TDP-43 Mediates Degeneration in a Novel Drosophila Model of Disease Caused by Mutations in VCP/p97 (original) (raw)
Abstract
Inclusion body myopathy associated with Paget's disease of bone and frontotemporal dementia (IBMPFD) is a dominantly inherited degenerative disorder caused by mutations in the valosin-containing protein (VCP7) gene. VCP (p97 in mouse, TER94 in Drosophila melanogaster, and CDC48 in Saccharomyces cerevisiae) is a highly conserved AAA+ (ATPases associated with multiple cellular activities) ATPase that regulates a wide array of cellular processes. The mechanism of IBMPFD pathogenesis is unknown. To elucidate the pathogenic mechanism, we developed and characterized a Drosophila model of IBMPFD (mutant-VCP-related degeneration). Based on genetic screening of this model, we identified three RNA-binding proteins that dominantly suppressed degeneration; one of these was TBPH, the Drosophila homolog of TAR (trans-activating response region) DNA-binding protein 43 (TDP-43). Here we demonstrate that VCP and TDP-43 interact genetically and that disease-causing mutations in VCP lead to redistribution of TDP-43 to the cytoplasm in vitro and in vivo, replicating the major pathology observed in IBMPFD and other TDP-43 proteinopathies. We also demonstrate that TDP-43 redistribution from the nucleus to the cytoplasm is sufficient to induce cytotoxicity. Furthermore, we determined that a pathogenic mutation in TDP-43 promotes redistribution to the cytoplasm and enhances the genetic interaction with VCP. Together, our results show that degeneration associated with VCP mutations is mediated in part by toxic gain of function of TDP-43 in the cytoplasm. We suggest that these findings are likely relevant to the pathogenic mechanism of a broad array of TDP-43 proteinopathies, including frontotemporal lobar degeneration and amyotrophic lateral sclerosis.
Introduction
Inclusion body myopathy associated with Paget's disease of bone and frontotemporal dementia (IBMPFD; MIM167320) is a rare, complex, and ultimately lethal autosomal dominant disorder. Affected individuals exhibit variable penetrance of progressive degeneration of muscle, bone and brain caused by mutations in the gene encoding valosin-containing protein (VCP) (Watts et al., 2004) (Kimonis et al., 2008). The molecular chaperone VCP (also known as p97, TER94, and CDC48) is a member of the AAA+ family of proteins (ATPases associated with multiple cellular activities) that segregates ubiquitinated substrates from multimeric protein complexes or structures (Ye, 2006). VCP activity is essential for multiple cellular processes, including ubiquitin-dependent protein degradation, nuclear envelope construction, Golgi and endoplasmic reticulum assembly, and autophagosome maturation (Halawani and Latterich, 2006; Ju et al., 2009; Tresse et al., 2010). The molecular basis of degeneration resulting from VCP mutations is unknown, although ubiquitin-positive pathology is prominent in affected tissues (Guinto et al., 2007; Weihl et al., 2008). TAR (trans-activating response region) DNA-binding protein 43 (TDP-43) has been identified as a major component of the ubiquitin pathology (Neumann et al., 2007; Salajegheh et al., 2009).
TDP-43 is a predominantly nuclear heterogeneous nuclear ribonucleoprotein (hnRNP) that undergoes nucleocytoplasmic shuttling and associates with translation machinery in the cytoplasm (Ayala et al., 2008; Wang et al., 2008; Freibaum et al., 2009). TDP-43 is redistributed to the cytoplasm after neuronal injury where it associates with stress granules (Colombrita et al., 2009; Moisse et al., 2009a). TDP-43 redistribution to the cytoplasm is recognized as a pathological feature of several sporadic and inherited human diseases including IBMPFD, frontotemporal dementia, and amyotrophic lateral sclerosis (ALS), although the significance of this is unclear (Neumann et al., 2007; Geser et al., 2009; Salajegheh et al., 2009). TDP-43 redistribution has also been observed in vitro in cells expressing mutant VCP, although the role of TDP-43 in mediating disease has not been explored (Gitcho et al., 2009). The recent identification of disease-associated mutations in TDP-43 strongly implicates this protein in disease pathogenesis (Gitcho et al., 2008; Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008), although it is not known whether disease-associated cytoplasmic accumulation of TDP-43 is a mediator of pathology or a physiological response to it.
Here we present the first Drosophila melanogaster model of IBMPFD. Through genetic screening, we identified three RNA-binding proteins that suppress degeneration. One of these was TBPH, the fly orthologue of TDP-43. We show in vitro that expression of disease-causing VCP mutants leads to cytotoxicity and coincidental redistribution of TDP-43. To determine the significance of TDP-43 redistribution, we generated transgenic flies expressing wild-type (WT) and mutant forms of TDP-43. We demonstrate that VCP and TDP-43 interact genetically, that disease-causing mutations in VCP lead to redistribution of TDP-43 to the cytoplasm in vivo, and that redistribution of TDP-43 is sufficient to induce degeneration in vivo. Thus, our study provides the first evidence that toxic gain of function of TDP-43 in the cytoplasm plays a primary role in mediating the pathogenesis initiated by mutations in VCP.
Materials and Methods
Plasmids.
To generate pUAST-dVCP constructs, Drosophila VCP orthologue (dVCP) cDNA in the pBluescript SK− (pBluescript sequence variants) was generated using Stratagene's QuikChange Site-Directed Mutagenesis (SDM) Kit (Agilent Technologies), changing R to H at amino acid 152 (R152H) and A to E at amino acid 229 (A229E), and were subsequently subcloned into pUAST. To generate DsRed VCP, VCP cDNA was obtained from Origene and sequence variants R95G, R155H, R155C, R191Q, and A232E were generated using Stratagene's QuikChange SDM Kit. Wild-type and mutant VCP were then subcloned into the BglII/BamH1 cloning site of the pDsRed monomer-C1 vector (Clontech). TBPH cDNA was obtained from Origene and subcloned into the EcoRI/XhoI site of pUAST. WT, nuclear localization sequence (NLS)-mutant, and nuclear export sequence (NES)-mutant TDP-43 cDNAs were amplified from the previously described mammalian expression constructs (Winton et al., 2008). Using Stratagene's QuikChange SDM Kit, TDP-43 M337V (M to V at amino acid 337) was generated from WT TDP-43. All TDP-43 constructs were subcloned into the NotI/XhoI cloning site of pUAST.
Fly culture.
All Drosophila stocks were maintained on standard media in 25°C incubators. Double-strand RNA interference (RNAi) lines targeting TBPH (ID38377), xl6 (ID31202, ID31203), and Hrb27C (ID16040, ID16041) were obtained from the Vienna Drosophila RNAi Center. Flies transgenic for UAS-dVCP (WT or mutant), UAS-TBPH, and UAS-TDP-43 (WT or mutant) were generated by injecting the constructs described above into embryos of w1118 using standard techniques.
Dominant modifier screen.
Deficiency (Df) lines for all four chromosomes obtained from the Bloomington Stock Center were used to identify dominant modifiers of mutant dVCP in a genetic screen. For the primary screen, balanced virgin female dVCP R152H (recombined with GMR-GAL4) flies were crossed with Df/balancer males from 270 deficiency lines, and progeny were examined for changes in eye phenotype (including color, ommatidia structure, and bristle formation). At least 10 progeny were examined and scored on a 20-point scale. Eyes were examined for the presence of: supernumerary interommatidial bristles (IOBs), IOBs with abnormal orientation, necrotic patches, a decrease in size, retinal collapse, fusion of ommatidia, disorganization of ommatidial array, and loss of pigmentation. Points were added if there was complete loss of IOBs (+1), more than three small or one large necrotic patch (+1), retinal collapse extended to the midline of the eye (+1) or beyond (+2), loss of ommatidial structure in <50% (+1) or >50% (+2) of the eye, and if pigmentation loss resulted in change of eye color from red to orange (+1) or pale orange/white (+2). GMR-GAL4; UAS-dVCP R152H/Balancer served as an internal control. In a secondary screen, to filter nonspecific modifiers of cell death, deficiencies defined as hits (either enhancing or suppressing the dVCP mutant phenotype) were then crossed with flies expressing the proapoptotic gene Reaper (recombined with GMR-GAL4). Any hits that similarly modified dVCP R152H and Reaper in the secondary screen were excluded from additional study because of the possibility of nonspecific anti-apoptotic effects. As regions of interest were identified from the primary and secondary screens, additional Df lines were obtained that overlapped with interacting deficiencies to verify and refine the position of potential modifiers. For the final step of gene identification, individual RNAi lines corresponding to the genes within the candidate intervals were obtained from the Vienna Drosophila RNAi Center. GMR-GAL4; UAS-dVCP R152H females were crossed with males from the RNAi lines, and the progeny eyes were evaluated for changes. A modifier was defined as an RNAi line that replicated the enhancement or suppression of the corresponding deficiency.
Cell culture and transfection.
Human embryonic kidney 293T (HEK293T) cells were grown in DMEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% l-glutamate. HEK293T cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Primary cortical neurons were cultured from postnatal day zero C57BL/6J pups. Briefly, pups were decapitated into Hanks medium without Ca2+ and Mg2+, and cortices were dissected in Neurobasal-A medium supplemented with 10 mm HEPES. After dissection, cortices were trypsinized for 25 min at 37°C and dissociated. Neurons were plated in Neurobasal-A with B27 supplement at a density of 2.5 × 105 cells per well in four-well chamber CC2 slides (Nalge-Nunc). After 4 d in vitro, cells were transfected with 1 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer.
Immunoblots.
To examine soluble and insoluble fractions of TDP-43 in cells, HEK293T cells were harvested at the indicated times, and radioimmunoprecipitation assay (RIPA)-soluble and -insoluble fractions were prepared as described previously (Winton et al., 2008). To examine proteins expressed in flies, three fly heads (or thoraces) of the appropriate genotype were lysed in 20 μl of RIPA. For immunoblots, 20 μg of cell lysates or lysate from 3 fly head equivalents were resolved on 10% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad), and immunoblotting was performed as described previously (Pandey et al., 2007). Mouse monoclonal anti-VCP antibody (Affinity BioReagents) was used at 1:10,000, rabbit polyclonal anti-TDP-43 antibody raised against recombinant TDP-43 (Protein Tech Group) at 1:1000, mouse monoclonal anti-tubulin (Sigma) at 1:10,000, and rabbit polyclonal anti-actin (Santa Cruz Biotechnology) at 1:3000. Primary antibodies were detected with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch), and proteins were visualized using Immobilon Western Chemiluminescent AP Substrate (Millipore).
Real-time quantitative PCR.
Total RNA was isolated from 10 animals of the appropriate genotype with TRIzol reagent (Invitrogen), and cDNA was generated using the iScript cDNA Synthesis kit (Bio-Rad) following the manufacturer's protocol. The concentration of each primer probe set was individually optimized. Quantitative real-time PCRs were performed in a total reaction volume of 25 μl of iQ Supermix (Bio-Rad) using Bio-Rad iCycler iQ5. Transcript levels were normalized to Drosophila glyceraldehyde 3 phosphate dehydrogenase 2 (dGAPDH2). The primer/probe sets for genes purchased from Applied Biosystems were as follows: Hrb27C, Dm01803323_g1; xl6, Dm01803314_m1; TBPH, Dm01820181_g1; and GAPDH2 control, Dm01843776_S1.
Cytotoxicity assays.
To assess toxicity in HEK293T cells, cells were harvested at the indicated times with Versene, rinsed with PBS, and resuspended in PBS plus 1% FBS. TO-PRO-3 (Invitrogen) in 1 mm DMSO was diluted 1:50,000 in PBS, and 50 μl was added to 450 μl of the cell suspension. After incubation for 1–2 min, cells were analyzed by fluorescence-activated cell sorting. Transfected cells were identified by DsRed fluorescence, and dead cells were identified by TO-PRO-3 fluorescence. Cell viability was calculated and expressed as described previously (Taylor et al., 2003). To assess toxicity in primary cortical neurons, we transfected these cells on day 4 in vitro with DsRed-conjugated WT or mutant VCP. Twenty-four or forty-eight hours after transfection, neurons were immunostained for MAP2 and stained with 4′,6′-diamidino-2-phenylindole (DAPI) to visualize nuclear morphology as described above. A blinded investigator scored VCP-expressing neurons for the presence or absence of toxicity. Only neurons with both condensed nuclei and loss of MAP2 staining were scored positive for cytotoxicity. More than 100 neurons from at least three trials were analyzed and results were compared by using the Student's t test with a significance threshold of p < 0.05.
Transcription assays.
HEK293T cells were transfected with firefly luciferase reporter constructs [HIV-1 long terminal repeat—luciferase reporter plasmid pLTR, pSP-10, or pSP-10D] combined with 100 ng of Renilla luciferase reporter pRL-CMV and 0.5 μg of VCP WT, VCP R155H, VCP A232E, or empty vector control (pcDNA 3.1) by using Fugene (Roche) according to the manufacturer's protocol. Forty-eight hours after transfection, cell lysates were analyzed using the Stop and Glo dual reporter system (Promega) in a 96-well format with a Vector3 luminometer, as directed by the Promega protocol. Firefly luciferase activity was then normalized to the Renilla luciferase activity to control for transfection efficiency. Data were then normalized to activity in cells transfected with empty vector control, which was given a value of 1.
Histology.
For immunofluorescence analysis in cell culture, HEK293T and HeLa cells were washed twice with PBS, fixed with 4% PFA for 10 min, washed twice with PBS, then permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked with 0.2% Triton X-100/5% goat serum in PBS for 30 min. Cells were incubated with primary antibody (rabbit polyclonal anti-TDP-43 antibody at 1:200 and mouse monoclonal anti-MAP2 at 1:500, diluted in PBS containing 0.2% Triton X-100/5% goat serum) for 1.5 h and washed twice with PBS (15 min each). After the final wash, the cells were incubated with secondary antibody (diluted in PBS containing 0.2% Triton X-100/5% goat serum) for 1 h, washed three times with PBS (5 min each), and mounted with Vectashield plus DAPI (Vector Laboratories). Digital imaging was performed with a Leica DMIRE2 fluorescent microscope using IP-LAB or Slidebook 5.1 software. To quantify fluorescence, regions of interest were drawn around the nuclei or cytoplasm of transfected (DsRed-positive) cells or untransfected neighboring cells, and the intensity of FITC fluorescence emission from >100 cells was measured in at least three experiments. For immunofluorescence analysis in Drosophila salivary glands, at least five third instar wandering larvae expressing dVCP (WT or mutant) and TDP-43 (WT or mutant) under control of the driver fkh-GAL4 were collected. Salivary glands from these larvae were dissected in PBS and fixed in 4% PFA and heptane at room temperature. After 20 min, the PFA was removed and 100% methanol added. After vigorous shaking for 1 min, the heptane was removed, and the samples were washed in methanol three times. After three washes in PBST (PBS/0.1% Tween 20) and three washes in PBSBT (PBS/0.1% Tween 20/1% BSA), the samples were blocked for 2 h in PBSBT at room temperature. Samples were incubated with primary antibody (rabbit polyclonal anti-TDP-43 antibody at 1:300, diluted in PBSBT) overnight and washed four times with PBSBT (30 min each). After the final wash, the cells were incubated with secondary antibody (diluted in PBSBT) for 2 h, washed three times with PBSBT (10 min each), and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen). Digital imaging was performed with a Leica DMIRE2 fluorescent microscope using Slidebook software. All cells from the glands were evaluated for the absence of nuclear TDP-43 staining. Using Slidebook software, DAPI was pseudocolored in red.
For immunohistochemistry of fly eyes, heads of the appropriate genotype were collected and fixed in 4% buffered paraformaldehyde in PBS for 2 h at room temperature. Samples were serially dehydrated in ethanol (1 h each in 50, 70, 80, 90, and 95, and twice in 100%). Samples were then either (1) embedded in JB-4 according to the manufacturer's protocol (JB-4 Plus Embedding Kit; Polysciences) and sectioned at 1 micron for staining with Richardson's or toluidine blue stains, or (2) embedded into paraffin, sectioned at 5 μm, and placed on glass slides. Immunohistochemistry was performed using (ProteinTech) rabbit anti-TDP43 at 1:200, Vector Rabbit HRP ImmPress detection, and Vector AEC substrate.
Results
Exogenous expression of disease-related VCP mutants causes toxicity in a novel Drosophila melanogaster model
To develop an in vivo model of VCP-mediated degeneration, we used Drosophila melanogaster, in which the gene TER94 encodes the highly conserved orthologue of VCP. dVCP is 92% similar and 83% identical to human VCP at the amino acid level. Conservation is even higher (95% similar, 84% identical) across the 250 aa N terminus, which hosts most known disease-causing mutations, and all amino acid residues altered in disease are perfectly conserved. We introduced R152H and A229E mutations into dVCP to create homologs of the R155H and A232E mutations, which cause the most common and most severe forms of IBMPFD, respectively, and generated transgenic flies. When these mutants were expressed in the fly eye or brain using the UAS/GAL4 system (Brand and Perrimon, 1993), we observed mutation-dependent degenerative phenotypes despite equivalent levels of dVCP expression (Fig. 1, supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In the eye, expression of WT dVCP caused a very modest phenotypic change, whereas matched expression of the R152H and A229E mutants caused severe external rough eye phenotypes with necrotic patches and histologically evident marked vacuolar degeneration. The more severe phenotype seen in flies expressing the A229E mutant is consistent with the more severe disease in patients with the A232E mutation.
Figure 1.

Exogenous expression of disease-related VCP mutants enhances toxicity in vivo. A, Top row, Stereomicrographs of 1-d-old adult fly eyes in which expression of WT or mutant dVCP is driven by GMR-GAL4. The eyes of control flies (GMR-GAL4/+) and flies expressing WT dVCP have a highly organized ommatidial array. The eyes of flies expressing mutant dVCP (R152H) show loss of individual ommatidia, partial collapse, and small necrotic patches. The eyes of flies expressing mutant dVCP (A229E) are severely degenerated with large necrotic patches. Bottom row, Corresponding Richardson's stained frontal sections. B, Western blot showing expression levels of WT and mutant forms of dVCP in the eyes of 1-d-old flies. Actin served as a loading control. C, Quantitative analysis of eye phenotypes. Flies from each genotype were randomly selected for objective scoring according to the criteria described in Materials and Methods. Values are the mean results; error bars represent SEM.
When expressed broadly in the CNS, mutant dVCP reduced viability by diminishing eclosion rates (supplemental Fig. 1_A_, available at www.jneurosci.org as supplemental material), and surviving male flies had a significantly reduced lifespan indicative of neurodegeneration (supplemental Fig. 1_B_,C, available at www.jneurosci.org as supplemental material). Thus, this fly model recapitulates the degeneration associated with disease-causing mutations in VCP in tissue known to be affected (brain) and in a nonessential tissue (eye), which allows screening to identify modifying genes.
A dominant modifier screen identifies RNA-binding proteins, including TBPH, as modifiers of mutant dVCP toxicity
VCP participates in a range of cellular activities and, thus, in diverse biological pathways. The specific pathways underlying pathogenesis of IBMPFD, however, are unknown. As an unbiased approach to identify molecules and pathways involved in mutant-VCP-mediated degeneration, we performed a dominant-modifier screen, using the moderate dVCP mutant phenotype of dVCP R152H to maximize the likelihood of visualizing enhancement or suppression of the mutant dVCP phenotype. To carry out the screen, we obtained a deficiency collection representing all four chromosomes from the Bloomington Drosophila Stock Center to scan the maximum proportion of genome (∼80%) with the smallest number of lines. From the primary screen, 74 deficiencies were identified as dominant modifiers (enhancers or suppressors) of the dVCP R152H eye phenotype. After the secondary screen, validation studies, and individual gene interrogation by double-strand RNAi lines, we identified three related genes, TBPH (CG10327), xl6 (CG10203), and Hrb27C (CG10377), that dominantly suppress the degenerative phenotype (Fig. 2 A,B). RNAi-mediated knockdown of these genes in the eye did not result in a phenotypic change independent of mutant dVCP expression (supplemental Fig. 2_A–D_, available at www.jneurosci.org as supplemental material). However, in flies expressing dVCP R152H, knockdown of these genes suppressed mutation-dependent degeneration (Fig. 2 B). Suppression of degeneration in VCP mutant flies was corroborated by seeing a significant reduction in the blinded phenotypic severity score (Fig. 2 C) and by using additional RNAi lines and classical alleles (supplemental Fig. 2_E_,F, available at www.jneurosci.org as supplemental material). We also generated transgenic lines overexpressing TBPH, which resulted in a degenerative phenotype evident externally and histologically when targeted to the eye. When exogenous TBPH was coexpressed with dVCP R152H, degeneration associated with mutant VCP was enhanced, confirming the genetic interaction (Fig. 2 D). Hrb27C, xl6, and TBPH correspond to the human genes DAZAP1, 9G8, and TDP-43, respectively. These are all RNA recognition motif-containing RNA-binding proteins that shuttle between the nucleus and cytoplasm (Huang and Steitz, 2001; Lin and Yen, 2006; Ayala et al., 2008). Furthermore, all three have been shown to regulate multiple aspects of RNA metabolism, including transcription, export, splicing, and translation (Elvira et al., 2006; Swartz et al., 2007; Yang et al., 2009). We focused on TDP-43 for additional assessment, because cytoplasmic deposition of this protein is a prominent feature of IBMPFD and other degenerative diseases (Geser et al., 2009).
Figure 2.
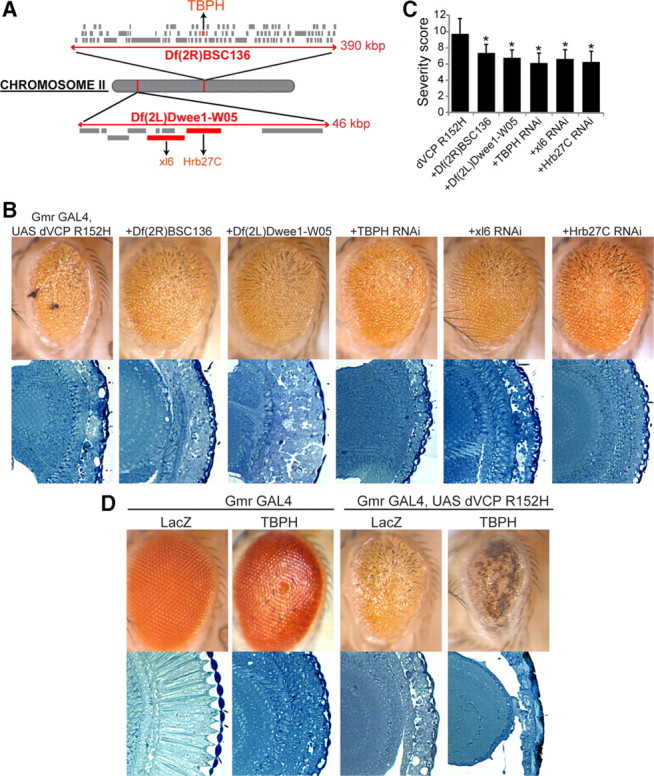
A dominant modifier screen identifies TBPH as a modifier of dVCP toxicity. A, The positions of the two deficiency lines from the Bloomington Deficiency Kit Df(2L)Dwee1-W05 and Df(2R)BSC136 and the genes within them in the Drosophila genome. The size of each deletion region is noted. Genes in red were identified by genetic screening to suppress the degenerative phenotype of mutant dVCP. B, Stereomicrographs of eyes of 1-d-old adult flies in which dVCP R152H and suppressors identified in a dominant modifier screen were expressed by using the driver GMR-GAL4. Top row, dVCP R152H alone and coexpressed with two deficiency lines (Df(2R)BSC136 and Df(2L)Dwee1–W05) or the individual RNAi lines corresponding to the genes within the regions defined by the deficiency lines, which all suppress the degenerative phenotype associated with mutant dVCP. Bottom row, Corresponding toluidine blue-stained frontal sections. C, Quantitative analysis of eye phenotypes. Flies from each genotype were randomly selected for objective scoring according to the criteria described in Materials and Methods. Quantitation of the phenotype shows a significant decrease in severity score when the deficiency and RNAi lines are coexpressed with mutant dVCP compared to mutant dVCP alone. Comparisons were made using Student's t test. Data show mean phenotype severity score (error bars represent SD; *p < 0.05). D, Top row, Stereomicrographs of eyes of 1-d-old flies expressing WT TBPH alone and with dVCP R152H. Overexpression of WT TBPH causes a profound degenerative phenotype and enhances the degenerative phenotype associated with mutant dVCP. Bottom row, Corresponding toluidine blue-stained frontal sections.
Disease-related mutations in VCP cause cytotoxicity and lead to TDP-43 redistribution
In the brain, TDP-43 is found predominantly in the nuclei of neurons and some glial cells (Fig. 3 A), although dynamic studies in vitro have shown TDP-43 to shuttle between the nucleus and cytoplasm (Ayala et al., 2008). In IBMPFD, affected brain regions show gross abnormalities in TDP-43 localization, including clearance of TDP-43 from many nuclei and accumulation in the cytoplasm; some neurons show dense nuclear inclusions of TDP-43 (Fig. 3 B). To examine the subcellular localization of TDP-43 in vitro, we first investigated whether VCP mutation-dependent toxicity could be recapitulated in primary mouse cortical neurons transfected with FLAG-conjugated WT VCP or mutant VCP (R155H and A232E). At 24 or 48 h after transfection, a blinded examiner scored VCP-positive neurons for changes in MAP2 staining and nuclear morphology (changes in DAPI staining) to assess cytotoxicity (Fig. 3 C). Quantitative analysis of the data revealed significant mutation-dependent toxicity in primary neurons 48 h after transfection (Fig. 3 D). This time-dependent cytotoxicity caused by mutant VCP was also demonstrated by fluorescence-activated cell sorting for living versus dead HEK293T cells. Exogenous expression of mutant but not WT VCP caused cell death despite equivalent levels of VCP expression (supplemental Fig. 3_A_,B, available at www.jneurosci.org as supplemental material).
Figure 3.
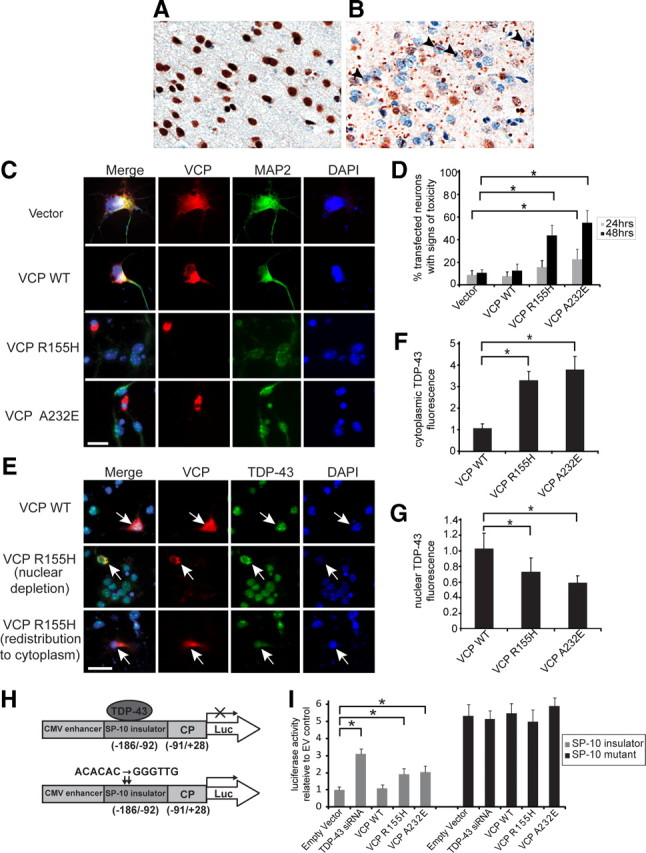
Exogenous expression of disease-related VCP mutants in vitro causes cytotoxicity and redistribution of TDP-43. A, B, Localization of TDP-43 in a patient with IBMPFD was first confirmed by immunohistochemistry. In an unaffected region (occipital cortex) (A), TDP-43 is predominantly nuclear, whereas in the affected frontal cortex (B), TDP-43 is largely redistributed to the cytoplasm and some nuclei show dense inclusions (arrowheads). C, Mouse primary cortical neurons were transfected on day 4 in vitro with DsRed-conjugated WT or mutant VCP, then immunostained 24 or 48 h later for MAP2 (green). DAPI staining (blue) shows nuclear morphology. Arrows indicate neurons expressing mutant VCP that have lost MAP2 staining and have condensed nuclei. Scale bar, 20 μm. D, A blinded investigator scored VCP-expressing neurons for cytotoxicity (condensed nuclei plus altered MAP2 staining). Values are the mean results from three trials; error bars represent SEM; *p < 0.05, Student's _t_ test. E**, Mouse primary cortical neurons transfected with DsRed-conjugated WT or mutant VCP were fixed and immunostained with anti-TDP-43 and DAPI 24 h after transfection. Neurons expressing mutant VCP showed reduced nuclear TDP-43 and increased cytoplasmic TDP-43. Representative examples are shown (arrows). Scale bar, 20 μm. F, G, A blinded examiner scored >100 VCP-expressing neurons from three separate cultures for the presence or absence of TDP-43 in the cytoplasm (F) and, independently, in the nucleus (G**). Expression of mutant VCP (R155H or A232E) was associated with significantly less nuclear TDP-43 and significantly more cytoplasmic TDP-43 (values are the means from three trials; error bars represent SEM; *p < 0.05, Student's t test). H, The firefly luciferase reporter constructs used to detect TDP-43 transcriptional repression activity. I, Luciferase values relative to control values (values are the means from three trials; error bars represent SEM; *p < 0.05, Student's t test). The SP-10 insulator inserted between the CMV enhancer and the core promoter (CP) repressed luciferase expression (Luc). Knockdown of TDP-43 by siRNA or mutation of the TDP-43 binding sites (ACACAC to GGGTTG) released the enhancer-blocking effect of the insulator. Cotransfection of VCP R155H and A232E, but not of WT VCP, with pSP-10 also significantly compromised the enhancer-blocking ability. Cells cotransfected with VCP constructs and mutant pSP-10 did not show greater luciferase activity than cells transfected with pSP-10-mutant alone.
To examine TDP-43 subcellular localization, we fixed and immunostained primary mouse cortical neurons 24 h after transfection, before occurrence of the changes in MAP2 staining or nuclear morphology. In neurons expressing FLAG vector alone or FLAG-conjugated WT VCP, TDP-43 was consistently nuclear. By contrast, in cells transfected with mutant VCP, we observed significant clearance of endogenous TDP-43 from nuclei and accumulation in the cytoplasm (Fig. 3 E–G). Redistribution of endogenous TDP-43 to the cytoplasm was also observed in HEK293T cells expressing mutant but not wild-type VCP (supplemental Fig. 3_C_,D, available at www.jneurosci.org as supplemental material). Exogenous expression of TDP-43 increased the amount of cytoplasmic redistribution (supplemental Fig. 3_E_,F, available at www.jneurosci.org as supplemental material). The nuclear depletion of TDP-43 in primary neurons and HEK293T cells was corroborated by an associated loss of TDP-43's known nuclear function as a transcriptional repressor of testis-specific SP-10 gene (Acharya et al., 2006). TDP-43 has been shown to interact with the SP-10 insulator, which prevents luciferase expression by repressing the interaction between the CMV enhancer and SP-10 core promoter in the pSP-10 construct (Fig. 3 H). Mutation of the two TDP-43 binding sites (ACACAC to GGGTTG) compromises the enhancer-blocking ability of the SP-10 insulator, allowing luciferase expression (Fig. 3 H). HEK293T cells were transfected with pSP-10 or pSP-10-mutant luciferase reporter constructs, and luciferase expression was quantified 48 h after transfection as an indicator of TDP-43 nuclear function. When TDP-43 binding to SP-10 was disrupted by use of the pSP-10 mutant or by siRNA knockdown of TDP-43, the normal repression of luciferase expression was attenuated. When mutant VCP, but not WT VCP, was cotransfected with pSP-10, the repression was released, allowing a significant increase in luciferase expression. Cells cotransfected with VCP constructs and mutant pSP-10 did not show greater luciferase activity than cells transfected with pSP-10 mutant alone. Similar results were found using the HIV-1 luciferase reporter plasmid (pLTR) (supplemental Fig. 3_G_, available at www.jneurosci.org as supplemental material), a construct shown previously to rely on TDP-43 as a transcriptional repressor (Ou et al., 1995). Thus, mutation-dependent VCP toxicity in primary neurons and HEK293T cells was coincident with TDP-43 redistribution from the nucleus to the cytoplasm, as was also recently observed in immortalized SH-SY5Y and U20S cells (Gitcho et al., 2009; Ju et al., 2009). TDP-43 redistribution is also a conspicuous feature of numerous TDP-43 proteinopathies. However, these observations do not tell us whether TDP-43 redistribution represents an adaptive response to neuronal injury or, alternatively, whether change in subcellular localization mediates disease. Furthermore, if TDP-43 redistribution mediates disease, it leaves unclear whether the mechanism involves loss of nuclear function or toxic gain of cytoplasmic function.
Mislocalization of TDP-43 to the cytoplasm causes degeneration
To further investigate the potential role of TDP-43 mislocalization in mutant-VCP-associated degeneration in vivo, we generated multiple transgenic flies expressing human WT or mutant TDP-43 using the UAS/GAL4 system. To target TDP-43 specifically to the nucleus or the cytoplasm, we used TDP-43 with previously characterized mutations that disrupt the NES or the NLS (Winton et al., 2008). Five independent transgenic lines expressing WT TDP-43 in the fly eye showed a modest degenerative phenotype (Fig. 4, supplemental Figs. 4_A_,B, 6_A_, available at www.jneurosci.org as supplemental material). A normal eye phenotype was observed in five independent transgenic lines expressing NES-mutant TDP-43 (Fig. 4, supplemental Figs. 4_C_,D, 6_A_, available at www.jneurosci.org as supplemental material). By contrast, expression of NLS-mutant TDP-43 in the fly eye in six independent lines resulted in a strong degenerative phenotype and high-molecular-weight smears of TDP-43 in immunoblots (supplemental Figs. 5_A_,B, 6, available at www.jneurosci.org as supplemental material). Immunostaining of NLS-mutant TDP-43 confirmed predominantly cytoplasmic localization, whereas WT and NES-mutant TDP-43 remained predominantly nuclear (Fig. 4). To confirm that the degenerative phenotype was caused by cytoplasmic localization of TDP-43 rather than another effect of the NLS mutation, we also generated lines with truncated TDP-43 (80-414). Deletion of the first 79 aa, which disrupts the NLS, led to cytoplasmic localization of TDP-43 and a degenerative phenotype indistinguishable from that observed with NLS-mutant TDP-43 (supplemental Fig. 5_C_, available at www.jneurosci.org as supplemental material) (data not shown). These findings indicate that accumulation of TDP-43 in the cytoplasm is sufficient to cause degeneration. Together with the observation that knockdown of TBPH in flies causes no phenotypic change alone but suppresses the mutant dVCP phenotype, these results indicate that a toxic gain of cytoplasmic TDP-43 function underlies pathogenesis. However, these observations do not preclude the possibility that a loss of TDP-43 nuclear function contributes to toxicity.
Figure 4.

Mislocalization of exogenous TDP-43 to the cytoplasm causes degeneration in vivo. Stereomicrographs of the eyes of 1-d-old adult flies in which LacZ (control), WT, or mutant TDP-43 was expressed by using the driver GMR-GAL4. Shown beside each panel is immunohistochemical analysis of a longitudinal section with anti-TDP-43. WT and NES-mutant TDP-43 are predominantly nuclear, and eye morphology is normal (see also supplemental Figs. 5, 6, available at www.jneurosci.org as supplemental material). NLS-mutant TDP-43 is predominantly cytoplasmic and causes a severe rough eye phenotype (see also supplemental Fig. 7, available at www.jneurosci.org as supplemental material).
An ALS-causing mutation expressed in vivo leads to toxicity associated with cytoplasmic redistribution of TDP-43
Missense mutations in TDP-43 have recently been identified in association with dominantly inherited and sporadic ALS. To assess the effect of mutant TDP-43 in vivo, we generated transgenic lines expressing TDP-43 with the ALS-causing mutation M337V (Sreedharan et al., 2008). In six independent lines evaluated, we observed degenerative phenotypes ranging from modest to severe (Fig. 5 A, supplemental Fig. 6_A_, available at www.jneurosci.org as supplemental material). The severity of the phenotype correlated with the amount of abnormal TDP-43 species, including a C-terminal 25 kDa fragment and high-molecular-weight bands (Fig. 5, supplemental Fig. 6_B_, available at www.jneurosci.org as supplemental material). This is similar to the pathogenic biochemical signature seen in patient tissue with TDP-43 proteinopathy (Neumann et al., 2007). Immunohistochemistry for TDP-43 in these eyes showed a marked cytoplasmic distribution of TDP-43, consistent with a recent report that this mutation increases cytoplasmic TDP-43 inclusions in vitro (Nonaka et al., 2009).
Figure 5.
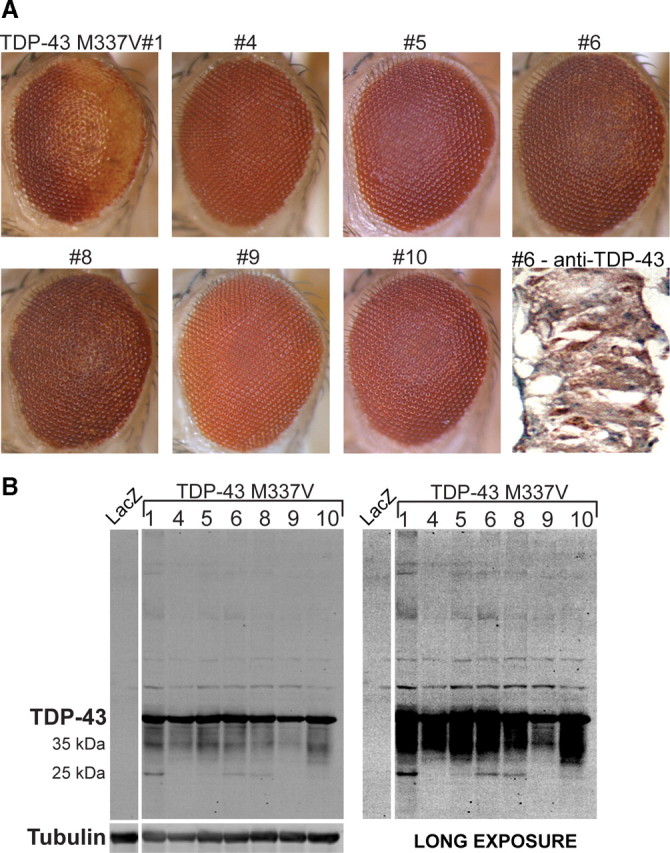
Exogenous expression of ALS-causing mutant M337V TDP-43 results in degeneration in vivo. A, Stereomicrographs of the eyes of 1-d-old adult flies in which expression of TDP-43 M337V is driven by GMR-GAL4. All seven of the independent transgenic lines generated are shown. The eyes of some lines exhibit a mild rough eye phenotype. Immunohistochemistry with anti-TDP-43 reveals cytoplasmic TDP-43 in M337V#6. B, Western blot analysis of head homogenates from TDP-43 M337V flies showing TDP-43 expression in all transgenic lines generated. All lines with a degenerative phenotype show both monomeric TDP-43 and a ∼25 kDa fragment of TDP-43. Staining for tubulin served as a loading control.
Mutation-dependent genetic interaction of dVCP and TDP-43 is associated with cytoplasmic redistribution of TDP-43 in vivo
To assess the relationship between dVCP and TDP-43 in vivo, we performed an epistasis study by coexpressing dVCP (WT or mutant) with TDP-43 (WT or mutant) and analyzing changes to external eye phenotype by light microscopy (Fig. 6 A), and in subcellular localization of TDP-43 by immunohistochemistry (Fig. 6 B). Crosses with most of our NLS-mutant TDP-43 lines resulted in lethality; we therefore used line 5, which exhibits a very mild phenotype (all lines are shown in supplemental Fig. 5_A_, available at www.jneurosci.org as supplemental material). Because of the severe degradative loss of internal eye tissue in some genotypes, immunohistochemistry of TDP-43 in the eye (Fig. 6 B) was corroborated with quantitative immunofluorescence analysis of TDP-43's subcellular distribution in larval salivary glands (Fig. 6 C,D).
Figure 6.
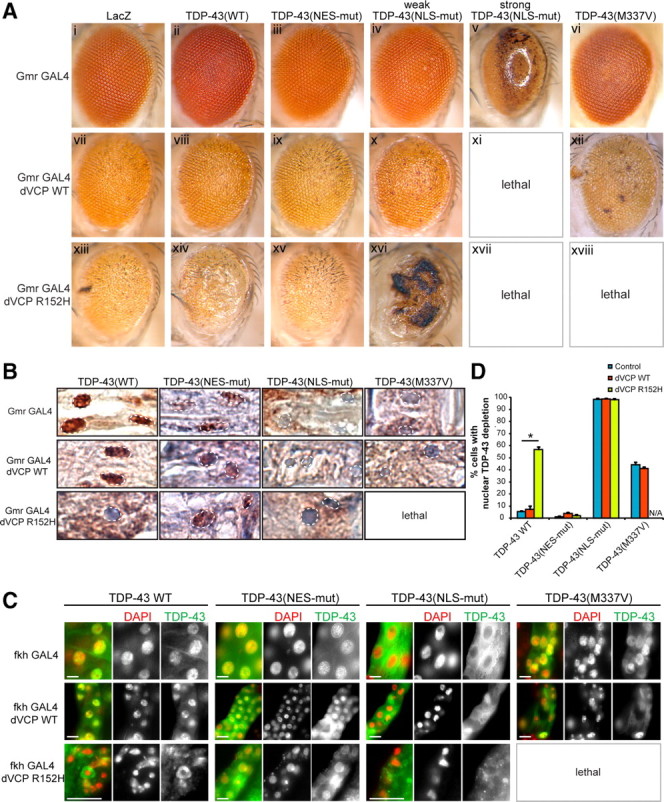
Mutation-dependent interaction of dVCP with TDP-43. A, Stereomicrographs of the eyes of 1-d-old adult flies coexpressing WT or mutant (R152H) dVCP and WT or mutant (NES-mutant, NLS-mutant or ALS-causing mutant M337V) TDP-43. Coexpression with LacZ served as a negative control. B, Immunohistochemical detection of the subcellular distribution of TDP-43 in longitudinal eye sections, with nuclei highlighted. C, Immunohistochemical detection of the subcellular distribution of TDP-43 in whole-mount salivary glands from Drosophila larvae. Scale bar, 10 μm. D, Quantitation of nuclear depletion of TDP-43 in salivary glands (values are the means from at least five larvae of each genotype; error bars represent SEM; *p < 0.05, Student's t test). WT TDP-43 coexpressed with WT dVCP did not modify the phenotype and remained predominantly nuclear. By contrast, WT TDP-43 enhanced the rough eye phenotype in flies expressing mutant dVCP and was significantly redistributed to the cytoplasm. NES-mutant TDP-43 did not modify the phenotype of WT or mutant dVCP and remained predominantly nuclear. The phenotype associated with mutant dVCP (but not WT dVCP) was strongly enhanced in a weakly degenerative NLS-mutant TDP-43 line, and the TDP-43 remained predominantly cytoplasmic. In the strongly degenerative NLS-mutant TDP-43 line, coexpression of either WT or mutant dVCP caused lethality. The ALS-causing mutant M337V enhanced the dVCP WT phenotype and resulted in lethality when coexpressed with mutant dVCP.
This epistasis study indicated no interaction between WT dVCP and WT or NES-mutant TDP-43 (Fig. 6 A,C). However, the combination of WT dVCP with a weak NLS mutant resulted in mild enhancement of the pathologic phenotype. The combination of WT dVCP with a strong NLS-mutant TDP-43 resulted in lethality. Notably, the subcellular distribution of WT and mutant TDP-43 was not altered by coexpression with WT dVCP (Fig. 6 B–D). By contrast, the degenerative phenotype of mutant dVCP R152H was strongly enhanced by coexpression of WT TDP-43 (Fig. 6 A,C), and this result was associated with significant clearance of WT TDP-43 from nuclei and accumulation in cytoplasm (Fig. 6 B–D). Importantly, the dVCP R152H phenotype was not enhanced by coexpression with NES-mutant TDP-43 (Fig. 6 A,C), and the subcellular distribution of NES-mutant TDP-43 remained nuclear (Fig. 6 B–D). Not surprisingly, the dVCP R152H phenotype was enhanced by NLS-mutant TDP-43 (Fig. 6 A,C), and NLS-mutant TDP-43 was found predominantly in the cytoplasm (Fig. 6 B–D). When the ALS-associated mutation TDP-43 M337V was coexpressed with WT dVCP, the phenotype was enhanced, and lethality resulted when the M337V mutant was coexpressed with mutant dVCP (Fig. 6 A,C). Thus, genetic interaction between dVCP and WT TDP-43 was dependent on a disease-causing mutation in dVCP and was associated with redistribution of TDP-43 from the nucleus to the cytoplasm. TDP-43 restricted to the nucleus failed to interact with mutant VCP. The interaction of VCP with a known ALS-causing TDP-43 mutant further underscores the importance of cytoplasmic TDP-43 in VCP mutation-dependent degeneration.
Discussion
We have developed and characterized a highly tractable Drosophila model of IBMPFD that exhibits VCP mutation-dependent degeneration (Fig. 1, supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Using this model, we identified multiple related RNA-binding proteins that genetically modified degeneration, and one of these was TBPH, the Drosophila orthologue of TDP-43. We further demonstrated that VCP and TDP-43 interacted genetically, that disease-causing mutations in VCP led to redistribution of TDP-43 to the cytoplasm in vitro and in vivo, and that this redistribution was sufficient to cause degeneration in vivo. We also determined that a pathogenic mutation in TDP-43 enhanced the genetic interaction with VCP. Together, our results show that toxic gain of function of TDP-43 in the cytoplasm contributes to degeneration initiated by mutations in VCP.
TDP-43 pathology is a prominent pathological feature in a broad array of sporadic and inherited human diseases including ALS, frontotemporal dementia–TDP, Perry syndrome, and IBMPFD (Neumann et al., 2007; Geser et al., 2009; Salajegheh et al., 2009). In these diseases, TDP-43 is found to be redistributed from the nucleus to the cytoplasm in affected neurons, although the significance of this has been unclear. In brain and muscle of IBMPFD patients, TDP-43 redistributes to the cytoplasm where it colocalizes with ubiquitin immunopositivity, and is also present in lenticular nuclear inclusions (Guinto et al., 2007). Expression of mutant VCP in SH-SY5Y cells (Gitcho et al., 2009), U20S cells (Ju et al., 2009), and in primary neurons (Fig. 3) resulted in redistribution of TDP-43 to the cytoplasm, although no nuclear inclusions were observed. TDP-43 also redistributes to the cytoplasm in response to neuronal injury where it colocalizes with stress granules (Colombrita et al., 2009; Moisse et al., 2009a). Therefore, it was unclear whether the redistribution of TDP-43 in the setting of mutant-VCP-related disease was a mediator of pathogenesis or an indicator of cytotoxic stress caused by disease. The results presented here clarify this issue, indicating that TDP-43 is a mediator of toxicity initiated by disease-causing mutations in VCP. This is illustrated by suppression of degeneration in the IBMPFD model when endogenous TBPH is depleted (Fig. 2 B,C), and enhancement of degeneration when TBPH or TDP-43 is overexpressed (Figs. 2 D, 6). Although our results indicate that accumulation of TDP-43 in the cytoplasm contributes to cytotoxicity, they do not exclude the possibility that depletion of nuclear TDP-43 also contributes to cytotoxicity.
The mechanism by which mutations in VCP influence the subcellular distribution of TDP-43 is unknown, but we outline several possibilities here (Fig. 7). First, the nuclear depletion and cytoplasmic accumulation of TDP-43 might reflect a defect in the well established role of VCP in ubiquitin-dependent segregation of substrates from multiprotein complexes. It is known that TDP-43 is present in the cytoplasm at low levels normally, where it is found in ribonucleoprotein complexes implicated in translation regulation (Elvira et al., 2006; Wang et al., 2008; Freibaum et al., 2009; Moisse et al., 2009b). Perhaps VCP activity is necessary for removal of TDP-43 from ribonucleoprotein complexes to permit recycling, and impairment of this activity by disease-causing mutations leads to progressive accumulation of TDP-43 in the cytoplasm. A second possibility is that VCP directly participates in nuclear import of TDP-43. VCP was shown previously to regulate the nuclear import of the TSAd protein (T-cell-specific adaptor protein) in T cell signal transduction (Marti and King, 2005). This aspect of VCP function may involve the adaptor Npl4 (nuclear protein localization-4), originally discovered in a yeast screen for mutants deficient in nuclear protein import (DeHoratius and Silver, 1996; Fabre and Hurt, 1997). Perhaps VCP regulates nucleocytoplasmic shuttling of additional proteins, including TDP-43, and that disease mutations impair this activity. A third possibility relates to the recently discovered role of VCP in autophagy, and the finding that disease-causing mutations in VCP impair autophagy (Ju and Weihl, 2010; Tresse et al., 2009). Since autophagy may be important for turnover of cytoplasmic TDP-43 (Wang et al., 2010), accumulation of TDP-43 in the cytoplasm may simply reflect a defect in this degradation pathway. These three possibilities are not mutually exclusive, and it is also possible that redistribution of TDP-43 in IBMPFD reflects defects in VCP functions that are presently unknown.
Figure 7.

VCP's role in the nucleocytoplasmic balance of TDP-43 The mechanism whereby VCP mutations lead to cytoplasmic accumulation of TDP-43 is unknown. We speculate that the possibilities include, but are not limited to, dysfunction in the following: (1) VCP's segregase activity for extracting TDP-43 from RNP complexes in the cytoplasm, (2) a role for VCP in nuclear import of TDP-43 (analogous to the role of VCP in nuclear import of TSAd), and (3) degradation of TDP-43 by VCP, a known component of the autophagic pathway.
The present study shows that degeneration initiated by mutations in VCP is mediated in part through toxic gain of function of TDP-43 in the cytoplasm. The basis for toxicity associated with excess cytoplasmic TDP-43 is unclear, but this observation is consistent with the recent report showing that (1) cytoplasmic mislocalization of TDP-43 is toxic to neurons, and (2) mutations in TDP-43 that cause familial ALS promote cytoplasmic mislocalization (Barmada et al., 2010). There is some evidence to suggest that TDP-43, or a fragment of TDP-43, is intrinsically prone to aggregation resulting in the formation of a toxic species (Johnson et al., 2009; Zhang et al., 2009). Indeed, we have observed a correlation between TDP-43 toxicity in vivo and the presence of TDP-43 cleavage products or high-molecular-weight species of TDP-43 (Fig. 5, supplemental Figs. 4–6, available at www.jneurosci.org as supplemental material), although this study does not address whether there is a cause-and-effect relationship between these abnormal species and toxicity. Whether or not TDP-43 aggregation promotes toxicity, we are particularly intrigued by the possibility that excess cytoplasmic TDP-43 perturbs some aspect of cytoplasmic RNA metabolism. The notion that a defect in RNA metabolism contributes to IBMPFD pathogenesis is supported in the present study by the identification of xl6 (the Drosophila ortholog of human SR protein 9G8) and Hrb27C (the fly ortholog of the human hnRNP DAZAP1) as dominant modifiers of mutant VCP toxicity in vivo. This notion is further supported by the high frequency with which inherited neurodegenerative diseases are caused by mutations that impair RNA metabolism, either through mutations in RNA-binding proteins or through mutations in RNA that impair the function of RNA-binding proteins (Cooper et al., 2009; La Spada and Taylor, 2010). The extent to which perturbation in RNA metabolism contributes to TDP-43 proteinopathies in general, and IBMPFD in particular, will be fascinating to learn as the field moves forward.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grant AG10124 to J.Q.T., NIH Grant AG17586 to V.M.-Y.L., and grants from the Association of Frontotemporal Dementias, the Dana Foundation, the Packard Foundation for ALS Research at Johns Hopkins University, the Comprehensive Neuroscience Center at the University of Pennsylvania, and the American Syrian Lebanese Associated Charities to J.P.T. G.P.R. and J.B.G. were supported by NIH Research Training Grant T32 AG000255. We are indebted to the Cell and Tissue Imaging Core of the Hartwell Center at St. Jude Children's Research Hospital for assistance with electron microscopy.
References
- Acharya KK, Govind CK, Shore AN, Stoler MH, Reddi PP. cis-requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295:781–790. doi: 10.1016/j.ydbio.2006.04.443. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHoratius C, Silver PA. Nuclear transport defects and nuclear envelope alterations are associated with mutation of the Saccharomyces cerevisiae NPL4 gene. Mol Biol Cell. 1996;7:1835–1855. doi: 10.1091/mbc.7.11.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvira G, Wasiak S, Blandford V, Tong XK, Serrano A, Fan X, del Rayo Sanchez-Carbente M, Servant F, Bell AW, Boismenu D, Lacaille JC, McPherson PS, DesGroseillers L, Sossin WS. Characterization of an RNA granule from developing brain. Mol Cell Proteomics. 2006;5:635–651. doi: 10.1074/mcp.M500255-MCP200. [DOI] [PubMed] [Google Scholar]
- Fabre E, Hurt E. Yeast genetics to dissect the nuclear pore complex and nucleocytoplasmic trafficking. Annu Rev Genet. 1997;31:277–313. doi: 10.1146/annurev.genet.31.1.277. [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Research. 2009;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurology. 2009;256:1205–1214. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM, Cairns NJ. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem. 2009;284:12384–12398. doi: 10.1074/jbc.M900992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinto JB, Ritson GP, Taylor JP, Forman MS. Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol. 2007;114:55–61. doi: 10.1007/s00401-007-0224-7. [DOI] [PubMed] [Google Scholar]
- Halawani D, Latterich M. p97: the cell's molecular purgatory? Mol Cell. 2006;22:713–717. doi: 10.1016/j.molcel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Huang Y, Steitz JA. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol Cell. 2001;7:899–905. doi: 10.1016/s1097-2765(01)00233-7. [DOI] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Weihl CC. p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy. 2010;6:283–285. doi: 10.4161/auto.6.2.11063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008;1782:744–748. doi: 10.1016/j.bbadis.2008.09.003. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Yen P. A novel nucleocytoplasmic shuttling sequence of DAZAP1, a testis-abundant RNA-binding protein. RNA. 2006;12:1486–1493. doi: 10.1261/rna.42206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti F, King PD. The p95-100 kDa ligand of the T cell-specific adaptor (TSAd) protein Src-homology-2 (SH2) domain implicated in TSAd nuclear import is p97 Valosin-containing protein (VCP) Immunol Lett. 2005;97:235–243. doi: 10.1016/j.imlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ. Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009a;1296:176–186. doi: 10.1016/j.brainres.2009.07.023. [DOI] [PubMed] [Google Scholar]
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009b;1249:202–211. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Arai T, Buratti E, Baralle FE, Akiyama H, Hasegawa M. Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 2009;583:394–400. doi: 10.1016/j.febslet.2008.12.031. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31. doi: 10.1002/mus.21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz J, Bor Y, Misawa Y, Rekosh D, Hammarskjold M. The shuttling SR protein 9G8 plays a role in translation of unspliced mRNA containing a constitutive transport element. J Biol Chem. 2007;282:19844–19853. doi: 10.1074/jbc.M701660200. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:217–227. doi: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469:112–116. doi: 10.1016/j.neulet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HT, Peggie M, Cohen P, Rousseau S. DAZAP1 interacts via its RNA-recognition motifs with the C-termini of other RNA-binding proteins. Biochem Biophys Res Commun. 2009;380:705–709. doi: 10.1016/j.bbrc.2009.01.166. [DOI] [PubMed] [Google Scholar]
- Ye Y. Diverse functions with a common regulator: Ubiquitin takes command of an AAA ATPase. J Struct Biol. 2006;156:29–40. doi: 10.1016/j.jsb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]