Betulinic Acid Suppresses STAT3 Activation Pathway Through Induction of Protein Tyrosine Phosphatase SHP-1 in Human Multiple Myeloma Cells (original) (raw)
. Author manuscript; available in PMC: 2011 Jul 15.
Published in final edited form as: Int J Cancer. 2010 Jul 15;127(2):282–292. doi: 10.1002/ijc.25059
Abstract
STAT3 activation has been associated with survival, proliferation and invasion of various human cancers. Whether betulinic acid, a pentacyclic triterpene, can modulates the STAT3 pathway, was investigated in human multiple myeloma (MM) cells. We found that betulinic acid inhibited constitutive activation of STAT3, Src kinase, JAK1 and JAK2. Pervanadate reversed the betulinic acid -induced down regulation of STAT3 activation, suggesting the involvement of a protein tyrosine phosphatase (PTP). Furthermore, betulinic acid induced the expression of the PTP SHP-1 and silencing of the SHP-1 gene abolished the ability of betulinic acid to inhibit STAT3 activation and rescues betulinic acid-induced cell death. Betulinic acid also downregulated the expression of STAT3-regulated gene products such as bcl-xL, bcl-2, cyclin D1, and survivin. This correlated with an increase in apoptosis as indicated by an increase in the sub-G1 cell population and an increase in caspase-3–induced PARP cleavage. Consistent with these results, over expression of constitutive active STAT3 significantly reduced the betulinic acid-induced apoptosis. Betulinic acid also enhanced the apoptosis induced by thalidomide (from 10% to 55%) and bortezomib (from 5% to 70%) in MM cells. Overall, our results suggest that betulinic acid down regulates STAT3 activation through upregulation of SHP-1 and this may have potential in sensitization of STAT3 over expressing tumors to chemotherapeutic agents.
Keywords: Betulinic acid, STAT3, JAK1, JAK2, SHP-1, apoptosis
Introduction
Betulinic acid, a pentacyclic triterpene discovered in 1995 from the stem bark of the plant Zizyphus mauritiana, was initially reported to be a melanoma-specific cytotoxic agent1. Betulinic acid is now known to be a component of various other plants that are widespread in tropical regions (e.g., Tryphyllum peltaum, Ancistrocladus heyneaus, Zizyphus joazeiro, Diospyoros leucomelas, Tetracera boliviana, and Syzygium formosanum). In addition, extensive work over the past decade has shown that betulinic acid can induce apoptosis in neuroblastomas2; glioblastomas3; rabdomyosarcoma-medulloblastomas; gliomas4; prostate5, thyroid, breast, lung, and colon carcinomas; and leukemia, as well as multiple myeloma (MM)4. Betulinic acid also appears to be active against HIV6. However, it is still poorly understood how betulinic acid mediates its anticancer effects.
Signal transducer and activator of transcription (STAT) proteins are a family of transcription factors that have been shown to play an important role in tumor cell survival and proliferation7. STAT3, one of the STAT family members, is often constitutively active in human cancer cells, including MM, lymphoma, leukemia, head and neck squamous cell carcinoma, breast, colon, esophageal, gastric, nasopharyngeal, ovarian, prostate, and pancreatic cancers, gliomas, and Ewing sarcoma8. Genes regulated by STAT3 have been linked with tumor survival and chemoresistance (e.g., bcl-xL, mcl-1, survivin), tumor cell proliferation (e.g., cyclin D1), and angiogenesis (e.g., vascular endothelial growth factor [VEGF]).
According to a Surveillance Epidemiology and End Results (SEER) report, 16,570 new cases of MM were diagnosed and 7,320 MM-related deaths were reported in the United States in 2006 alone. MM is a B cell malignancy characterized by the latent accumulation in the bone marrow of secretory plasma cells with a low proliferative index and an extended life span9. Patients with MM often develop resistance to conventional therapies, which is one of the major challenges associated with this disease. Despite several important and exciting advances with novel biologic agents, such as bortezomib and thalidomide, MM remains incurable, as most patients develop relapsed/refractory disease. MM is commonly diagnosed in patients older than 70 years of age. However, because most therapies currently available for MM are highly toxic, treatment options for these older patients are limited. A possible target, STAT3, has been shown to be constitutively active in MM cells10. In addition, STAT3 activation is involved in tumor growth mediated by cytokines such as interleukin-6 (IL-6), which is a growth factor for MM cells11. Therefore, a novel and safe therapeutic agent is urgently needed to improve clinical outcome.
Here, we investigated whether betulinic acid could modulate the constitutive and IL-6–inducible STAT3 pathway in MM cells, leading to suppression of tumor cell survival, proliferation, and chemo resistance. Specifically, we tested the hypothesis that betulinic acid can suppress both constitutive and IL-6–inducible STAT3 activation, leading to the down-regulation of cell survival and proliferative gene product expressions, suppression of proliferation, induction of apoptosis, and enhancement of the response to the cytotoxic effects of bortezomib (a proteasome inhibitor, also called PS341) and thalidomide (an inhibitor of tumor necrosis factor [TNF] expression) in MM cells.
Materials and Methods
Reagents
We prepared a 50-mM solution of betulinic acid (purity >98%; Alexis, San Diego, CA) in ethanol, stored the solution as small aliquots at −20°C, and then diluted the solution as needed in cell culture medium. We purchased Hoechst 33342, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), Tris, glycine, NaCl, sodium dodecyl sulfate (SDS), and bovine serum albumin from Sigma-Aldrich (St. Louis, MO), and we obtained Roswell Park Memorial Institute (RPMI) medium 1640, fetal bovine serum (FBS), 0.4% trypan blue vital stain, and antibiotic-antimycotic mixture from Life Technologies (Grand Island, NY). We obtained rabbit polyclonal STAT3 antibodies and mouse monoclonal antibodies against phospho-STAT3 (Tyr705), bcl-2, bcl-xL, SHP-1, cyclin D1, procaspase-3, and poly(ADP-ribose) polymerase (PARP) from Santa Cruz Biotechnology (Santa Cruz, CA), and we purchased goat anti-rabbit horseradish peroxidase (HRP) conjugate from Bio-Rad (Hercules, CA). We also purchased antibodies to phospho-specific Src (Tyr416), Src, phospho-specific JAK1 (Tyr1022/1023), JAK1, and JAK2 from Cell Signaling Technology (Beverly, MA), goat anti-mouse HRP from Transduction Laboratories (Lexington, KY), and goat anti-rabbit Alexa 594 from Molecular Probes (Eugene, OR). We obtained bacteria-derived recombinant human IL-6 from Novartis Pharmaceuticals (East Hanover, NJ). We obtained bortezomib (PS-341) from Millennium (Cambridge, MA) and thalidomide from Tocris Cookson (Ellisville, MO). The glutathione S-transferase (GST)-JAK2 substrate was provided by Dr. Zhizhuang Joe Zhao (Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK). The constitutive active STAT3 construct was kindly provided by Dr. John DiGiovanni from The University of Texas MD Anderson Cancer Center, Smithville, Texas.
Cell lines
We obtained the human MM cell lines U266 and MM.1S (melphalan-sensitive) from the American Type Culture Collection (Manassas, VA). The U266, MM.1S, prostate carcinoma PC-3 and Du-145 cells were cultured in RPMI 1640 medium containing 1X antibiotic-antimycotic solution with 10% FBS. Human breast carcinoma MCF-7 and MDA-MB-231 were cultured in DMEM/F12 supplemented with 10% FBS. Human embryonic kidney A293 cells were cultured in DMEM with 10% FBS. Human squamous cell carcinoma SCC-4 cells were cultured in DMEM containing 10% FBS, 100 μmol/L nonessential amino acids, 1 mmol/L pyruvate, 6 mmol/L L-glutamine, and 1× vitamins. Cells were maintained at 37°C in an atmosphere of 5% CO2-95% air.
Electrophoretic mobility shift assay for STAT3-DNA binding
We analyzed STAT3-DNA binding with the electrophoretic mobility shift assay (EMSA) using a 32P-labeled high-affinity sis-inducible element (hSIE) probe (5'-CTTCATTTCCCGT AAATCCCT AAA GCT-3' and 5'-AGCTTTAGGGATTTACGGGAAATGA-3') as previously described12. Briefly, we prepared the nuclear extracts from betulinic acid-treated cells and incubated the extracts with the hSIE probe. We separated the DNA-protein complex formed from the free oligonucleotides on 5% native polyacrylamide gels. We then visualized the dried gels and quantitated the radioactive bands using Storm 820 and Image quant software (Amersham Biosciences, Piscataway, NJ).
Western blot analysis
To detect various proteins, we treated the U266 cells (2 × 106/mL) with betulinic acid. The cells were then washed and extracted by incubation for 30 min on ice in buffer containing 20 mM HEPES (pH 7.4), 2 mM ethylenediaminetetraacetic acid, 250 mM NaCl, 0.1% NP-40, 2 μg/mL leupeptin, 2 μg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, 0.5 μg/mL benzamidine, 1 mM dithiothreitol (DTT), and 1 mM sodium vanadate. The lysate was centrifuged, and the supernatant was collected. Whole-cell extract protein (30 μg) was resolved on 7.5%–12% SDS-polyacrylamide gel electrophoresis (PAGE), electro transferred onto a nitrocellulose membrane, blotted with antibodies, and then detected by electrochemiluminescence (Amersham Biosciences).
Immunocytochemistry for STAT3 localization
Betulinic acid-treated MM cells were plated on a glass slide by centrifugation using a cytospin 4 (ThermoShendon, Pittsburgh, PA), air-dried for 1 h at room temperature, and fixed with 4% formaldehyde. After a brief washing in phosphate-buffered saline (PBS), slides were blocked with 5% normal goat serum for 1 h and then incubated with a rabbit polyclonal antihuman STAT3 antibody (dilution 1/100). After overnight incubation, the slides were washed and then incubated with goat anti-rabbit immunoglobulin (Ig)G-Alexa 594 (dilution 1/100) for 1 h and counterstained for nuclei with Hoechst 33342 (50 ng/mL) for 5 min. We stained the slides with mounting medium (Sigma-Aldrich) and analyzed them under an epifluorescence microscope (Labophot-2; Nikon, Tokyo, Japan). We used a Photometrics Coolsnap CF color camera (Nikon) and MetaMorph version 4.6.5 software Molecular Devices, Downingtown, PA) to capture the pictures.
STAT3 luciferase reporter assay
A293 cells were plated in six-well plates with 5 × 105 per well in DMEM containing 10% FBS. The STAT3-responsive elements linked to a luciferase reporter gene were transfected with wild-type or dominant-negative STAT3-Y705F (STAT3F). Transfections were done according to the manufacturer's protocols using Fugene-6 (Roche). At 24 h post transfection, cells were pretreated with betulinic acid for 4 h and then induced by IL-6 for additional 24 h before being washed and lysed in luciferase lysis buffer (Promega). Luciferase activity was measured with a luminometer by using a luciferase assay kit (Promega) and was normalized to β-galactosidase activity. All luciferase experiments were done in triplicate and repeated three times. The data show the mean and the SD of the mean of the experiments.
MTT assay
We determined the anti-proliferative effect of betulinic acid against the MDA-MB231, MCF-7, MM cells, Du-145, PC-3 line using the MTT dye uptake method.
Live/Dead assay
We determined the viability of the cells using the Live/Dead assay (Invitrogen, Carlsbad, CA), which measures intracellular esterase activity and plasma membrane integrity.
Flow cytometric analysis
To determine the effect of betulinic acid on the cell cycle, we first synchronized the U266 cells by serum starvation and then exposed the cells to betulinic acid. We washed the cells with PBS, and fixed with 70% ethanol, and then incubated for 30 min at 37°C with 0.1% RNase A in PBS. We then washed the cells again, resuspended and stained them with PBS containing 25 °g/mL propidium iodide for 30 min at room temperature. We analyzed the cell distribution across the cell cycle with a FACS Calibur flow cytometer (BD, Franklin Lakes, NJ).
Kinase assay
To determine the effect of betulinic acid on JAK2 activation, we performed an immuno complex kinase assay using GST-JAK2 as the substrate. Briefly, we precipitated the JAK complex from the whole-cell extracts with an antibody against JAK2 with protein A/G-Sepharose beads (Pierce Biotechnology, Rockford, IL). After 2 h, we washed the beads with whole-cell extract buffer and then resuspended the complex in a kinase assay mixture containing 50 mM _N_-(2-hydroxyethyl)piperazine-_N'_-2-ethanesulfonic acid (pH 7.4), 20 mM MgCl2, 2 mM DTT, 20 μCi [-32P]adenosine triphosphate (ATP), 10 μM unlabeled ATP, and 2 μg substrate GST-JAK2. After incubation at 30°C for 30 min, we terminated the reaction by boiling the complex-kinase mixture with SDS sample buffer for 5 min. Finally, the protein was resolved on 10% SDS-PAGE, the gel was dried, and the radioactive bands were visualized with the Storm 820 imaging system. To determine the total amounts of JAK2 in each sample, we resolved 30 μg of the whole-cell proteins on 10% SDS-PAGE, electro transferred the proteins to a nitrocellulose membrane, and then blotted with anti-JAK2 antibody.
Transfection with SHP-1 siRNA
SCC4 cells were plated either in six-well plates or chamber slides allowed to adhere for 24 h. On the day of transfection, 12 μl of HiPerFect transfection reagent (QIAGEN) was added to 50 nM SHP-1 siRNA in a final volume of 100 μl of culture medium. After 48 h of transfection, cells were treated with betulinic acid for 4 h, and whole-cell extracts were prepared for SHP-1, STAT3, and phospho-STAT3 analysis by Western blot. Cells transfect in the chamber slides were treated with betulinic acid for 24 h and viability of the cells were determined by Live-Dead assay.
Transfections with Constitutive STAT3 Construct
A293 cells were plated in chamber slides in DMEM containing 10% FBS. After 24 h, the cells were transfected with constitutive STAT3-plasmid by FuGene 6 according to manufacturer's protocol (Roche, Indianapolis, IN). Twenty-four hours after the transfection, cells were treated with betulinic acid for 24 h, and viability of the cells was determined by Live/Dead assay.
Results
We investigated the effect of betulinic acid on both constitutive and IL-6-inducible STAT3 activation in MM cells. We also evaluated the effect of betulinic acid on various mediators of cellular proliferation, cell survival, and apoptosis. The structure of betulinic acid is shown in Fig. 1A.
Fig. 1.
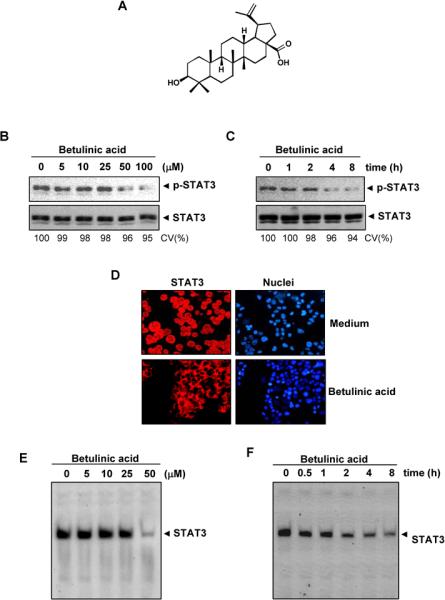
Effect of betulinic acid on constitutively active STAT3. A, The structure betulinic acid. B, Betulinic acid suppresses p-STAT3 levels. U266 cells (2 × 106/mL) were treated with the indicated concentrations of betulinic acid for 4 h, then whole-cell extracts were prepared, and 30 μg of protein was resolved on 7.5% SDS-PAGE gel, electro transferred onto nitrocellulose membranes, and probed for p-STAT3 C, Betulinic acid suppresses p-STAT3 levels in a time-dependent manner. U266 cells (2 × 106/mL) were treated with 50 μM betulinic acid for the indicated durations and analyzed for p-STAT3 levels. D, Betulinic acid causes inhibition of STAT3 translocation to the nucleus. U266 cells (1 × 105/mL) were incubated with or without 50 μM betulinic acid for 4 h and then analyzed for the intracellular distribution of STAT3 by immuno-cytochemistry. The same slides were counterstained for nuclei with Hoechst (50 ng/mL) for 5 min. E, Betulinic acid inhibits STAT3 DNA binding. U266 cells (2 × 106/mL) were treated with the indicated concentrations of betulinic acid for 4 h and analyzed for STAT3 DNA binding by EMSA. F, Cells were treated with 50 μM betulinic acid for the indicated durations and analyzed for nuclear STAT3 levels by EMSA. CV, cell viability
Betulinic acid inhibits constitutive STAT3 phosphorylation in MM cells
We investigated whether betulinic acid modulates constitutive STAT3 activation in MM cells. We incubated U266 cells with different concentrations of betulinic acid for 4 h, and we prepared whole-cell extracts and examined for phosphorylated STAT3 by Western blot analysis using an antibody that recognized STAT3 phosphorylated at the tyrosine 705 site. As shown in Fig. 1B, betulinic acid inhibited constitutive STAT3 activation in the U266 cells, with maximum inhibition occurring at 50 μM betulinic acid. Betulinic acid had no effect on STAT3 protein expression (Fig. 1B, lower panel). We also determined the betulinic acid incubation time required for suppression of STAT3 activation in U266 cells. As shown in Fig. 1C, STAT3 inhibition was time-dependent, with maximum inhibition occurring 8 hrs after the beginning of betulinic acid treatment (Fig. 1C). Under these conditions betulinic acid had no significant effects on cell viability (CV).
Betulinic acid depletes the nuclear pool of STAT3 in MM cells
Because tyrosine phosphorylation causes STAT3 dimerization leading to STAT3 translocation to the nucleus, we used immuno cytochemical analysis to determine whether betulinic acid suppresses nuclear translocation of STAT3. Figure 1D clearly demonstrates that betulinic acid inhibited the translocation of STAT3 to the nucleus in U266 cells.
Betulinic acid inhibits STAT3 DNA-binding activity in MM cells
Since STAT3 translocation to the nucleus leads to DNA binding that results in gene transcription12, we sought to determine whether betulinic acid suppresses STAT3 DNA-binding activity. An EMSA analysis of nuclear extracts prepared from U266 cells showed that betulinic acid caused a decrease in STAT3 DNA-binding activity in a dose- (Fig. 1E) and time-dependent manner (Fig. 1F)
Betulinic acid inhibits IL-6-induced STAT3 activation in human MM cells
Because IL-6 is a growth factor for MM and induces STAT3 phosphorylation13, we investigated whether betulinic acid could inhibit IL-6-induced STAT3 phosphorylation. MM.1S cells, which lack constitutively active STAT3, were treated with different concentrations of IL-6 and then examined for p-STAT3. IL-6 rapidly induced STAT3 activation in MM.1S cells in a time-dependent manner, with maximum activation occurring at 10 min (Fig. 2A). When we incubated the MM.1S cells with betulinic acid for different times, IL-6-induced STAT3 activation was suppressed. Exposure of the MM.1S cells to betulinic acid for 4 h was sufficient to optimally suppress IL-6-induced STAT3 phosphorylation (Fig. 2B).
Fig. 2.
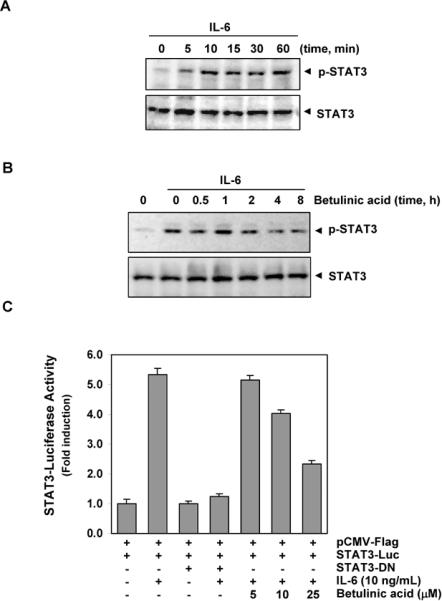
Effect of betulinic acid on IL-6-inducible p-STAT3. A, MM1.S cells (2 × 106/mL) were stimulated with IL-6 (10 ng/mL) for the indicated time, whole-cell extracts were prepared, and p-STAT3 was detected by Western blot analysis. The same blots were stripped and reprobed with STAT3 antibody to verify equal protein loading. B, MM1.S cells (2 × 106/mL) were pre-treated with 50 μM betulinic acid for the indicated times and then stimulated with IL-6 (10 ng/mL) for 10 min. Whole-cell extracts were then prepared and analyzed for p-STAT3 by Western blotting. The same blots were stripped and reprobed with STAT3 antibody to verify equal protein loading. C, A293 cells (5 × 105/mL) were transfected with STAT3-luciferase (STAT3-Luc) plasmid, incubated for 24 h, and treated with indicated concentrations of betulinic acid for 4 h and then stimulated with IL-6 (10 ng/mL) for 24 h. Whole-cell extracts were then prepared and analyzed for luciferase activity. Cells were co-transfected with β-gal and the data were normalized with β-galactosidase assay (data not shown). The results shown are representative of three independent experiments.
Betulinic acid suppresses IL-6-induced STAT3-dependent reporter gene expression
Our results showed that betulinic acid inhibited the phosphorylation, nuclear translocation, and DNA-binding activity of STAT3. We next determined whether betulinic acid affects STAT3-dependent gene transcription. When cells transiently transfected with the pSTAT3-Luc construct were stimulated with IL-6, STAT3-mediated luciferase gene expression significantly increased. Dominant-negative STAT3 blocked this increase, indicating specificity. When the cells were pretreated with betulinic acid, IL-6-induced STAT3 activity was inhibited in a dose-dependent manner (Fig 2C).
Betulinic acid suppresses constitutive c-Src , JAK1 and JAK2 activation
STAT3 has been reported to be activated by soluble tyrosine kinases of the Src kinase families and Janus family14. Hence, we examined the effect of betulinic acid on constitutive Src kinase, JAK1 and JAK2 activation in U266 cells. We found that betulinic acid suppressed the constitutive phosphorylation of the c-Src kinase (Fig. 3), while the levels of total c-Src kinase remained unchanged. We also found that betulinic acid suppressed the constitutive phosphorylation of JAK1. However, the levels of total JAK1 remained unchanged under the same conditions. We further investigated whether betulinic acid affects JAK2 activity in U266 cells using immuno-complex kinase assays with GST-JAK2 acting as the substrate. We found that betulinic acid suppressed the constitutive facilitation of JAK2 in a time-dependent manner (Fig. 3).
Fig. 3.

Effect of betulinic acid on c-Src, p-JAK1 and JAK2 activity. U266 cells (2 × 106/mL) were treated with betulinic acid (50 μM) for indicated time points, whole-cell extracts were prepared, and 30 μg aliquots of those extracts were resolved on 10% SDS-PAGE, electro transferred onto nitrocellulose membranes, and probed for either p-Src or p-JAK1 antibody. The same blots were stripped and reprobed with Src or JAK1 antibody to verify equal protein loading. For JAK2 kinase activity U266 cells (4 × 106/mL) were treated betulinic acid, whole-cell extracts were prepared, and kinase assay was performed.
Tyrosine phosphatase and betulinic acid -induced inhibition of STAT3 activation
Because protein tyrosine phosphatases (PTPase) have also been implicated in STAT3 activation15, we investigated whether betulinic acid -induced inhibition of STAT3 tyrosine phosphorylation could be due to PTPase activation. Treating U266 cells with the broad-acting tyrosine phosphatase inhibitor sodium pervanadate prevented the betulinic acid -induced inhibition of STAT3 activation (Fig. 4A), suggesting that tyrosine phosphatase are involved in betulinic acid -induced inhibition of STAT3 activation.
Fig. 4.
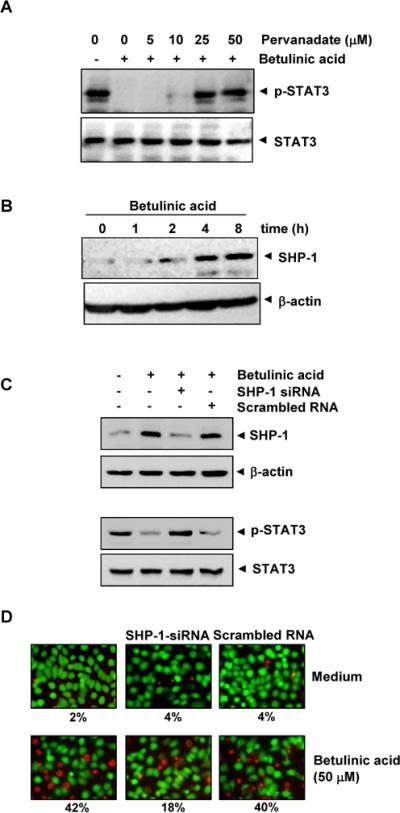
A, Pervanadate reverses the p-STAT3 inhibitory effect of betulinic acid. U266 cells (2 × 106/mL) were first treated with the indicated concentration of pervanadate for 30 min followed by 50 μM betulinic acid for 4 h, and whole-cell extracts were subjected to Western blot analysis for p-STAT3 and STAT3. B, Betulinic acid induces the levels of SHP-1 in U266 cells. U266 cells (2 × 106/mL) were treated with betulinic acid (50μM) for indicated time points. After treatment, whole-cell extracts were prepared and 30 μg portions of those extracts were subjected to Western blot analysis for SHP-1. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. C, Effect of SHP-1 knockdown on betulinic acid -induced expression of SHP-1. SCC4 cells (1 × 105/ml) were transfected with either SHP-1-specific or scrambled siRNA (50 nM). After 48 h, cells were treated with 50 μM betulinic acid for 4 h, and whole-cell extracts were subjected to Western blot analysis for SHP-1. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. Transfection with SHP-1 siRNA reverses betulinic acid -induced suppression of STAT3 activation. The same whole-cell extracts were subjected to p-STAT3 and STAT3. D,Effect of SHP-1 knockdown on betulinic acid -induced cell death. SCC4 cells were transfected with either SHP-1-specific or scrambled siRNA (50 nM). After 48 h, cells were treated with 25 μM betulinic acid for 24 h, live and dead assay was performed and 20 random fields were counted.
Betulinic acid induces SHP-1 expression in U266 cells
SHP-1 is a non-transmembrane PTPase expressed most abundantly in hematopoietic cells16. PTPases have been shown to be involved in the negative regulation of JAK/STAT signaling in leukemia and lymphoma17. Therefore, we examined whether betulinic acid modulates SHP-1 expression in U266 cells. We incubated cells betulinic acid for various time points. As shown inFig. 4B, betulinic acid induced SHP-1 protein expression in U266 cells. Our results suggest that stimulation of SHP-1 expression by betulinic acid was associated with the down regulation of constitutive STAT3 activation in U266 cells.
SHP-1 siRNA down-regulates the expression of SHP-1 and reverses the inhibition of STAT3 activation and attenuates cell death by betulinic acid
We determined whether the silencing of SHP-1 expression by siRNA would abrogate the inhibitory effects of betulinic acid on STAT3 activation and attenuate cell death. Western blotting showed that betulinic acid-induced SHP-1 expression was effectively abolished in the cells treated with SHP-1 siRNA; treatment with scrambled siRNA had no effect (Fig. 4C). We also found that betulinic acid failed to suppress STAT3 activation in cells treated with SHP-1 siRNA (Fig. 4C). Furthermore, betulinic acid has minor effects on cell death in SHP-1 silenced cells when compared with control (Fig 4D). These results suggest the critical role of SHP-1 in the suppression of STAT3 phosphorylation by betulinic acid.
Betulinic acid downregulates bcl-2, bcl-xL, survivin and cyclin D1 expression
We found that expression of antiapoptotic proteins, such as bcl-2, bcl-xL, survivin and cell cycle regulator proteins, such as cyclin D1, all reported to be regulated by STAT38, 18, were modulated by betulinic acid treatment. Betulinic acid treatment downregulated expression of these proteins in a time-dependent manner, with maximum suppression observed 24 h after the beginning of treatment (Fig. 5A).
Fig. 5.

A, Betulinic acid downregulates the expression of antiapoptotic proteins. U266 cells (2 × 106/mL) were treated with 25 μM betulinic acid for indicated time intervals, whole-cell extracts were prepared, and 30 μg portions of those extracts were resolved on 10% SDS-PAGE, and probed against bcl-2, bcl-xL, survivin, and cyclin D1 antibodies. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. B, U266 cells were treated with 25 μM betulinic acid for the indicated times, and whole-cell extracts were prepared, separated on SDS-PAGE, and subjected to Western blot analysis against the caspase-3 and PARP antibody. The same blot was stripped and reprobed with β-actin antibody to show equal protein loading. C, Betulinic acid causes accumulation of cells in the sub-G1 phase. U266 cells (2 × 106/mL) were synchronized by incubation overnight in the absence of serum and then treated with 25 μM betulinic acid for the indicated times, after which the cells were washed, fixed, stained with propidium iodine, and analyzed for DNA content by flow cytometry.
Betulinic acid causes caspase-3activation and PARP cleavage
Treatment of U266 cells with betulinic acid induced caspase-3–dependent cleavage of a 118 kDa PARP protein into an 87 kDa fragment (Fig. 5B).
Betulinic acid causes accumulation of cells in the sub-G1 cell cycle phase
Because D-type cyclins are required for cell progression from the G1 to the S phase of the cell cycle19 and because we observed a rapid decline in cyclin D1 levels in betulinic acid -treated cells, we sought to determine the effect of betulinic acid on cell cycle phase distribution. We found that betulinic acid caused significant accumulation of cells in the sub-G1 phase, an indicator of apoptosis (Fig. 5C).
Betulinic acid inhibits survival of breast carcinoma, MM and prostate carcinoma
Because betulinic acid suppressed the activation of STAT3 and STAT3-regulated gene products linked to cell survival, we next determined whether betulinic acid modulates survival of constitutively and non-constitutively expressed STAT3 cells. Results in Fig. 6A show that betulinic acid suppressed the survival of breast carcinoma (MDA-MB-231, MCF-7), MM (U266, MM .1S) and prostate carcinoma (Du-145, PC-3). The results indicate that effects of betulinic acid are not only specific to MM cells, indeed betulinic acid may be used in solid tumors.
Fig. 6.

A, Cytotoxic effects of betulinic acid on MDA-MB-231, MCF-7, U266, MM.1S and Du-145, PC-3 cells. Cells were plated in triplicate, treated with indicated concentrations of betulinic acid for 72 h, and then subjected to MTT assay to analyze the viability of cells. B, Over expression of constitutive STAT3 rescues A293 cells from betulinic acid -induced cytotoxicity. First, A293 cells were transfected with constitutive STAT3 plasmid. After 24 h of transfection, the cells were treated with indicated concentration of betulinic acid for 24 h, and then the cytotoxicity was determined by Live/Dead assay and 20 random fields were counted. C, Betulinic acid potentiates the apoptotic effect of bortezomib and thalidomide. U266 cells (5000 cells/well) were treated with indicated amounts of betulinic acid for 4h followed by indicated amounts of thalidomide or bortezomib for 24 h at 37°C. Cell viability was assessed by MTT uptake method. For live and dead assay, U266 cells (1 × 106/mL) were treated with 25 μM betulinic acid and 10 μg/mL thalidomide or 20 nM bortezomib alone or in combination for 24 h at 37°C. Cells were stained with a Live/Dead assay reagent for 30 min and then analyzed under a fluorescence microscope and 20 random fields were counted.
Over expression of constitutively active STAT3 rescues betulinic acid-induced apoptosis
We assessed whether the over expression of constitutive active STAT3 can rescue betulinic acid-induced apoptosis. A293 cells were transfected with constitutively active STAT3 plasmid for 24 h, and cells were incubated with betulinic acid for the next 24 h and examined for apoptosis by esterase staining assay. The results show that the forced expression of STAT3 significantly reduces the betulinic acid-induced apoptosis (Fig. 6B).
Betulinic acid potentiates the apoptotic effect of bortezomib and thalidomide in MM cells
We examined whether betulinic acid potentiates the effects of bortezomib and thalidomide. We first determined the optimum dose of betulinic acid required potentiating apoptotic effects of either bortezomib or thalidomide (Fig 6C, lower panel). We treated U266 cells with betulinic acid combined with either bortezomib or thalidomide and examined the apoptosis using the Live/Dead assay, which determines plasma membrane stability using esterase staining. As shown in Figure 6C, betulinic acid significantly enhanced the apoptotic effects of bortezomib (from 5% to 70%) and thalidomide (from 10% to 55%), suggesting that betulinic acid can be used in combination with both drugs.
Discussion
Agents which can suppress STAT3 activation pathway, have potential for cancer prevention and treatment. In the present report we ascertained that betulinic acid, derived from variety of fruits, exerts its anticancer effects through the abrogation of the STAT3 signaling pathway in MM cells. We found that betulinic acid could suppress both constitutive and inducible STAT3 activation in MM cells by STAT3 phosphorylation, STAT3 nuclear translocation, and STAT3 DNA binding. The effects of betulinic acid on STAT3 phosphorylation were associated with JAK1 and JAK2 suppression.
Our results show that betulinic acid can suppress activation of c-Src. The phosphorylation of c-Src is known to play a critical role in tumor cell transformation and proliferation8, 18. Most Src-transformed cell lines have persistently activated STAT3, and dominant-negative STAT3 blocks transformation20, 21. Constitutive STAT3 activation is associated with various types of carcinoma, sarcoma, lymphoma and leukemia8. Thus, the suppression of constitutively active STAT3 in MM cells raises the possibility that betulinic acid might also inhibit constitutively active STAT3 in other types of cancer cells4. Furthermore, we observed that betulinic acid inhibited the growth of both, breast carcinoma and human prostate carcinoma cells.
We observed that betulinic acid-induced inhibition of STAT3 activation involves a PTPase. Numerous PTPases have been implicated in STAT3 signaling, including SHP-1, SHP-2, TC-PTPase, PTEN, PTPase-1D, CD45, PTPase-epsilon, and low molecular weight PTPase22, 23. It is not clear what type of PTPase was involved in the downregulation of STAT3 phosphorylation. Loss of SHP-1 has been shown to enhance JAK/STAT3 signaling in ALK-positive anaplastic large-cell lymphoma15. We also found that the betulinic acid-induced inhibition of STAT3 activation specifically involves a protein tyrosine phosphatase SHP-1. Our results clearly show that SHP-1 is induced by betulinic acid, and its knocking down with siRNA abolished its STAT3 inhibitory effects and rescues betulinic acid-induced cell death.
We previously reported that betulinic acid suppresses nuclear factor (NF)-κB activation24. However, it is not clear whether suppression of STAT3 activation by betulinic acid is linked to inhibition of NF-κB activation. Interestingly, studies of Digicaylioglu and Lipton showed that erythropoietin activate NF-κB through JAK2 kinase activation25. Thus, it is possible that suppression of JAK activation is the critical target for inhibition of both NF-κB and STAT3 activation by betulinic acid. However, it is still unclear how betulinic acid upregulates SHP-1 and inhibits the JAK2 kinase.
We observed that betulinic acid induces apoptosis through caspase-3–mediated PARP cleavage in MM cells. Our results are consistent with earlier studies of Fulda et al that mitochondria activation leading to caspase activation by betulinic acid3, 26. We also found that betulinic acid suppresses the expression of STAT3-regulated genes, including cyclin D1, and antiapoptotic gene products, including survivin, bcl-2, and bcl-xL. We found a decrease in bcl-2 and cyclin D1 expression by betulinic acid, which was similar to that seen in previous reports27. Constitutively active STAT3 contributes to oncogenesis by protecting cancer cells from apoptosis, which implies that suppression of STAT3 activation by agents such as betulinic acid could facilitate apoptosis. Bcl-xL expression is regulated by STAT328 and is over expressed in MM cells29. Bcl-xL can also block cell death induced by a variety of chemotherapeutic agents in parallel with an increase in chemoresistance30. Down regulation of bcl-xL expression is likely linked with betulinic acid's ability to induce apoptosis in MM cells. Betulinic has been shown to induce proteasome-dependent degradation of other transcription factors, specificity protein 1 (Sp1), Sp3, and Sp4, which regulate VEGF expression5. VEGF expression is also regulated by STAT3, suggesting that betulinic acid may mediate anti-angiogenesis through the down regulation of VEGF. Furthermore, we observed over expression of STAT3 also rescues the apoptotic effects of betulinic acid, strengthening our hypothesis that anti-proliferative effects of betulinic acid are mediated through the abrogation of the STAT3 signaling pathway.
Recently, bortezomib and thalidomide were approved for the treatment of MM31, 32. We found that betulinic acid sensitizes MM cells to the apoptotic effects of bortezomib and thalidomide, which is in agreement with other reports using different chemotherapeutic agents in different tumor cell types3.
Betulinic acid and several other analogues have also been tested in animals and were found to be well-tolerated and efficacious5, 33. Furthermore, a betulinic acid derivative, DSB [PA-457], is currently in phase II clinical trial as a first-in-class HIV maturation inhibitor34. We believe that the pharmacologic safety of betulinic acid and its ability to down regulate the expression of several genes involved in cell survival and chemoresistance provides a sufficient rationale for testing betulinic acid further in patients with MM.
Statement of Significance.
Patients with multiple myeloma (MM), often develop resistance to conventional therapies. Furthermore, existing therapies are highly toxic, hence, a novel and safe therapeutic agent is urgently needed to improve clinical outcome. Constitutive activation of STAT3 is associated with MM. We demonstrate here that betulinic acid (a pentacyclic triterpene), component of Ber, Jamun and Persimmons, can inhibits both the constitutive and IL-6–induced STAT3 activation pathway and enhance apoptotic effects of thalidomide and bortezomib in MM cells.
Acknowledgment
We would like to thank Alyson Todd for carefully editing this manuscript and providing valuable comments. Dr. Aggarwal is the Ransom Horne, Jr., Professor of Cancer Research.
Grant support: This work was supported by a grant from the Clayton Foundation for Research (B.B.A.), a program project grant from National Institutes of Health (NIH CA-124787-01A2), and grant from Center for Targeted Therapy of M.D. Anderson Cancer Center.
References
- 1.Pisha E, Chai H, Lee IS, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CW, Fong HH, Kinghorn AD, Brown DM, et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nature medicine. 1995;1:1046–51. doi: 10.1038/nm1095-1046. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt ML, Kuzmanoff KL, Ling-Indeck L, Pezzuto JM. Betulinic acid induces apoptosis in human neuroblastoma cell lines. Eur J Cancer. 1997;33:2007–10. doi: 10.1016/s0959-8049(97)00294-3. [DOI] [PubMed] [Google Scholar]
- 3.Fulda S, Jeremias I, Steiner HH, Pietsch T, Debetulinic acidtin KM. Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. International journal of cancer. 1999;82:435–41. doi: 10.1002/(sici)1097-0215(19990730)82:3<435::aid-ijc18>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Rzeski W, Stepulak A, Szymanski M, Sifringer M, Kaczor J, Wejksza K, Zdzisinska B, Kandefer-Szerszen M. Betulinic acid decreases expression of bcl-2 and cyclin D1, inhibits proliferation, migration and induces apoptosis in cancer cells. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:11–20. doi: 10.1007/s00210-006-0090-1. [DOI] [PubMed] [Google Scholar]
- 5.Chintharlapalli S, Papineni S, Ramaiah SK, Safe S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer research. 2007;67:2816–23. doi: 10.1158/0008-5472.CAN-06-3735. [DOI] [PubMed] [Google Scholar]
- 6.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. Journal of medicinal chemistry. 1996;39:1016–7. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 7.Darnell JE., Jr. Transcription factors as targets for cancer therapy. Nature reviews. 2002;2:740–9. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, Sung B, Ichikawa H. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Annals of the New York Academy of Sciences. 2006;1091:151–69. doi: 10.1196/annals.1378.063. [DOI] [PubMed] [Google Scholar]
- 9.Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 10.Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, Estrov Z, Talpaz M, Aggarwal BB. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–84. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 11.Zhong Z, Wen Z, Darnell JE., Jr. Science. Vol. 264. New York, N.Y: 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6; pp. 95–8. [DOI] [PubMed] [Google Scholar]
- 12.Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Science. Vol. 269. New York, N.Y: 1995. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein; pp. 81–3. [DOI] [PubMed] [Google Scholar]
- 13.Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–5. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 14.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–4. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 15.Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108:2796–803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- 16.Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 17.Oka T, Ouchida M, Koyama M, Ogama Y, Takada S, Nakatani Y, Tanaka T, Yoshino T, Hayashi K, Ohara N, Kondo E, Takahashi K, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer research. 2002;62:6390–4. [PubMed] [Google Scholar]
- 18.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nature reviews. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 19.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 20.Brierley MM, Fish EN. Stats: multifaceted regulators of transcription. J Interferon Cytokine Res. 2005;25:733–44. doi: 10.1089/jir.2005.25.733. [DOI] [PubMed] [Google Scholar]
- 21.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 22.Woetmann A, Nielsen M, Christensen ST, Brockdorff J, Kaltoft K, Engel AM, Skov S, Brender C, Geisler C, Svejgaard A, Rygaard J, Leick V, et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc Natl Acad Sci U S A. 1999;96:10620–5. doi: 10.1073/pnas.96.19.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim H, Betulinic acidumann H. Dual signaling role of the protein tyrosine phosphatase SHP-2 in regulating expression of acute-phase plasma proteins by interleukin-6 cytokine receptors in hepatic cells. Molecular and cellular biology. 1999;19:5326–38. doi: 10.1128/mcb.19.8.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takada Y, Aggarwal BB. Betulinic acid suppresses carcinogen-induced NF-kappa B activation through inhibition of I kappa B alpha kinase and p65 phosphorylation: abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J Immunol. 2003;171:3278–86. doi: 10.4049/jimmunol.171.6.3278. [DOI] [PubMed] [Google Scholar]
- 25.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–7. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 26.Fulda S, Friesen C, Los M, Scaffidi C, Mier W, Benedict M, Nunez G, Krammer PH, Peter ME, Debetulinic acidtin KM. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer research. 1997;57:4956–64. [PubMed] [Google Scholar]
- 27.Ji ZN, Ye WC, Liu GG, Hsiao WL. 23-Hydroxybetulinic acid-mediated apoptosis is accompanied by decreases in bcl-2 expression and telomerase activity in HL-60 Cells. Life sciences. 2002;72:1–9. doi: 10.1016/s0024-3205(02)02176-8. [DOI] [PubMed] [Google Scholar]
- 28.Niu G, Heller R, Catlett-Falcone R, Coppola D, Jaroszeski M, Dalton W, Jove R, Yu H. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer research. 1999;59:5059–63. [PubMed] [Google Scholar]
- 29.Tu Y, Renner S, Xu F, Fleishman A, Taylor J, Weisz J, Vescio R, Rettig M, Berenson J, Krajewski S, Reed JC, Lichtenstein A. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer research. 1998;58:256–62. [PubMed] [Google Scholar]
- 30.Simonian PL, Grillot DA, Nunez G. Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood. 1997;90:1208–16. [PubMed] [Google Scholar]
- 31.Cavo M. Proteasome inhibitor bortezomib for the treatment of multiple myeloma. Leukemia. 2006;20:1341–52. doi: 10.1038/sj.leu.2404278. [DOI] [PubMed] [Google Scholar]
- 32.Glasmacher A, Hahn C, Hoffmann F, Naumann R, Goldschmidt H, von Lilienfeld-Toal M, Orlopp K, Schmidt-Wolf I, Gorschluter M. A systematic review of phase-II trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. British journal of haematology. 2006;132:584–93. doi: 10.1111/j.1365-2141.2005.05914.x. [DOI] [PubMed] [Google Scholar]
- 33.Liby K, Honda T, Williams CR, Risingsong R, Royce DB, Suh N, Dinkova-Kostova AT, Stephenson KK, Talalay P, Sundararajan C, Gribble GW, Sporn MB. Novel semisynthetic analogues of betulinic acid with diverse cytoprotective, antiproliferative, and proapoptotic activities. Molecular cancer therapeutics. 2007;6:2113–9. doi: 10.1158/1535-7163.MCT-07-0180. [DOI] [PubMed] [Google Scholar]
- 34.Yu D, Morris-Natschke SL, Lee KH. New developments in natural products-betulinic acidsed anti-AIDS research. Med Res Rev. 2007;27:108–32. doi: 10.1002/med.20075. [DOI] [PubMed] [Google Scholar]