The AIM2 inflammasome is essential for host-defense against cytosolic bacteria and DNA viruses (original) (raw)
. Author manuscript; available in PMC: 2010 Nov 1.
Published in final edited form as: Nat Immunol. 2010 Mar 28;11(5):395–402. doi: 10.1038/ni.1864
Abstract
Inflammasomes regulate the activity of capase-1 and maturation of interleukin-1β and interleukin-18. Recently, AIM2 was shown to bind DNA and engage ASC to form a caspase-1 activating inflammasome. Using _Aim2_-deficient mice, we reveal a central role for AIM2 in regulating caspase-1-dependent maturation of IL-1β and IL-18, as well as pyroptosis in response to synthetic dsDNA. AIM2 is essential for inflammasome activation in response to Fransicella tularensis, vaccinia virus, mouse cytomegalovirus and plays a partial role in sensing Listeria monocytogenes. Moreover, production of IL-18 and NK cell-dependent IFN-γ production, events critical in early control of virus replication were dependent on AIM2 during mCMV infection in vivo. Collectively, these observations reveal the importance of AIM2 in sensing both bacterial and viral pathogens and triggering innate immunity.
Optimal protection from infection requires complex immune responses involving both the innate and adaptive immune system. The innate immune system plays a key role in initiating and orchestrating host defenses by regulating the production of pro-inflammatory cytokines, type I interferons and anti-microbial effectors1. Several distinct classes of germline-encoded pattern recognition receptors (PRRs) have been discovered and implicated in the sensing of microbial products. These include the Toll-like receptors (TLRs)2, the C-type lectin receptors (CLRs)3, the RIG-like helicases (RLRs)4, the NOD-like receptors (NLRs)5 and cytosolic DNA sensors; DNA dependent activator of IRFs (DAI)6, RNA polymerase III7, 8 and Absent in melanoma-2 (AIM2) (ref 9-12). These individual PRRs recognize microbial products from bacteria, viruses, fungi and parasites and in most cases trigger signaling pathways which culminate in the transcription of immune response genes1.
The pro-inflammatory cytokine IL-1β is among the arsenal of defense measures deployed by the innate immune system13. The activity of IL-1β is regulated at the level of expression, processing, secretion and antagonism by a naturally occurring inhibitor, IL-1 receptor antagonist (IL1RA)13. An initial microbial stimulus acting through innate PRRs results in accumulation of intracellular stores of pro-IL-1β. A second stimulus then controls the activation of caspase-1, a proteolytic enzyme, which in turn leads to cleavage of pro-IL-1β, followed by release of the active mature 17-kDa cytokine. Interleukin-18 (IL-18) is structurally similar to IL-1β, and like IL-1β is first synthesized as a leaderless precursor requiring caspase-1 for cleavage into an active molecule14.
Multiprotein complexes containing one or more Nod-like receptors (NLRs) commonly referred to as “inflammasomes” regulate caspase-1 activity. The NLRP1 inflammasome is activated by anthrax lethal toxin15. The NLRP3 inflammasome is activated by a broad range of stimuli including bacteria, bacterial pore forming toxins, viruses, endogenous danger signals such as ATP and monosodium urate and indigestible particulates, like silica, asbestos, alum and β-amyloid (reviewed in 16). The IPAF inflammasome is activated by flagellin or by the basal body rod component of the type 3 secretion system (T3SS) found in Salmonella typhimurium and other bacteria17. A fourth inflammasome, the AIM2 inflammasome was also recently identified. AIM2 is a member of the IFI20X/IFI16 (PYHIN) protein family18. AIM2 binds to DNA via a HIN200 domain, while the PYD domain associates with the adapter molecule ASC to activate caspase-1 (ref 9-12). The role of AIM2 in orchestrating immune responses to DNA or in regulating immune responses to viral or bacterial pathogens in vivo has not been examined further.
Here we investigate the role of AIM2 in vivo by generating mice lacking Aim2. We show that Aim2 regulates caspase-1-dependent processing of pro-IL-1β and pro-IL-18 in response to dsDNA in both dendritic cells and macrophages. Aim2 does not mediate type I IFN responses to dsDNA but rather acts to negatively regulate this pathway. We also reveal a central role for Aim2 in sensing the cytosolic bacterial pathogen, Fransicella tularensis, live vaccine strain (LVS), as well as vaccinia virus and mouse cytomegalovirus (mCMV). Additionally, we reveal a partial role for Aim2 in sensing _Listeria monocytogenes. In vivo, Aim2_- and _Asc_-deficient mice infected with mCMV have reduced IL-18 concentrations in the serum, and severely attenuated IFN-γ production by NK cells, events, which are critical for the early control of viral replication. Collectively, these observations establish Aim2 as an important anti-microbial sensor and a key determinant of protective immunity to viral pathogens.
Results
Generation of AIM2-/- mice
To study the role of the Aim2 gene in vivo, we generated _Aim2_-deficient mice using a gene-trap ES cell line carrying a pGT0lxf vector insertion (CSG445;http://www.genetrap.org) (Fig. 1a). In this ES cell line, the pGT0lxf gene-trap vector has inserted into intron 6 of the Aim2 gene. The fusion transcript generated by the gene-trap vector insertion directs the expression of a truncated Aim2 protein carrying only the N-terminal PYD domain fused to the β-geo cassette (Fig. 1a). The presence of the pGT0lxf vector insertion within the Aim2 gene in the CSG445 ES cells was confirmed by RT-PCR (Supplementary Fig. 1) and the site of the pGT0lxf integration was mapped by long range PCR and sequencing and found to reside within intron 6 (Supplementary Fig. 2). Mice harboring 2 copies of the trapped Aim2 gene were bred to homozygosity and deletion of Aim2 confirmed by RT-PCR (Fig. 1b) and immunoblotting (Fig. 1c). Aim2 was absent in thioglycollate-elicited macrophages (TEM), bone marrow-derived macrophages (BMDM), bone marrow-derived DCs (BMDC) and splenocytes from _Aim2_-/- mice (Fig. 1c, and data not shown). The mutant mice were born at the expected Mendelian ratios. They developed and bred normally and displayed no apparent abnormalities. Aim2+/+ and _Aim2_-/- mice were compared throughout our studies and were generated from breedings of heterozygous parents.
Figure 1.
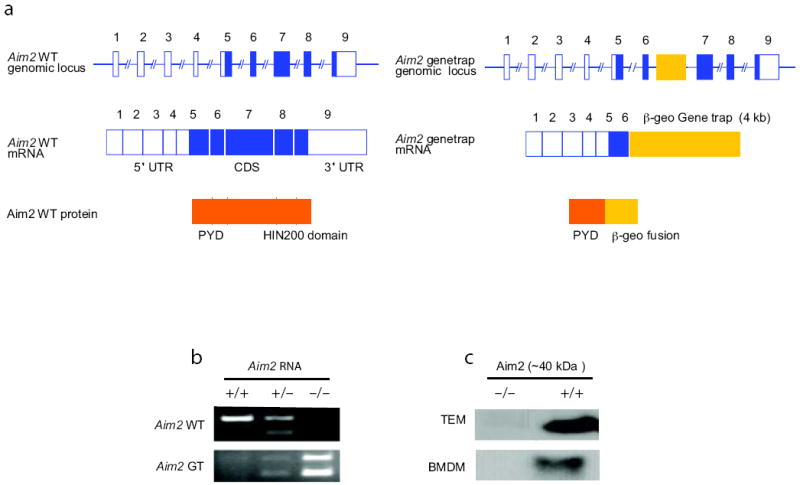
Characterization of _Aim2_-genetrap mice. a Schematic of wild-type and gene-trapped Aim2 allele. b. RT-PCR analysis shown the absence of wild-type (WT) Aim2 transcripts in the _Aim2_-genetrap mice. WT Aim2 RNA was detected by primers F and R1 and gene-trapped Aim2 RNA was detected by primers F and R2. c. Immunoblotting of Aim2 protein in peritoneal and bone marrow derived macrophages from Aim2+/+ and _Aim2_-/- mice. Equal amounts of protein were loaded for all the samples and Aim2 was detected using a rabbit polyclonal antibody.
Aim2 is critical for responses to cytosolic dsDNA
Consistent with previous studies, which identified human AIM2 as a cytosolic sensor for dsDNA and a key regulator of IL-1β production (ref 9-12), transfection studies with the murine Aim2 gene also led to the formation of a functional inflammasome complex. As an initial screen for the ability of Aim2 to associate with Asc and elicit signaling, we tested the ability of transfected Aim2 to trigger Asc-dependent NFκB signaling. Overexpression of mouse Aim2 led to Asc-dependent NFκB reporter gene activation (Fig. 2a). We next examined whether the murine Aim2-Asc complex could lead to the caspase-1-dependent maturation of pro-IL-1β using a transient transfection assay with Aim2 and Asc in the presence of caspase-1 and flag-tagged pro-IL-1β in 293T cells. Aim2 led to maturation of pro-IL-1β in an Asc-dependent manner (Fig. 2b).
Figure 2.

Aim2 is essential for inflammasome activation by DNA. a. 293T cells were transfected with indicated plasmids together with a NF-κB reporter and TK Renilla-luciferase reporter gene. After 48 h, luciferase activity was measured. b. 293T cells were transfected with the indicated plasmids together with murine pro-IL-1β-Gluc/Flag. After 24 h, cell lysates were immunoblotted for pro-IL1β-Flag, IL-1β-Flag and β-actin. c. F4/80 and CD11b expression by BMDM from Aim2+/+ and _Aim2_-/- mice. d-f. LPS (200 ng/ml) primed _Aim_2+/+ and _Aim_2-/- BMDM, thioglycollate-elicited macrophages (TEM), and BMDCs were transfected with poly(dA-dT) (0.75 – 3 μg/106 cells) for 6 h. The secreted IL-1β (d-e) and the cleaved forms of IL-1β and caspase 1 (f) in the culture supernatants were analyzed. g. _Aim_2+/+ and _Aim_2-/- BMDM were treated as above and the IL-18 levels in the supernatants at 6 h were measured by ELISA. (h). _Aim_2+/+ and _Aim_2-/- TEM were treated as indicated for 24 h and cell viability was reported as % of viability of medium treated cells. Asterisks indicate P values of < 0.05 for _Aim_2+/+ versus _Aim_2-/-. Data are presented as mean ± SD from one experiment representative of 3 experiments.
To evaluate the role of Aim2 in the innate response to DNA, BMDM were generated from Aim2+/+ and _Aim2_-/- littermate controls. Macrophage differentiation was examined by staining cells for CD11b and F480. Both Aim2+/+ and _Aim2_-/- macrophages had equivalent F480/CD11b staining patterns indicating normal macrophage differentiation (Fig. 2c). Synthetic B-form dsDNA, poly(dA:dT) induced a robust IL-1β response in BMDM as measured by ELISA and this response was abolished in _Aim2_-/- macrophages (Fig. 2d). Similar results were obtained when thioglycollate-elicited macrophages and BMDCs were examined (Fig. 2e). DNA treatment also led to the proteolytic cleavage of caspase-1 and production of the mature 17kDa IL-1β cytokine and in both cases these responses were impaired in _Aim2_-/- cells (Fig. 2f). Macrophages lacking Asc or caspase-1 had a similar phenotype (data not shown). Poly(dA:dT) also induced IL-18 production and this was also Aim2-dependent (Fig. 2g). Finally, poly(dA:dT) induced an inflammatory form of cell death (pyroptosis) in Aim2+/+ macrophages and this response was also attenuated in _Aim2_-/- cells (Fig. 2h). Collectively, these data indicate that Aim2 is a non-redundant sensor regulating caspase-1 dependent maturation of IL-1β, IL-18 and pyroptosis in response to DNA19.
Aim2-deficient cell responses to other inflammasome activators
To define the specificity of Aim2 for DNA, we examined inflammasome activity in response to agonists of other NLRs. Aim2+/+ and _Aim2_-/- BMDM responded equivalently to anthrax lethal toxin which signals via Nlrp115 (Fig. 3a). IL-1β release in response to ATP and nigericin, activators of the Nlrp3 inflammasome was also normal (Fig 3b). Analysis of caspase-1 cleavage and IL-1β cleavage showed similar results (Fig 3c). Aim2-deficient cells also responded normally to Salmonella typhimurium, which activates the Nlrc4/Ipaf inflammasome (Fig. 3d). Rig-I regulates caspase-1-dependent processing of IL-1β in response to some RNA viruses20. To evaluate this pathway we monitored IL-1β release in BMDCs from Aim2+/+ and _Aim2_-/- mice that were infected with Sendai virus, a paramyxovirus which signals through RIG-I. Sendai virus induced IL-1β response was not impaired in _Aim2_-/- DCs (Fig. 3e). Collectively, these data indicate that Aim2 is a receptor for cytosolic dsDNA and does not contribute to inflammasome activation in response to agonists of the Nlrp1, Nlrp3, Nlrc4 or Rig-I inflammasomes.
Figure 3.
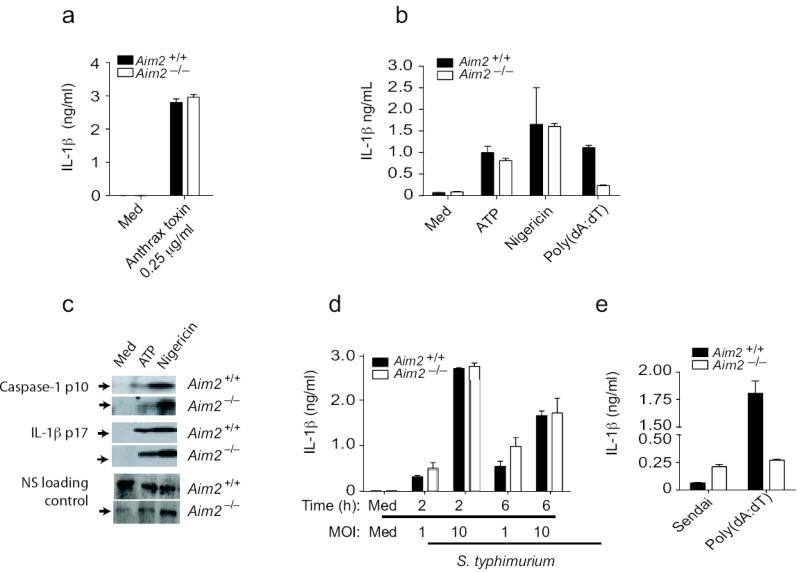
Aim2-deficient cells respond normally to other inflammasome activating stimuli. a. BMDM from _Aim_2+/+, _Aim_2-/-, and C57Bl/6 mice were primed with LPS (200 ng/ml) for 3 h and stimulated with anthrax toxin units PA and LF. Supernatants were harvested after 6 h and analyzed by ELISA for IL-1β production. b-c. LPS primed _Aim_2+/+ and _Aim_2-/- TEM were treated with ATP (5 mM) and Nigericin (5 μM) for 1 h and secretion of IL-1β and cleavage of IL-1β and caspase 1 were analyzed. d. _Aim_2+/+ and _Aim_2-/- BMDM were primed with LPS (200 ng/ml) for 3 h and infected with S. typhimurium at MOI 1 and MOI 10. Supernatants were harvested at 2 and 6 h post infection and IL-1β levels were analyzed by ELISA. e. BMDCs were treated with Sendai virus (200 HA units/ml) for 18 h and IL-1β concentrations in the supernatant were measured by ELISA. NS; nonspecific band. Asterisks indicate P values of < 0.05 for _Aim_2+/+ versus _Aim_2-/-. Data are presented as mean ± SD from one experiment representative of 3 experiments.
IFN responses in Aim2-deficient macrophages
Cytosolic DNA is also a potent trigger of type I IFN production. To evaluate the role of Aim2 in this response, we measured IFN-β production in response to poly(dA:dT) in Aim2+/+ and _Aim2_-/- mice. Poly(dA:dT) induced IFN-β in Aim2+/+ splenocytes and thioglycollate-elicited macrophages and this response was enhanced in cells from _Aim2_-/- mice (Fig. 4a and b). These observations indicate that Aim2 is not a sensor for IFN-β production but it may exert a negative regulatory effect on IFN responses. We also examined the responsiveness of _Aim2_-/- BMDM to type I IFNs themselves. Aim2+/+ and _Aim2_-/- macrophages were stimulated with IFN-β and induction of viperin, a well-characterized IFN stimulated gene was measured by immunoblotting. IFN-β induced high levels of viperin expression in Aim2+/+ cells and this response was still induced in _Aim2_-/- cells (Fig. 4c).
Figure 4.
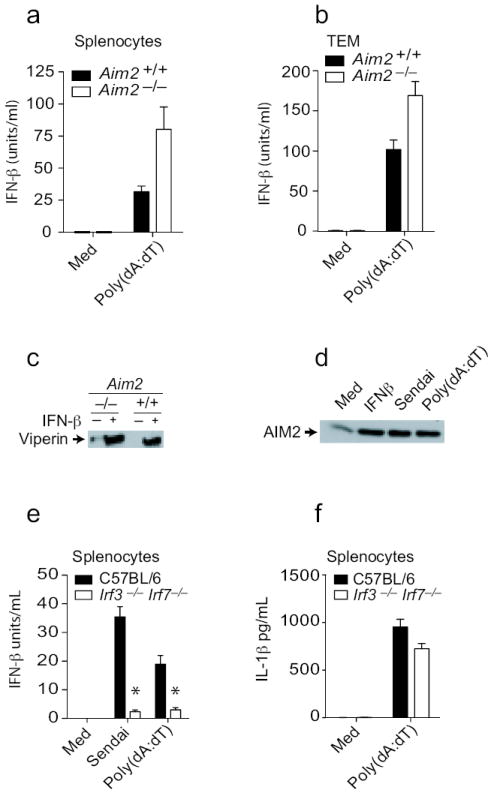
Type I interferon production and signaling remain intact in Aim2-deficient cells. A and e-f. Splenocytes from _Aim_2+/+, _Aim_2-/-, Irf3-/- Irf7-/- and C57Bl/6 mice were plated at 2 × 105 cells/well in 96-well plates in antibiotic free DMEM and were treated as indicated above. After 24 h, supernatants were collected and analyzed for IFN-β concentrations. b. TEM from _Aim_2+/+, _Aim_2-/-, _Irf_3-/- _Irf_7-/-, and C57BL/6 mice were treated as indicated above and supernatants were analyzed for IL-1β and IFN-β concentrations 6 h later. c. BMDM from _Aim_2+/+ and _Aim_2-/- mice were treated with 1000 units/ml IFNβ for 18 h and the viperin protein level in the lysates was analyzed by immunoblotting. d. TEM from C57Bl/6 mice were treated with 1000 units/ml IFNβ, 200 HA units/ml Sendai virus, and poly(dA-dT)1.5 μg/106 cells for 18 h and the Aim2 protein amounts in lysates was analyzed. Asterisks indicate P values of < 0.05 for Aim2+/+ versus _Aim2_-/-. Data are presented as mean ± SD from one experiment representative of 3 experiments.
AIM2 is a type I IFN inducible gene21. While human monocytes do not express AIM2, except after IFN induction10, mouse macrophages and splenocytes express Aim2 constitutively (Fig. 4d and data not shown) and the expression of Aim2 was further increased upon treatment of BMDM with IFN-β, poly(dA:dT) or after infection with Sendai virus (Fig. 4d). Previous work had suggested that type I IFN signaling was essential for inflammasome activation in response to F. tularensis and L. monocytogenes22, 23. To define the requirement for IFN production and/or signaling in Aim2 inflammasome activation, we monitored DNA induced IL-1β responses under conditions where IFN-β was not induced. Poly(dA:dT) induced a robust IFN-β response which was completely dependent on Irf3 and Irf7 (Fig. 4e). To determine if IFN-β production following poly(dA:dT) treatment was required for Aim2 inflammasome activation, we monitored IL-1β release in these cells. In contrast to IFN-β production, IL-1β production was not defective in Irf3 and Irf7-double-deficient cells in response to poly(dA:dT) (Fig. 4f). These observations indicate that although DNA treatment leads to type I IFN production and elevated expression of Aim2, these events are dispensable for Aim2 inflammasome activity.
Aim2–dependent IL-1β responses to bacteria
We next examined the role of Aim2 in regulating inflammasome activation in response to microbial pathogens. F. tularensis (Ft) is a pathogenic bacterium whose virulence is linked to its ability to replicate within the host cell cytosol24. Entry of the organism into the cytosol from the phagosome triggers the release of type I IFNs, IL-1β and IL-18, as well as caspase-1–dependent cell death22, 23, 25. Although production of inactive pro-IL-1β in response to Ft Live Vaccine Strain (LVS) is dependent on Tlr222, 26, cleavage of pro-IL1β into its active mature form requires both entry into the cytoplasm and engagement of a previously unknown cytosolic receptor22, 23, 25. These events are dependent upon Asc25 but independent of Nlrp127, 28, Nlrc425 or Nlrp327, 28. Additionally, an intact type I IFN response was shown to be critical for this response22, 23. To test the role of Aim2 as a candidate mediator of this response, we infected Aim2+/+ and _Aim2_-/- macrophages with Ft LVS and assayed cleavage of caspase-1 and release of IL-1β. While Ft LVS strongly activated caspase-1 and IL-1β release in Aim2+/+ cells, the loss of Aim2 ablated these responses (Fig. 5a and b). Priming of macrophages with LPS or other TLR ligands was not required in order to observe Ft LVS-induced caspase-1 activation, as Ft recognition by Tlr2 regulates pro-IL1β gene induction22, 26, 29. To confirm that the lack of secreted IL-1β by _Aim2_-/- cells was due to impaired inflammasome activation and not due to reduced production of pro-IL-1β̣ we also evaluated the induction of IL-1β mRNA by qPCR. Infection with Ft LVS led to equivalent induction of pro-IL-1β mRNA in Aim2+/+ and _Aim2_-/- cells (Fig. 5c).
Figure 5.
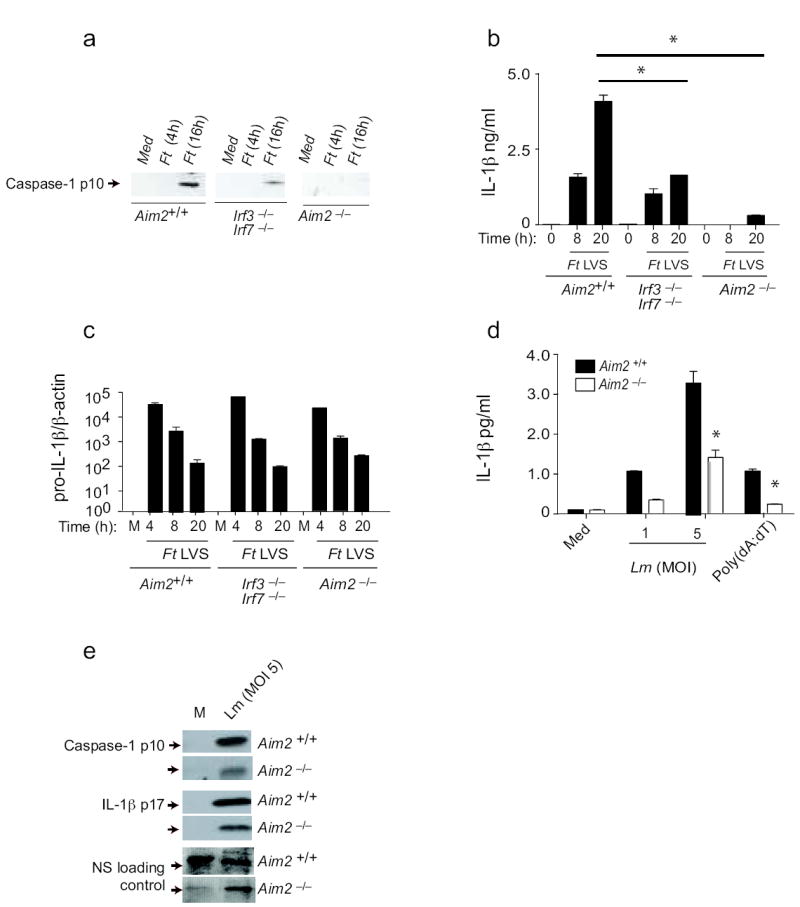
Aim2 mediates inflammasome activation in response to bacterial pathogens. TEM from _Aim_2+/+, _Aim_2-/-, _Irf_3-/- _Irf_7-/- and C57Bl/6 mice were infected with F. tularensis LVS (MOI = 50) as indicated in the materials and methods. At indicated time points after infection, cleaved caspase 1 (a) and IL-1β (b) in the supernatants and the mRNA levels of pro-IL-1β (c) were measured. d-e. LPS primed _Aim_2+/+ and _Aim_2-/- TEM were infected with L. monocytogenes at MOI of 1 and 5 and supernatants were analyzed for secreted IL-1β by ELISA (d) and cleaved caspase 1 and IL-1β by immunoblotting (e). NS; nonspecific band. Asterisks indicate P values of < 0.05 for Aim2+/+ versus _Aim2_-/-. Data are presented as mean ± SD from one experiment representative of 2 or 3 experiments.
Ft is known to induce IFN-β and previous work has shown that IFN®-deficient22 or IFNα/βR-deficient23 macrophages failed to induce inflammasome activation by Ft, indicating a requirement for an intact IFN response. We therefore also examined _Ft_-induced caspase-1 cleavage and IL-1β release in macrophages lacking Irf3 and Irf7. These cells had a blunted but not a defective response (Fig. 5a and b), in contrast to published studies with IFN-β– or IFNα/βR-deficient mice22, 23. While these combined data clearly indicate that Ft LVS-mediated activation of the inflammasome is dependent on type I IFN signaling and Aim2, they also suggest that in the absence of Irf3 and Irf7, Ft can still induce some IFN-β production, which is sufficient to trigger Aim2. Indeed, Ft LVS still induced IFN-β production albeit reduced in macrophages lacking Irf3 and Irf7 (data not shown). Precisely, how Ft induces type I IFNs is still unclear, although DNA sensing mechanisms have been proposed. The sensor responsible for these events has not been identified.
L. monocytogenes can also trigger inflammasome activity, although the mechanisms involved are not well established. Like _Ft, L. monocytoge_nes also triggers type I IFN responses and IL-1β upon entry to the cytosol. The Nlrp3, Nlrc4 and another as yet-to-be-identified Asc-dependent inflammasome pathway have all been implicated 30-33. To define the role of Aim2 in response to Listeria, macrophages from Aim2+/+ and _Aim2_-/- mice were infected and caspase-1 cleavage as well as IL-1β maturation and release measured. In all cases, these responses were reduced but not abrogated in Aim2-deficient cells (Fig. 5d and e). We observed similar findings in DCs (data not shown). These data indicate therefore that inflammasome activation by L. monocytogenes is only partially dependent on Aim2 signaling and additional pathways also contribute to this activation, consistent with published studies30-33.
Aim2-dependent IL-1β responses to DNA viruses
To examine the role of Aim2 in inflammasome activation following DNA virus infection, we first examined responses in cells infected with mCMV. mCMV induced IL-1β release in BMDCs and this response was entirely dependent on Aim2 (Fig. 6a). Similar data were obtained when thioglycollate elicited macrophages were examined (data not shown). Activation of caspase-1 and maturation of IL-1β in response to mCMV were also Aim-2-dependent (Fig. 6b). mCMV also induced IFN-β and this response was not affected in _Aim_2-deficient BMDM (Fig. 6c), but was ablated in Irf3 and Irf7 double-deficient BMDM (Fig. 6d).
Figure 6.
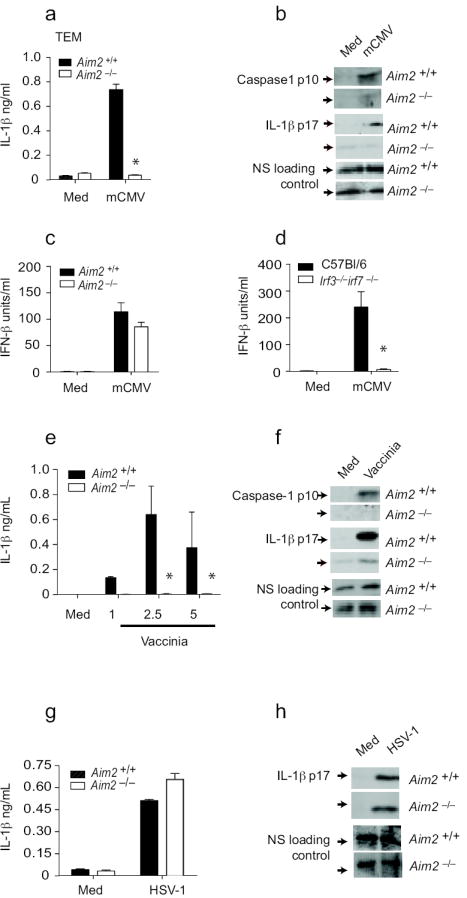
Aim2 is essential for inflammasome activation by DNA viruses. a-b. _Aim_2-/- and _Aim_2-/- TEM were primed with LPS for 3 – 4 h and stimulated with mCMV at an MOI of 10 for 20 h. IL-1β concentrations were measured by ELISA and cleaved caspase 1 p10 and IL-1β p17 in the supernatants by immunoblotting. c-d. TEM from _Aim_2+/+, _Aim_2-/-, _Irf_3-/- _Irf_7-/- and C57Bl/6 mice were stimulated with CMV as indicated above and IFN-β concentrations were measured by ELISA. e-f. _Aim_2+/+ and _Aim_2-/- BMDM and BMDCs were infected with vaccinia virus as described for mCMV and the supernatants were analyzed for caspase 1 and IL-1β. g-h. _Aim_2+/+ and _Aim_2-/- TEM were LPS primed and treated with HSV-1 at an MOI of 40 and at 18 h post infection, IL-1β concentrations in the supernatants were determined by ELISA and immunoblotting. NS; nonspecific band. Asterisks indicate P values of < 0.05 for _Aim_2+/+ versus _Aim_2-/-. Data are presented as mean ± SD from one experiment representative of 3 experiments.
Vaccinia virus (VV) also triggered the Aim2 inflammasome. VV-induced IL-1β release, caspase-1 cleavage and IL-1β maturation were all attenuated in macrophages lacking Aim2 (Fig. 6e and f). Similar findings were made when BMDCs were tested (data not shown). Herpes Simplex Virus-1 (HSV-1) is also a strong inducer of IL-1β in macrophages, however, in contrast to VV and mCMV, maturation and release of IL-1β in response to HSV-1 was normal in cells from _Aim2_-/- mice (Fig. 6g and h). Collectively these data indicate that Aim2 is important in driving IL-1β maturation and release in response to DNA viruses. How Aim2 differentially senses mCMV and VV but not HSV-1 is unclear.
Aim2-dependent responses to mCMV infection in vivo
IL-18 possesses biochemical similarities to IL-1β̣ but functionally it is more similar to IL-12. IL-18 and IL-12 synergize to stimulate IFN-γ production from NK cells34, events that are crucial in early defense against mCMV infection. IL-18R-deficient mice have dramatically reduced IFN-γ amounts in both the systemic circulation and the spleen following Mcmv infection35. To define the role of Aim2 in vivo, we infected C57BL/6, _Asc_-/-, Aim2+/+ and _Aim2_-/- mice with mCMV and first monitored IL-18 production. In agreement with published reports35, IL-18 was readily detectable in the sera of wild-type C57BL/6 mice 36 hours after infection with mCMV (Fig. 7a). IL-18 was not detected in the sera of mCMV-infected mice deficient in Asc (the serum concentrations in _Asc_-/- mice were below the limit of detection of this assay, 25.6 pg/ml). Serum concentrations of IL-18 during mCMV infection were reduced 6-fold (p = 0.026, n = 4) in _Aim2_-/- mice relative to Aim2+/+ littermate controls (Fig. 7b), whereas systemic TNF-α concentrations were unaffected (66 ± 30 pg/mL (Aim2+/+) vs. 56 ± 16 pg/mL (_Aim2_-/-), p = 0.56, n = 4) (data not shown). These results suggest that Aim2 and Asc are required for systemic induction of IL-18 but not TNF-α during mCMV infection in vivo.
Figure 7.

Aim2- and Asc-dependent IL-18 production and NK cell IFN-γ production. a-b. Serum IL-18 concentrations in C57BL/6, _Asc_-/-, _Aim_2+/+ and _Aim_2-/- mice infected with MCMV. c Proportions of NK1.1+ NKp46+ CD3- NK cells in the spleens of infected mice as well as an uninfected C57BL/6 control. d and e At 36 h p.i., splenocytes were cultured ex vivo in medium containing brefeldin A for 4 h with no additional stimuli. Intracellular IFN-γ (d) and surface CD69 (e) expressions were determined on total splenic NK cells. f-g. Splenocytes cultured in either medium without additional stimuli or medium containing PMA (50 ng/mL) and ionomycin (500 ng/mL) (denoted P + I) were stained for surface Ly49H and intracellular IFN-γ. Representative staining of gated NK cells is shown for (f) C57BL/6 and _Asc_-/- as well as (g) _Aim_2+/+ and _Aim_2-/- mice. Numbers in upper quadrants are proportion of Ly49H+ or Ly49H- NK cells that are IFN-γ+. Proportions of Ly49H+ and Ly49H- NK cells expressing IFN-γ are shown for individual mice in lower plots. In lower panels, black squares are C57BL/6 mice and white squares are _Asc_-/-, while black circles are _Aim_2+/+ and white circles are _Aim_2-/- (h-i) Viral titers in the spleen at 36 h p.i. nd; not detectable. P values were determined by unpaired two-tailed Student’s T test. Data are from one experiment representative of 2 experiments.
Based on the important role of IL-18 in the induction of systemic and splenic IFN-γ production during mCMV infection, we next evaluated ex vivo IFN-γ production by NK cells in the spleens of mCMV-infected mice36. The percentages of NK1.1+ NKp46+ CD3ε- NK cells in the spleens of C57BL/6, _Asc_-/-, Aim2+/+ and _Aim2_-/- mice were similar following mCMV infection (Fig. 7c and supplemental fig. 3). However, C57BL/6 and Aim2+/+ mice had significantly higher numbers of IFN-γ producing NK cells ex vivo, as determined by intracellular staining, _Asc_-/- and _Aim2_-/- mice had substantially reduced numbers of IFN-γ producing NK cells (29% and 52% less than controls, respectively) (Fig. 7d). Diminished numbers of IFN-γ producing NK cells could not be attributed to reduced NK cell activation as similar proportions of splenic NK cells expressed CD69, an activation marker in response to infection in _Asc_-/- and _Aim2_-/- relative to C57BL/6 and Aim2+/+ controls, respectively (Fig. 7e). Of note, NK cell CD69 expression was enhanced in Aim2+/+ and _Aim2_-/- mice (mixed 129×B6 background) relative to mice on a pure C57/Bl6 background (supplemental fig. 3).
In C57BL/6 mice, which are resistant to mCMV, NK cell control of early virus replication is mediated in large part through recognition of mCMV m157 protein by the activating NK cell receptor, Ly49H. Although the expression (MFI) of Ly49H expression was slightly reduced in both Aim2+/+ and _Aim2_-/- littermates on a mixed 129×B6 background compared to C57BL/6 and _Asc_-/- mice (supplemental Fig. 3), there were no significant differences in the proportion of Ly49H+ NK cells within paired groupings of 129×B6 littermates (48 ± 9 (Aim2+/+) vs 52 ± 4 (_Aim2_-/-), p = 0.45, n = 4) or C57BL/6 background mice (68 ± 4 (Aim2+/+) vs 68 ± 1 (_Aim2_-/-), p = 0.98, n = 4). Importantly, both the Ly49H+ and Ly49H- subsets of splenic NK cells displayed reduced spontaneous ex vivo IFN-γ production in mCMV-infected _Asc_-/- (Fig. 7f) and _Aim2_-/- (Fig.7g) mice relative to C57BL/6 and Aim2+/+, respectively. Thus, the reduced IFN-γ response from NK cells in Aim2 inflammasome defective mice (_Asc_-/- and _Aim2_-/-) could not be attributed to the reduction of cells that can be activated by mCMV-encoded ligands for Ly49H. Following in vitro PMA and ionomycin stimulation, NK cells (both Ly49H+ and Ly49H-) from spleens of mCMV-infected C57BL/6 and _Asc_-/- (Fig. 7f) as well as Aim2+/+ and _Aim2_-/- littermate mice produced similarly high amounts of IFN-γ (Fig. 7g). These data indicate that NK cells in _Asc_-/- and _Aim2_-/- mice are not intrinsically defective, but rather that their impaired IFN-γ responses are likely due to the decreased systemic IL-18 production during mCMV infection.
As NK cell-derived IFN-γ is critical for early control of mCMV replication in vivo, we next determined viral loads in the spleens of infected mice at 36 hours post-infection. Relative to C57BL/6 mice, _Asc_-/- mice demonstrated elevated viral titers at this early time point (Fig. 7h). Since mice of mixed background have higher viral titers relative to B6 mice, to evaluate the role of Aim2 in viral control, we used a lower dose (1 × 105, rather than 1 × 106 PFUs of mCMV). Like the _Asc_-/- mice, Aim2-/- mice demonstrated dramatically elevated viral titers at this early time point (Fig. 7i). Importantly, IL-18 levels in the serum and NK cell-dependent IFNγ production were reduced in _Aim_2-/- relative to _Aim_2+/+ at this lower dose (data not shown). Collectively, these data reveal the essential role of the Aim2-Asc inflammasome in early control of mCMV infection through IL-18 and NK-cell dependent IFN-γ production.
Discussion
Considerable progress has been made in defining the molecular basis for the immune stimulatory activity of microbial DNA. TLR9 recognizes CpG-DNA and resides in the endosomal compartment (reviewed in 37). Additional TLR9-independent pathways have also emerged. These include an IFN inducing-pathway and a pathway leading to the conversion of the zymogens pro-IL-1β and IL-18 into the active mature cytokines. The IFN inducing pathway is complex and involves RNA polymerase III, and one or more redundant sensors, which collectively converge on a signaling axis involving STING38, TANK Binding Kinase-139 and the Interferon Regulatory Factors (IRF3 and IRF7) (reviewed in 37). The IL-1-IL-18 pathway was recently shown to involve AIM2, which binds its ligand DNA directly and engages ASC to form a caspase-1-activating inflammasome (ref 9-12). Whether AIM2 is the sole sensor for this pathway is unknown.
Herein, we have characterized the role of Aim2 in DNA responses by generating mice lacking Aim2. By monitoring IL-1β responses, our studies reveal the essential and non-redundant role for Aim2 in DNA-dependent caspase-1 activation and IL-1β-IL-18 maturation in both macrophages and dendritic cells. Although overproduction of IL-1β can have deleterious consequences for the host if not well-regulated13, the importance of IL-1β in anti-microbial defenses is highlighted by the enhanced susceptibility of IL-1R-deficient mice to various pathogens40, 41 and by the ability of viruses to evade the production and/or action of these cytokines42-44. By studying IL-1β responses in _Aim2_-deficient mice, we revealed a central role for Aim2 in regulating IL-1β in response to F. tularensis LVS. Previous work had revealed the importance of type I IFNs, ASC and caspase-1 in innate responses to Ft25. However, the precise mechanisms by which Ft triggered caspase-1 activation were unclear22, 45. Our studies identify Aim2 as the sole mediator of inflammasome activation by Ft. Ft also elicits IFN-β production, events which are not mediated by Aim2. The sensor regulating IFN-β induction and whether DNA is also the activating ligand is unknown. Our data also reveal a contribution of the Aim2 pathway in sensing L. monocytogenes. In this case the role of Aim2 is partial, consistent with redundant NLR sensing mechanisms. Indeed, Aim2 and Nlrp3 appear to collaborate for sensing L. monocytogenes, since knockdown of Aim2 in Nlrp3-deficient macrophages abolished all inflammasome activity (S. Kim, F., Bauernfeind, A. Ablasser, G. Hartmann, K. Fitzgerald, E., Latz and V., Hornung, manuscript in press E. J. I).
The most important finding from our data is the observation that the Aim2-Asc pathway is crucial for caspase-1 dependent signaling and innate immunity to DNA viruses, particularly mCMV. Using a mouse model of mCMV, we found a critical role for Aim2 and Asc in regulating IL-18 production and NK cell-dependent IFN-γ production. These events are critical for the early innate response to infection, as reflected by the higher viral titres in Aim2 and _Asc_-deficient mice relative to their wild type counterparts. It is important to note that _Aim2_-/- and _Aim2_-/- mice are on a mixed 129×B6 background, which are more susceptible to mCMV than C57BL/6 mice, which encode Cmv-1, an autosomal dominant gene on chromosome 6 conferring mCMV resistance46. Defects associated with DAP12 and possibly other signaling pathways on the 129 background may also contribute to higher susceptibility of 129×B6 mice to mCMV47. In our studies, the use of Aim2+/+ littermate controls (derived from breedings of heterozygous parents), which showed similar expression of the NK cell receptors (NK1.1 and Ly49H; the activating NK receptor encoded by the Cmv-1 locus on chromosome 636, 48, 49) on both Aim2+/+ and _Aim2_-/- littermates, permitted us to properly control for the influence of genetic background on resistance to mCMV and assess the importance of Aim2 in early antiviral responses. Moreover, the similar reductions in systemic IL-18 and NK cell-dependent IFN-γ production as well as the enhanced viral burden in _Asc_-/- mice fully backcrossed onto the C57BL/6 background, support our argument that the Aim2- and Asc-dependent induction of IL-18 contributes to early control of mCMV replication. Moreoever, these data are consistent with published studies in IL-18R-deficient mice35. A key question rising from these studies relates to how Aim2, which is localized to the cytosolic compartment, gains access to mCMV DNA, since mCMV replicates in the nucleus. Whether the incoming virion or newly synthesized virions are being sensed is unclear at present. One possibility is that as virions are disassembled or assembled, DNA may gain access to the cytosol if the capsid proteins fail to perfectly protect/sequester the DNA.
While it is not surprising that organisms have evolved mechanisms to sense microbial DNA and trigger innate immunity, our understanding of how cells discriminate between self and non-self DNA is still limited. Normal defense strategies that discriminate between host and pathogenic nucleic acids are not perfect and fail under certain circumstances. This perturbation in normal control may contribute to the pathogenesis of autoinflammatory and autoimmune diseases such as Systemic Lupus Erythematosis (reviewed in 37). Normally, mammalian DNA is sequestered in the nucleus or mitochondria of cells. An exception however is during replication or cell death when the integrity of the nuclear envelope is compromised thereby exposing the cytosolic compartment to free DNA. DNA found free outside cells, within endosomes or within the cytosol is degraded by DNaseI, DNaseII and DNaseIII (also called TREX1). Failure of these systems to function (due to mutation, deletion etc) can lead to aberrant accumulation of DNA and inappropriate immune activation. Whether Aim2 senses aberrant host DNA and thus contributes to the pathogenesis of SLE awaits investigation. Further characterization of the Aim2 inflammasome, will likely yield new insights into microbial pathogenesis as well as autoinflammatory and autoimmune diseases and enable the rational design of new therapies and treatments for a number of debilitating conditions.
Materials and Methods
Reagents
ATP, LPS, nigericin and poly(dA-dT) were from Sigma-Aldrich. Anthrax lethal toxin was from List Biologicals (5 μg·ml–1 PA and 5 μg·ml–1 LF). Vaccinia virus (Western Reserve strain) was from G. Barton (U. of Berkeley, CA). MCMV (Smith strain) was from R. Welsh (UMASS Medical School, MA). HSV-1 was from D. Knipe, (Harvard Medical School, Boston, MA). Sendai virus (Cantrell strain) was from Charles River Laboratories (Boston, MA). Francisella tularensis (Ft) Live Vaccine Strain (LVS) (ATCC 29684) was prepared as described50. Salmonella typhimurium (SL1344 lab strain) was from Mary O’Riordan (U. Of Michigan, Ann Arbor, MI). L. monocytogenes (clinical isolate 10403s) was from V. Boyartchuk (UMASS Medical School, MA). Anti-murine Aim2 polyclonal antibody was from E. Alnemri (Thomas Jefferson University, PA).
Plasmid constructs
Full length human AIM2 was as described10. Full length murine Aim2 and murine Asc were from Invivogen (San Diego, CA). Expression plasmids (pCI) encoding human Caspase-1 and ASC-HA were from Millenium Pharmaceuticals (Cambridge, MA)10.
Mice
ES cells harboring a genetrap insertion in the _Aim_2 gene were from the international genetrap consortium and were microinjected into C57BL/6 blastocysts. Chimeric offspring were backcrossed to C57Bl/6 mice and germline transmission was confirmed by PCR with genetrap specific primers. Long PCRs were performed with a 5’ exon5 primer combined and a 3’ genetrap vector primer to amplify the gene trap integration site in the Aim2 gene and the amplified bands were examined by sequencing. This revealed that the gene-trap vector inserted into intron 6 of the Aim2 gene. Due to duplication of part of Aim2 gene at the 3’ end of the integration site, we were unable to distinguish heterozygous from homozygous KO mice by PCR on genomic DNA. To definitively determine homozygous KOs, we performed reverse transcription PCR on RNA extracted from blood with following primers: F- CACACTCGACGTGGCAGATAGGAC; R1- CAGCACCGTGACAACAAGTGG; R2- TCGATGCGATCTGCGTTCTTC. Primers F and R1 amplified wild type _Aim_2 transcripts and F and R2 amplifies Genetrap-inserted _Aim_2 transcripts. _Pycard (Asc)_−/− mice were from Millennium Pharmaceuticals and were backcrossed into C57Bl/6 background. _Irf_3-/- _Irf_7-/- DKO mice were generated at UMASS Medical School from _Irf_3-/- and _Irf_7-/- mice obtained from T. Taniguichi (Tokyo, Japan). C57BL/6 mice were from Jackson Laboratories (Bar Harbor, ME) and were bred at UMASS. All mouse strains were bred and maintained under specific pathogen-free conditions in the animal facilities at the UMASS Medical School and were carried out in accordance with the guidelines set forth by the University of Massachusetts Medical School Department of Animal Medicine and the Institutional Animal Care and Use Committee.
Cell culture and stimulation
Thioglycollate elicited peritoneal macrophages (TEM) were isolated from mice 4 days after i.p. injection of sterile 3% thioglycollate (Remel, Lenexa, KS). BMDM and BMDCs were generated as described10, 51. Cells were first primed with 200 ng/ml LPS for 3-4 h prior to treatment with different agents. ATP (5 mM) or Nigericin (10 μM) was added one h prior to harvesting supernatants. Poly(dA-dT) DNA was transfected using Lipofectamine 2000 at a concentration of 1.5 μg/ml. The doses of viruses used were as follows unless otherwise stated; vaccinia virus, MOI of 0.5 – 5; CMV, MOI of 5 for splenocytes and 10 for macrophages and DCs; HSV, MOI of 10 or 40. For experiments involving F. tularensis (Ft) LVS, S. typhimurium or L. monocytogenes, cells were cultured in antibiotic-free medium unless otherwise indicated. Macrophages were infected with Ft LVS (MOI = 50) for 4 h. After washing twice with PBS, infected cells were incubated for 45 min in medium containing 50μg/ml gentamicin. Cells were washed twice with PBS and then incubated with serum free medium. For Listeria, cells were stimulated with MOI of 1 and 5 for 1 h and then medium was replaced with 100 μg/ml gentamicin containing medium. For Salmonella, 2 h post-infection, supernatants were collected and 50μg/ml gentamicin containing medium was added. After 1h, the medium was again replaced with 10μg/ml gentamicin containing medium and supernatants harvested after 3h.
ELISA
Cell culture supernatants were assayed for IL-1β (BD Biosciences, Franklin Lakes, NJ), IL-18 (R & D Systems, Piscataway, NJ) by ELISA. Murine IFNβ sandwich ELISA was used as previously described52.
Immunoblotting
Supernatants were harvested and precipitated by methanol chloroform extraction, and cell lysate was collected. Immunoblotting was conducted as previously described2,3, using anti-murine caspase-1 p10 (sc-514, Santa Cruz Biotechnology) or anti-murine IL-1β (R&D systerms, Minneapolis, MN), rabbit polyclonal anti-murine Aim2 (E. Alnemri), anti-Flag (M2, Sigma), anti-Viperin (P. Cresswell, Yale, CT), anti-human β-actin (Sigma).
Quantitative real-time PCR for pro-IL1b as described previously22.
Reporter assays
All reporter gene assays were conducted as described10.
Cell Viability Assay
_Aim_2+/+ and _Aim_2-/- cells were treated with poly(dA-dT) as described above and the cell viability was assessed at 24 h by calcein staining as described 10.
In vivo mCMV infection
The Smith strain of mCMV was propagated in vivo in salivary glands of Balb/c mice53. Infected salivary gland homogenates were diluted in HBSS and mice were inoculated intraperitoneally with 1 × 106 plaque-forming units (PFUs) or 1 × 105 PFUs (Fig 7i), as titrated on mouse alpha-1,3-galactosyltransferase knockout fibroblast monolayers.
Immunofluorescence Staining and Intracellular IFN-γ Assay
Leukocytes were isolated and stained for the intracellular IFN-γ using reagents and the protocol of BD PharMingen. Two million cells were cultured in medium containing GolgiPlug at 0.2 μl per sample in the presence or absence of 50 ng/ml PMA (Sigma-Aldrich) and 500 ng/ml ionomycin (Sigma-Aldrich) for 4 h at 37°C. Cells were washed using FACS® buffer and incubated for 5 min at 4°C in 100 μl of FACS® buffer plus 0.5 μl of anti-FcγII receptor Ab. Cells were then washed and incubated at 4°C for 30 min with combinations of anti-NK1.1-PerCPCy5.5 (BD PharMingen), anti-CD3-FITC (BD PharMingen), anti-NKp46-PE (eBioscience), anti-CD8a-Alexafluor700 (eBioscience), anti-CD69-PE-C y 7 (BD PharMingen), and anti-Ly49H-Alexafluor647 (eBioscience) mAb. Samples were washed twice, fixed and permeabilized using 100 μl of cytofix/cytoperm (BD PharMingen) solution for 20 min at 4°C. A_ft_er fixation the samples were washed twice using perm/wash solution and stained with rat IgG1-Pacific Blue mAb to mouse IFN-γ (BD PharMingen) for 30 min at 4°C. Samples were then washed twice using perm/wash and once using FACS® buffer for analysis on a LSRII. Data analysis was done using FlowJo (Treestar Inc.) software. For these analyses, 100–200,000 events were calculated, ensuring a sizable NK cell population for a valid analysis of NK subsets.
Plaque assay
Viral loads in the spleen were titrated by plaque assay on mouse alpha-1,3-galactosyltransferase knockout fibroblast monolayers.
Statistical analysis
Two-way analysis of variance followed by the Bonferroni post-test was performed unless otherwise stated using Prism Software. P values of < 0.05 were considered significant.
Supplementary Material
1
Acknowledgments
The authors would like to thank A. Cerny for animal husbandry and genotyping, A. Poltorak for guidance and help with genotyping mice, R. Barbalat and G. Barton for VV, S. Schattgen for assistance with cell viability assays, M. Pickering and T. Kowolik for helpful discussions, E. Alnemri and T. Fernandez-Alnemri (Thomas Jefferson University, Philadelphia) for helpful discussions and the anti-Aim2 antibody and E. Lien for critical reading of the manuscript. This work is supported by NIH grants AI083713 (to K.A.F and E.L), CA66644 (to E. T. S) AI07349 (to S. W), CA034461 (to R. W), U54 AI-157168 (to S.N. V) and a NERCE Fellowship to V.A.R.
Footnotes
Author Contributions; K.A.F oversaw the whole project. K.A.F, V.A.R and S.W conceived the research with assistance from S.N.V,, E.L. and E.T.S. V.A.R, B.M. and V. H characterized the genetrap integration. V.A.R and L.W conducted all genotyping, V.A.R., Z.J and S.W designed and conducted the experiments with help from S.S., L.W., S.K.V., S.G. and L.E.C who conducted the Ft experiments. K. A. F, V.A.R, S.S and S.W wrote the manuscript.
Competing financial interest. The authors declare no competing financial interest.
References
- 1.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 3.Huysamen C, Brown GD. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol Lett. 2009;290:121–128. doi: 10.1111/j.1574-6968.2008.01418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoneyama M, Fujita T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007;18:545–551. doi: 10.1016/j.cytogfr.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 6.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 7.Chiu YH, Macmillan JB, Chen ZJ. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell. 2009 doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ablasser A, et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009 doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts TL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 10.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009 doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burckstummer T, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 13.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 14.Dinarello CA, Novick D, Rubinstein M, Lonnemann G. Interleukin 18 and interleukin 18 binding protein: possible role in immunosuppression of chronic renal failure. Blood Purif. 2003;21:258–270. doi: 10.1159/000070699. [DOI] [PubMed] [Google Scholar]
- 15.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 16.Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao EA, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludlow LE, Johnstone RW, Clarke CJ. The HIN-200 family: more than interferon-inducible genes? Exp Cell Res. 2005;308:1–17. doi: 10.1016/j.yexcr.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 19.Fernandes-Alnemri T, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poeck H, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 21.Landolfo S, Gariglio M, Gribaudo G, Lembo D. The Ifi 200 genes: an emerging family of IFN-inducible genes. Biochimie. 1998;80:721–728. doi: 10.1016/s0300-9084(99)80025-x. [DOI] [PubMed] [Google Scholar]
- 22.Cole LE, et al. Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. J Immunol. 2008;180:6885–6891. doi: 10.4049/jimmunol.180.10.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elkins KL, Cowley SC, Bosio CM. Innate and adaptive immunity to Francisella. Ann N Y Acad Sci. 2007;1105:284–324. doi: 10.1196/annals.1409.014. [DOI] [PubMed] [Google Scholar]
- 25.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole LE, et al. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect Immun. 2007;75:4127–4137. doi: 10.1128/IAI.01868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009;227:95–105. doi: 10.1111/j.1600-065X.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- 28.Kanneganti TD, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J Leukoc Biol. 2006;80:766–773. doi: 10.1189/jlb.0406294. [DOI] [PubMed] [Google Scholar]
- 30.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 31.Meixenberger K, et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol. 184:922–930. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- 32.Franchi L, Kanneganti TD, Dubyak GR, Nunez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282:18810–18818. doi: 10.1074/jbc.M610762200. [DOI] [PubMed] [Google Scholar]
- 33.Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–7564. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 35.Pien GC, Biron CA. Compartmental differences in NK cell responsiveness to IL-12 during lymphocytic choriomeningitis virus infection. J Immunol. 2000;164:994–1001. doi: 10.4049/jimmunol.164.2.994. [DOI] [PubMed] [Google Scholar]
- 36.Daniels KA, et al. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med. 2001;194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 10:123–130. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 38.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 40.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labow M, et al. Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol. 1997;159:2452–2461. [PubMed] [Google Scholar]
- 42.Alcami A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–167. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
- 43.Smith GL, Chan YS. Two vaccinia virus proteins structurally related to the interleukin-1 receptor and the immunoglobulin superfamily. J Gen Virol. 1991;72(Pt 3):511–518. doi: 10.1099/0022-1317-72-3-511. [DOI] [PubMed] [Google Scholar]
- 44.Johnston JB, et al. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23:587–598. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Henry T, Monack DM. Activation of the inflammasome upon Francisella tularensis infection: interplay of innate immune pathways and virulence factors. Cell Microbiol. 2007;9:2543–2551. doi: 10.1111/j.1462-5822.2007.01022.x. [DOI] [PubMed] [Google Scholar]
- 46.Scalzo AA, et al. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics. 1995;27:435–441. doi: 10.1006/geno.1995.1074. [DOI] [PubMed] [Google Scholar]
- 47.McVicar DW, et al. Aberrant DAP12 signaling in the 129 strain of mice: implications for the analysis of gene-targeted mice. J Immunol. 2002;169:1721–1728. doi: 10.4049/jimmunol.169.4.1721. [DOI] [PubMed] [Google Scholar]
- 48.Brown MG, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 49.Lee SH, et al. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet. 2001;28:42–45. doi: 10.1038/ng0501-42. [DOI] [PubMed] [Google Scholar]
- 50.Elkins KL, Winegar RK, Nacy CA, Fortier AH. Introduction of Francisella tularensis at skin sites induces resistance to infection and generation of protective immunity. Microb Pathog. 1992;13:417–421. doi: 10.1016/0882-4010(92)90085-3. [DOI] [PubMed] [Google Scholar]
- 51.Rathinam VA, Hoag KA, Mansfield LS. Dendritic cells from C57BL/6 mice undergo activation and induce Th1-effector cell responses against Campylobacter jejuni. Microbes Infect. 2008;10:1316–1324. doi: 10.1016/j.micinf.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts ZJ, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. 2007;204:1559–1569. doi: 10.1084/jem.20061845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bukowski JF, Woda BA, Welsh RM. Pathogenesis of murine cytomegalovirus infection in natural killer cell-depleted mice. J Virol. 1984;52:119–128. doi: 10.1128/jvi.52.1.119-128.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1