δ[Ca2+]ER response of all 100–200 cells in response to 10 μm (G) and 100 nm (H) InsP3 in DT40-WT cells expressing Bcl-2 (red), Mcl-1 (green), or empty vector (black).
Bcl-2 and Mcl-1 Enhance the InsP3 Sensitivity of InsP3R-dependent ER Ca2+ Release
It was previously demonstrated in patch clamp studies that purified recombinant Bcl-xL binding to the InsP3R increases its single channel open probability and the number of activated channels in response to subsaturating [InsP3] (7, 8). This activation increases ER Ca2+ efflux in the presence of low [InsP3]. The low aqueous solubilities of full-length Bcl-2 and Mcl-1 proteins prevent comparable patch clamp studies. To determine if Bcl-2 and Mcl-1 have similar functional effects as Bcl-xL, InsP3-dependent ER Ca2+ fluxes were measured directly in permeabilized, mag-Fura-2-loaded DT40-WT cells. After steady-state [Ca2+]ER was achieved by exposure to MgATP and 100 nm Ca2, the perfusion solution was rapidly switched to one containing various [InsP3] and 1 μm free Ca2+ (optimal for InsP3R activation (16)), with no MgATP to prevent Ca2+ reuptake. A saturating [InsP3] (10 μm) (16) evoked a rapid decrease in [Ca2+]ER (Fig. 2E). As expected, the absolute Δ[Ca2+]ER was significantly smaller in both Bcl-2- and Mcl-1-expressing cells, because of reduced steady-state [Ca2+]ER (Fig. 2E, inset). In response to subsaturating [InsP3] (100 nm), a more complete store depletion was observed in the Bcl-2- and Mcl-1-expressing cells, which released about the same total Ca2+ as vector controls, despite the reduced pool size prior to stimulation (Fig. 2F). These data suggest that the sensitivity of InsP3R-dependent Ca2+ release is enhanced by the presence of Bcl-2 or Mcl-1.
The absence of MgATP during InsP3-induced Ca2+ release prevents Ca2+ reuptake into the ER (18). Accordingly, monitoring Ca2+ release under these conditions provides a measure of the rate of the InsP3-dependent Ca2+ efflux. Plotting the first order derivative d[Ca2+]ER/dt as a function of [Ca2+]ER during InsP3 stimulation revealed that both Bcl-2 and Mcl-1 increased the Ca2+ release rate in response to subsaturating (100 nm) InsP3 without affecting the responses to high [InsP3] (Fig. 2, G and H). Thus, Bcl-2 and Mcl-1 both increase the apparent sensitivity of InsP3R-dependent Ca2+ release to low levels of InsP3.
Bcl-2 and Mcl-1 Enhance the Probability of InsP3-dependent [Ca2+]i Oscillations/Spiking
Single cell imaging of Fura-2-loaded DT40-WT cells was performed to determine the effects of the interactions of Mcl-1 and Bcl-2 with the InsP3R on cellular Ca2+ signaling. In the first series of experiments, InsP3-mediated [Ca2+]i signals were evoked upon ligation of the B cell receptor (BCR) by application of a saturating concentration of IgM antibody (anti-IgM; 5 μg ml−1) (17). The majority (∼70%) of vector control cells responded with a single [Ca2+]i transient, while the remaining cells displayed sustained [Ca2+]i oscillations (Fig. 3A). In contrast, when Bcl-2 or Mcl-1 were overexpressed, the majority of cells displayed sustained [Ca2+]i oscillations/spiking (Fig. 3, A and D), with fewer cells responding with a single [Ca2+]i transient (∼55% for both Bcl-2 and Mcl-1; Fig. 3D). The amplitude of the initial [Ca2+]i peak following anti-IgM was reduced in cells expressing Bcl-2, although the total Ca2+ released during the 500 s after the initial [Ca2+]i peak was unchanged (Fig. 3, B and C). Interestingly, in Mcl-1-expressing cells the initial peak and total released Ca2+ was larger than vector control cells (Fig. 3, B and C).
FIGURE 3.

Bcl-2 and Mcl-1 enhance agonist-induced and spontaneous [ Ca2+] i signaling. A–D, agonist-induced signaling. A, typical single cell [Ca2+]i transients in response to 5 μg ml−1 anti-BCR antibody (anti-IgM) in DT40-WT cells expressing Bcl-2 (red), Mcl-1 (green), or empty vector (black). B–D, summary (mean ± S.E.) for >80 cells in multiple trials representing area under the [Ca2+]i response recordings during first 5 min of stimulation (B), peak amplitude of initial transient (C) and percentage of cells responding with spiking/oscillations (D) (*, p < 0.001, Student's unpaired t test (B and C) and z-test (D)). E–H, spontaneous [Ca2+]i signaling. E, representative recordings of spontaneous [Ca2+]i oscillations in DT40-WT cells expressing Bcl-2 (red), Mcl-1 (green), or empty vector (black). Bcl-2 and Mcl-1 increased percentage of cells with spontaneous oscillatory/spiking response from 25.7 ± 1.1% to 37.9 ± 1.3 and 48.6 ± 2.2%, respectively (p < 0.05; z-test). F, histograms of amplitudes and frequencies of spontaneous spiking/oscillations in DT40-WT cells expressing Bcl-2 (n = 368), Mcl-1 (n = 313), or vector (n = 409). Data pooled from at least three independent coverslips. G and H, summary of mean ± S.E. oscillation/spike amplitude (G) and frequency (H) in cells expressing empty vector (129 ± 2 nm; 3.45 ± 0.1 mHz), Bcl-2 (121 ± 1 nm; 6.18 ± 0.2 mHz) and Mcl-1 (215 ± 2 nm; 8.19 ± 0.3 mHz). *, p < 0.001; Mann-Whitney.
It was previously shown that expression of Bcl-xL enhances the frequency of spontaneous, InsP3R-dependent [Ca2+]i oscillations in resting DT40-WT cells (7, 8). This effect was attributed to the observed Bcl-xL-mediated enhanced sensitivity of InsP3R channels to low levels of InsP3 that exist in un-stimulated cells. In the absence of stimulation, ∼25% of DT40-WT cells expressing empty vector displayed spontaneous [Ca2+]i oscillations/spiking (Fig. 3E). Expression of Bcl-2 increased this to nearly 40% and enhanced the proportion of cells displaying higher frequency oscillations (Fig. 3F) that resulted in an average oscillation frequency increase by nearly 2-fold compared with vector controls (Fig. 3G). Similarly, Mcl-1 expression also increased the number of oscillating cells (to nearly 50%) and high frequency oscillators, resulting in the mean oscillation frequency enhanced by 2.4-fold compared with control (Fig. 3H). Expression of Mcl-1 also increased the proportion of higher amplitude [Ca2+]i spikes (Fig. 3F), elevating the mean spike amplitude by nearly 2-fold (Fig. 3G).
Taken together, the effects of Bcl-2 and Mcl-1 expression on InsP3-mediated [Ca2+]i signals are similar to those observed for Bcl-xL, including enhanced InsP3R-mediated Ca2+ flux (Fig. 2), InsP3R-dependent reduced steady-state [Ca2+]ER (Fig. 2), and enhanced [Ca2+]i oscillations in resting cells (Fig. 3). However, there were also distinctive features associated with the expression of each anti-apoptotic Bcl-2 protein. Bcl-2 expression reduced and Mcl-1 enhanced the amplitude of the initial Ca2+ response to anti-IgM, whereas Bcl-xL has no effect (8). Neither Bcl-2 nor Bcl-xL (7, 8) affected the amplitude of individual spontaneous [Ca2+]i spikes in resting cells, whereas Mcl-1 increased the spike amplitudes. These differences between the effects of Bcl-2, Mcl-1, and Bcl-xL may be due to their different expression levels. Despite these differences, the predominant [Ca2+]i signaling phenotypes induced by the expression of the three anti-apoptotic Bcl-2 proteins are qualitatively similar and consistent with their having a primary effect to increase the excitability of the InsP3R.
InsP3R Enhances Bcl-2- and Mcl-1-mediated Apoptosis Protection
To determine the significance of enhanced InsP3R excitability by Bcl-2 and Mcl-1, apoptosis was assessed in DT40 WT and InsP3-KO cells stably expressing Bcl-2 and Mcl-1. After exposure to staurosporine (STS; 0.5 μm) or etoposide (ETS; 50 μm), reagents that induce apoptosis in DT40 cells (19, 20), cells were assayed at various times to assess apoptotic cell death. Cells labeled positive for annexin V or positive for annexin V and the vital stain TOTO-3 were considered to represent the total apoptotic cell population (Fig. 4A). In vector-only expressing control cells, apoptosis initially progressed faster in WT than in InsP3R-KO cells, as indicated by a higher percentage of apoptotic cells at the 8-h and 2-h time points in STS- and ETS-treated samples, respectively (Fig. 4, B and C). This is consistent with previous reports identifying the InsP3R as a proapoptotic mediator during early apoptosis (7, 19). Bcl-2 and Mcl-1 provided apoptosis protection when expressed in either the WT or InsP3R-KO backgrounds (Fig. 4, B–D). Bcl-2 provided better protection against STS than ETS (Fig. 4, B and C), whereas MCl-1 was more effective against ETS (Fig. 4, D and E). In general, protection was more effective when the two proteins were expressed in WT compared with InsP3R-KO cells. These data demonstrate that expression of the InsP3R enables both Bcl-2 and Mcl-1 to provide greater apoptosis resistance. The lack of difference between WT and InsP3-KO in the ability of Mcl-1 to inhibit apoptosis in response to STS (Fig. 4D) may indicate that the efficacy of protection afforded by this mechanism is stimulus dependent, suggesting that InsP3R interactions with different Bcl-2 proteins may couple to different downstream targets.
FIGURE 4.

Effects of Bcl-2 and Mcl-1 expression on apoptosis. Apoptosis was induced with staurosporine (0.5 μm) or etoposide (50 μm). Cells were dual labeled with Annexin V and TOTO-3 and analyzed by flow cytometry. Cells positive for both Annexin V alone (early apoptotic) and Annexin V + TOTO-3 (early and late apoptotic) were counted as the total apoptotic population. A, representative dot plots showing DT40-WT vector control cells before and after treatment with staurosporine or etoposide for 16 and 4 h respectively. B and C, summary graphs (mean ± S.E., data pooled from multiple independent trials) showing the total apoptotic population in DT40 WT or InsP3-KO cells expressing Bcl-2 (red) or empty vector (black) at various times after staurosporine or etoposide treatment. D and E, summary graphs showing the percentage of apoptotic cells in DT40 WT or InsP3-KO cells expressing Mcl-1 (green) or empty vector (black) at various times after staurosporine or etoposide. For each time point, statistical comparisons were made between WT and InsP3-KO cells expressing Bcl-2 or Mcl-1 (*, p < 0.05; ANOVA).
DISCUSSION
We previously identified a biochemical and functional interaction of Bcl-xL with the InsP3R channel carboxyl terminus that impinged on InsP3R gating activity and apoptosis protection (8). In the current study, we have determined that Bcl-2 and Mcl-1 also interact with this same region in all three rat InsP3R isoforms. By semi-quantitative pull-down assays, Bcl-xL, Bcl-2, and Mcl-1 appear to bind to this region in each isoform with approximately equal affinities. The similar binding to the three isoforms may not have been unexpected, as there is considerable sequence homology in the InsP3R region studied. However, the similar binding affinities of the three Bcl-2 family proteins is unexpected, and suggests that shared structural features among them are important for their interaction with the InsP3R carboxyl terminus. Direct binding of Bcl-2 to the InsP3R has been previously reported (14), whereas this is the first report of an interaction of Mcl-1 with InsP3R. Previous studies of Bcl-2 binding suggested weak binding to the InsP3R carboxyl terminus (21). However, the weak binding can be accounted for by the use of a carboxyl-terminal fragment that lacked critical binding determinants further upstream.4 Bcl-2 was also reported to bind to a region within the channel regulatory domain (21). Both Bcl-xL and Mcl-1 bind weakly to this region,4 whereas we show here that they, as well as Bcl-2, bind with similar and much higher affinity to the carboxyl-terminal construct employed in our studies. It was reported that the BH4 domain was necessary and sufficient in mediating the interaction between Bcl-2 and the InsP3R (22), whereas we have observed robust binding between the InsP3R carboxyl terminus and Mcl-1 that lacks a BH4 domain (23). Thus, Bcl-2 protein interactions with the InsP3R are likely to be complex and involve multiple binding sites within the channel.
The interactions of Bcl-2 and Bcl-xL with the InsP3R have been previously demonstrated to affect cellular Ca2+ homeostasis, but the molecular mechanisms are controversial. Expression of Bcl-2 and Bcl-xL attenuate InsP3R-dependent Ca2+ release in response to maximal InsP3 stimulation, but this has been attributed either to inhibition (14, 22, 24) or activation (7–9) of InsP3R channel activity by these proteins. In support of the former, recombinant Bcl-2 inhibited reconstituted InsP3R gating in planar lipid bilayers (14). In the latter case, reduced InsP3-mediated Ca2+ signals were proposed to be caused by Bcl-2 protein-mediated reduction of steady-state [Ca2+]ER (reviewed in Ref. 6). We previously identified the mechanism of this reduction by demonstrating that full-length, soluble recombinant Bcl-xL activated InsP3R single channel gating in native ER membranes, by an allosteric mechanism that sensitized the channel to low [InsP3] (7, 8). This activation mechanism is robust, because it was observed for the three mammalian InsP3R isoforms as well as the Sf9 insect cell InsP3R. We have been unable to perform similar studies with Bcl-2 because of its well-recognized poor solubility. Sensitization by Bcl-xL of the InsP3 sensitivity of the InsP3R can account for reduced steady-state [Ca2+]ER observed in some studies (7, 8) because increased InsP3R activity resets the ER membrane Ca2+ pump-leak balance. Here, we demonstrate directly, using an ER-localized Ca2+ indicator, that both Bcl-2 and Mcl-1, like Bcl-xL, enhance the InsP3 sensitivity of InsP3R-mediated Ca2+ release that results in decreased steady-state [Ca2+]ER. These effects of Mcl-1 and Bcl-2 are InsP3R-dependent because store filling is normalized by InsP3R inhibition, and they are not observed when Bcl-2 or Mcl-1 is expressed in InsP3R-KO cells. These results are significant because they demonstrate that reduced [Ca2+]ER, observed when Bcl-2 is overexpressed in many studies (25–27), is caused by Bcl-2 interaction with the InsP3R. Our results therefore conflict with the reported inhibition of InsP3R channel gating by Bcl-2 (14, 15). Furthermore, they reveal for the first time that Mcl-1 interacts with the InsP3R to produce qualitatively similar effects to Bcl-2 and Bcl-xL on ER Ca2+ release and steady state [Ca2+]ER.
Whereas our results suggest a common mechanism by which anti-apoptotic Bcl-2 family proteins impinge on Ca2+ signaling, the mechanisms whereby enhanced InsP3R Ca2+ signaling impinges on apoptosis protection are themselves controversial. Evidence that ER Ca2+ release and subsequent uptake into mitochondria promotes cytochrome c release, and that reduced [Ca2+]ER can minimize this effect and provide apoptosis protection, have supported the hypothesis that Bcl-2/Bcl-xL impinges on apoptosis by modulating [Ca2+]ER (28). However, such a the model is not supported by observations in many studies of unchanged [Ca2+]ER during Bcl-2-mediated apoptosis protection (5). Furthermore, co-expression of Bcl-xL with each individual InsP3R isoform in an InsP3R null background reduced [Ca2+]ER only in cells expressing type-3 InsP3R whereas it provided InsP3R-dependent apoptosis protection in all of them. Thus, while lowering [Ca2+]ER can provide apoptosis protection, it may not be the only or even most important mechanism involved. In contrast, a consistent feature associated with Bcl-xL/InsP3R-dependent apoptosis protection was the presence of enhanced InsP3R-dependent spontaneous [Ca2+]i spiking or oscillations (7–9). We now show that expression of either Bcl-2 or Mcl-1 provides InsP3R-dependent apoptosis resistance and enhances InsP3R activity that is similarly manifested as an increase in the frequency and number of cells displaying of spontaneous [Ca2+]i spiking. Similar enhanced, low-level InsP3R-dependent [Ca2+]i signaling has been observed previously in other Bcl-2 expression cell models (15, 27).
Our data suggest that a primary mechanism by which the ER impinges on apoptosis involves enhanced low-level spontaneous [Ca2+]i signaling mediated similarly by Bcl-xL, Bcl-2, and Mcl-1 interactions with the InsP3R. The mechanisms that transduce enhanced spontaneous InsP3R [Ca2+]i spiking into apoptotic resistance are unknown but are of obvious importance. Frequency modulation of [Ca2+]i signals regulates numerous processes, including gene transcription (29). Periodic transient InsP3R-mediated [Ca2+]i oscillations activate NF-κB to afford apoptosis resistance (30). Thus, enhanced low-level spontaneous [Ca2+]i signaling mediated by Bcl-xL, Bcl-2, and Mcl-1 may impinge on the nucleus to alter gene expression that promotes apoptosis resistance. Periodic InsP3R-mediated Ca2+ release might also be an efficient mechanism to facilitate Ca2+ delivery to mitochondria at a rate and magnitude that is optimal for stimulating mitochondrial bioenergetics but insufficient to trigger irreversible mitochondrial permeability transition and membrane disruption. In this model, increased apoptosis protection is linked to enhanced cellular bioenergetics (8, 31). Future studies are necessary to identify the molecular mechanisms whereby constitutive low-level Ca2+ signaling affords apoptosis protection.
In summary, we have discovered that Bcl-2, Bcl-xL, and Mcl-1, three structurally related anti-apoptotic proteins with differential expression and responses to different stimuli, bind with similar affinity to the carboxyl terminus of all three isoforms of the InsP3R. We have determined that both Bcl-2 and Mcl-1 have similar functional effects to those reported for Bcl-xL (7, 8), in that they provide enhanced InsP3R activity and InsP3R-dependent spontaneous [Ca2+]i signaling and apoptosis resistance. We conclude that multiple anti-apoptotic Bcl-2 homologues function at the ER by interacting with the InsP3R to generate pro-survival Ca2+ signals that adapt cells to withstand apoptotic stimuli.
Acknowledgments
We thank the Calcium Imaging Research Support Laboratory, Dept of Physiology & Biophysics, Rosalind Franklin University.
*
This work was supported, in whole or in part, by National Institutes of Health Grant GM/DK56328 (to J. K. F.) and funding from the Schweppe Foundation and Rosalind Franklin University (to C. W.).
3
K. Foskett, unpublished observations.
4
K. Foskett, unpublished observations.
2
The abbreviations used are:
ER
endoplasmic reticulum
GST
glutathione _S_-transferase
WT
wild type
ANOVA
analysis of variance
InsP3R
inositol trisphosphate receptor
InsP3
inositol 1,4,5-trisphosphate
STS
staurosporine
ETS
etoposide
KO
knockout.
REFERENCES
- 1.Korsmeyer S. J., Wei M. C., Saito M., Weiler S., Oh K. J., Schlesinger P. H. (2000) Cell Death Differ. 7, 1166–1173 [DOI] [PubMed] [Google Scholar]
- 2.Wei M. C., Lindsten T., Mootha V. K., Weiler S., Gross A., Ashiya M., Thompson C. B., Korsmeyer S. J. (2000) Genes Dev. 14, 2060–2071 [PMC free article] [PubMed] [Google Scholar]
- 3.Decaudin D., Geley S., Hirsch T., Castedo M., Marchetti P., Macho A., Kofler R., Kroemer G. (1997) Cancer Res. 57, 62–67 [PubMed] [Google Scholar]
- 4.Yang J., Liu X., Bhalla K., Kim C. N., Ibrado A. M., Cai J., Peng T. I., Jones D. P., Wang X. (1997) Science 275, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 5.Distelhorst C. W., Shore G. C. (2004) Oncogene 23, 2875–2880 [DOI] [PubMed] [Google Scholar]
- 6.Pinton P., Rizzuto R. (2006) Cell Death Differ. 13, 1409–1418 [DOI] [PubMed] [Google Scholar]
- 7.Li C., Wang X., Vais H., Thompson C. B., Foskett J. K., White C. (2007) Proc. Natl. Acad. Sci. U. S. A. 104, 12565–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White C., Li C., Yang J., Petrenko N. B., Madesh M., Thompson C. B., Foskett J. K. (2005) Nat. Cell Biol. 7, 1021–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones R. G., Bui T., White C., Madesh M., Krawczyk C. M., Lindsten T., Hawkins B. J., Kubek S., Frauwirth K. A., Wang Y. L., Conway S. J., Roderick H. L., Bootman M. D., Shen H., Foskett J. K., Thompson C. B. (2007) Immunity 27, 268–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danial N. N. (2007) Clin. Cancer Res. 13, 7254–7263 [DOI] [PubMed] [Google Scholar]
- 11.Fadeel B., Orrenius S. (2005) J. Intern. Med. 258, 479–517 [DOI] [PubMed] [Google Scholar]
- 12.Willis S. N., Chen L., Dewson G., Wei A., Naik E., Fletcher J. I., Adams J. M., Huang D. C. (2005) Genes Dev. 19, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L., Willis S. N., Wei A., Smith B. J., Fletcher J. I., Hinds M. G., Colman P. M., Day C. L., Adams J. M., Huang D. C. (2005) Mol. Cell 17, 393–403 [DOI] [PubMed] [Google Scholar]
- 14.Chen R., Valencia I., Zhong F., McColl K. S., Roderick H. L., Bootman M. D., Berridge M. J., Conway S. J., Holmes A. B., Mignery G. A., Velez P., Distelhorst C. W. (2004) J. Cell Biol. 166, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong F., Davis M. C., McColl K. S., Distelhorst C. W. (2006) J. Cell Biol. 172, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foskett J. K., White C., Cheung K.-H., Mak D.-O. D. (2007) Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugawara H., Kurosaki M., Takata M., Kurosaki T. (1997) EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betzenhauser M. J., Wagner L. E., 2nd, Won J. H., Yule D. I. (2008) Methods 46, 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan M. T., Bhanumathy C. D., Schug Z. T., Joseph S. K. (2007) J. Biol. Chem. 282, 32983–32990 [DOI] [PubMed] [Google Scholar]
- 20.Korfali N., Ruchaud S., Loegering D., Bernard D., Dingwall C., Kaufmann S. H., Earnshaw W. C. (2004) J. Biol. Chem. 279, 1030–1039 [DOI] [PubMed] [Google Scholar]
- 21.Rong Y. P., Aromolaran A. S., Bultynck G., Zhong F., Li X., McColl K., Matsuyama S., Herlitze S., Roderick H. L., Bootman M. D., Mignery G. A., Parys J. B., De Smedt H., Distelhorst C. W. (2008) Mol. Cell 31, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rong Y. P., Bultynck G., Aromolaran A. S., Zhong F., Parys J. B., De Smedt H., Mignery G. A., Roderick H. L., Bootman M. D., Distelhorst C. W. (2009) Proc. Natl. Acad. Sci. U. S. A. 106, 14397–14402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day C. L., Chen L., Richardson S. J., Harrison P. J., Huang D. C., Hinds M. G. (2005) J. Biol. Chem. 280, 4738–4744 [DOI] [PubMed] [Google Scholar]
- 24.Hanson C. J., Bootman M. D., Distelhorst C. W., Wojcikiewicz R. J., Roderick H. L. (2008) Cell Calcium 44, 324–338 [DOI] [PubMed] [Google Scholar]
- 25.Foyouzi-Youssefi R., Arnaudeau S., Borner C., Kelley W. L., Tschopp J., Lew D. P., Demaurex N., Krause K. H. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 5723–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinton P., Ferrari D., Magalhães P., Schulze-Osthoff K., Di Virgilio F., Pozzan T., Rizzuto R. (2000) J. Cell Biol. 148, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer A. E., Jin C., Reed J. C., Tsien R. Y. (2004) Proc. Natl. Acad. Sci. U. S. A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hajnóczky G., Csordás G., Das S., Garcia-Perez C., Saotome M., Sinha Roy S., Yi M. (2006) Cell Calcium 40, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolmetsch R. E., Lewis R. S., Goodnow C. C., Healy J. I. (1997) Nature 386, 855–858 [DOI] [PubMed] [Google Scholar]
- 30.Li J., Zelenin S., Aperia A., Aizman O. (2006) J. Am. Soc. Nephrol. 17, 1848–1857 [DOI] [PubMed] [Google Scholar]
- 31.Hammerman P. S., Fox C. J., Thompson C. B. (2004) Trends Biochem. Sci 29, 586–592 [DOI] [PubMed] [Google Scholar]