YLoc—an interpretable web server for predicting subcellular localization (original) (raw)
Abstract
Predicting subcellular localization has become a valuable alternative to time-consuming experimental methods. Major drawbacks of many of these predictors is their lack of interpretability and the fact that they do not provide an estimate of the confidence of an individual prediction. We present YLoc, an interpretable web server for predicting subcellular localization. YLoc uses natural language to explain why a prediction was made and which biological property of the protein was mainly responsible for it. In addition, YLoc estimates the reliability of its own predictions. YLoc can, thus, assist in understanding protein localization and in location engineering of proteins. The YLoc web server is available online at www.multiloc.org/YLoc.
INTRODUCTION
Protein sorting is a complex and still poorly understood process. It is crucial for a protein’s function as a protein’s location is often correlated with its molecular function. Thus, knowledge of protein localization can help biologists to infer the function of a protein. However, experimental methods for determining a protein’s location are expensive and time consuming. In contrast, computational predictions rely only on the protein sequence, are fast, and fairly accurate. Over recent years, various prediction methods have been introduced. Most methods use sequence information, such as known sorting signals and amino acid composition (1–9). More advanced methods incorporate annotation information such as functional domains and motifs (10,11), homologous proteins (12,13), Gene Ontology (GO) terms (14) and textual information (15,16). Predictions based on annotated knowledge are often more accurate, but are less robust in cases where little is known about the protein. Hybrid prediction approaches combine the advantages of both information sources (17–21).
Although the prediction performance of subcellular localization predictors has increased significantly over recent years, their predictions are often not considered to be trustworthy. Very complex machine learning models of state-of-the-art prediction systems make it difficult to understand why a prediction was made. Consequently, the web interfaces of most methods are non-transparent and offer no explanation for a particular prediction. In addition, most methods do not offer confidence estimates for an individual prediction.
We present YLoc, an interpretable web server for predicting subcellular localization. Users are provided with the prediction itself, and also with an explanation why this prediction was made. The features contributing to the prediction are translated into natural language aiming at the most likely explanation of the localization. In addition, a confidence score helps the users to verify whether the prediction is reliable or not. YLoc is available in a low-resolution version, YLoc-LowRes, and a high-resolution version, YLoc-HighRes, covering 5 or 11 eukaryotic subcellular locations, respectively. YLoc+, the most general version, integrates multiple locations sites. All three predictors are available for animal, fungal and plant proteins.
METHODS AND MATERIALS
YLoc-LowRes was trained on the BaCelLo data set (6), which contains only globular proteins. The animal and fungal versions predict four locations: the nucleus (nu), cytoplasm (cy), mitochondrion (mi) and the secretory pathway (SP). The plant version additionally predicts the chloroplast (ch). YLoc-HighRes was trained on the Höglund data set (7). It covers 11 locations: nu, cy, mi, ch, endoplasmatic reticulum (er), Golgi apparatus (go), peroxisome (pe), plasma membrane (pm), extracellular space (ex), lysosome (ly) and vacuole (va). In the training of YLoc+, we used the Höglund data set and additional proteins with multiple locations from the DBMLoc database (22). The extracted 3054 proteins share <80% sequence similarity. Only dual locations with more than 100 representative proteins were included: cy and nu (cy_nu), ex and pm (ex_pm), cy and pm (cy_pm), cy and mi (cy_mi), nu and mit (nu_mi), er and ex (er_ex) and ex and nu (ex_nu). To our knowledge, this is currently the largest data set of proteins from multiple locations.
We derived about 30 000 features from our protein sequences using amino acid composition and pseudo composition (3) as well as properties such as hydrophobicity, charge and volume of amino acids. In addition, we included PROSITE motifs and GO terms from close homologs. For more details, we refer to Briesemeister et al. (26). To guarantee interpretable predictions, we first reduced the number of features using a backward best first search together with correlation-based feature selection (23) implemented in the Weka machine learning library (24). For YLoc-LowRes, we obtained 20 features; for YLoc-HighRes and YLoc+, we obtained 30 features. However, a small number of features is only the first step toward interpretable predictions. To provide meaningful explanations, we manually annotated all selected features in biological terms. Unfortunately, not every feature can be easily mapped to a biological property. In such cases, we carefully inspected the initial feature set and transferred the biological meaning of a highly correlated feature. A list of all selected and annotated features can be found in the Supplementary Data.
YLoc uses naïve Bayes alongside entropy-based discretization (25) to make predictions. Given a set of features  and a set of location classes
and a set of location classes  , the conditional distribution of class Cj can be expressed by:
, the conditional distribution of class Cj can be expressed by:
 |
(1) |
|---|
The final posterior probabilities 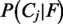 are calculated by normalizing the right term of Equation (1) such that all posteriors sum up to one. Based on the feature likelihoods
are calculated by normalizing the right term of Equation (1) such that all posteriors sum up to one. Based on the feature likelihoods  , we calculate a discrimination score which provides a simple and transparent understanding of the influence of a feature on the prediction, for details see Briesemeister et al. (26). A positive score indicates that this feature is typical for the predicted location, whereas a negative score indicates that this feature alone would suggest a different location. Secondly, the discrimination score shows how strongly a feature influenced the prediction.
, we calculate a discrimination score which provides a simple and transparent understanding of the influence of a feature on the prediction, for details see Briesemeister et al. (26). A positive score indicates that this feature is typical for the predicted location, whereas a negative score indicates that this feature alone would suggest a different location. Secondly, the discrimination score shows how strongly a feature influenced the prediction.
For multiple localization prediction, we assume that a protein present in multiple locations is equally distributed among those. Proteins labeled with two locations are assigned to a dual-location class, for example,  . YLoc+ then evenly distributes the posterior probabilities of the dual-location classes onto the probabilities of the two individual locations. For example,
. YLoc+ then evenly distributes the posterior probabilities of the dual-location classes onto the probabilities of the two individual locations. For example,  is added to
is added to  and to
and to 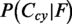 . All locations with a probability above a threshold of
. All locations with a probability above a threshold of  are predicted, where
are predicted, where 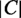 is the number of locations. If a location is less than half as probable as the next most probable one, this location and all less probable locations are not predicted.
is the number of locations. If a location is less than half as probable as the next most probable one, this location and all less probable locations are not predicted.
The probability of the predicted location shows only how likely a protein is to be found in this location compared with the other locations. A confidence estimate, however, tells how likely it is that this prediction is to be correct. For this purpose, we analyze whether the protein is typical for the predicted class or whether YLoc already extrapolates. If a feature vector is more likely for proteins from the predicted location than for proteins from all locations, i.e. 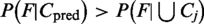 , we rate a prediction as being reliable. Since predicted locations with only a few training examples are often less reliable, we include the prior class probability in our confidence score:
, we rate a prediction as being reliable. Since predicted locations with only a few training examples are often less reliable, we include the prior class probability in our confidence score:
 |
(2) |
|---|
Confidence scores ranges from zero for unreliable predictions to one for very confident predictions. For more details on the YLoc methodology, refer to Briesemeister et al. (26).
EVALUATION
We have tested the performance of YLoc on two independent data sets (IDSs). The BaCelLo IDS (27) consists of animal, fungal and plant proteins from the nu, cy, mi and SP which have at most 30% sequence identity to proteins in the BaCelLo data set. The Höglund IDS (20) contains animals proteins from remaining locations, the er, go, pe, pm, ex and ly, and was constructed with the same restrictions as the BaCelLo data set. In addition, proteins from the same location which align with an _E_-value >10−3 are clustered and treated as one instance in the evaluation. We compared the YLoc predictors with five other state-of-the-art subcellular localization predictors: MultiLoc2 (20), BaCelLo (6), LOCTree (4), WoLF PSORT (9) and Euk-mPloc (19). All methods are available as web servers. The individual prediction performance was evaluated using the overall accuracy (ACC), which is the percentage of correctly predicted instances, and the average _F_1-score (_F_1), which is the average over the harmonic means of precision and recall of each location. Note that YLoc+, WoLF PSORT and Euk-mPloc are evaluated using the generalized ACC and _F_1 from multilabel classification (28).
The evaluation results are summarized in Table 1. In our benchmark study, we observed that YLoc shows comparable performance to current state-of-the-art methods. For the BaCelLo IDS, YLoc-LowRes and MultiLoc2-LowRes perform best since they are specialized in distinguishing globular proteins. The high-resolution predictors YLoc-HighRes, YLoc+, MultiLoc2-HighRes, WoLF PSORT and Euk-mPloc perform slightly worse on this data set, since they are more general predictors. On the Höglund IDS, MultiLoc2-HighRes, YLoc-HighRes and YLoc+ show comparable performance, whereas WoLF PSORT and Euk-mPloc perform worse. MultiLoc2 shows very good accuracy throughout the study. However, its architecture is very complex and the output is not interpretable. In contrast, YLoc uses a very simple model and its predictions are hence interpretable. The detailed location-wise performance of YLoc is shown in the Supplementary Data. When YLoc is applied without the use of GO-term-based features, the performance is only slightly reduced compared with the original predictors. In most cases, the performance drops only by 0.01 to 0.04. However, YLoc-LowRes plants shows a considerable performance loss on the BaCelLo plant IDS. In contrast, on the Höglund IDS, we observe a slight performance gain. For details see supplementary material of (26).
Table 1.
Performance of the YLoc and other state-of-the-art predictors on the BaCelLo IDS (27) (B) and Höglund IDS (20) (H) concerning _F_1 and ACC (in brackets)
| Data set | YLoc- LowRes | YLoc- HighRes | YLoc+ | MultiLoc2- LowRes | MultiLoc2- HighRes | BaCelLo | LOCTree | WoLF PSORT | Euk-mPloc |
|---|---|---|---|---|---|---|---|---|---|
| B Animals | 0.75 (0.79) | 0.69 (0.74) | 0.67 (0.58) | 0.76 (0.73) | 0.71 (0.68) | 0.66 (0.64) | 0.58 (0.62) | 0.67 (0.70) | 0.54 (0.61) |
| B Fungi | 0.61 (0.56) | 0.51 (0.56) | 0.51 (0.48) | 0.61 (0.60) | 0.58 (0.53) | 0.60 (0.57) | 0.43 (0.47) | 0.51 (0.50) | 0.56 (0.60) |
| B Plants | 0.58 (0.71) | 0.54 (0.58) | 0.49 (0.58) | 0.64 (0.76) | 0.54 (0.62) | 0.56 (0.69) | 0.58 (0.70) | 0.46 (0.57) | 0.37 (0.46) |
| H Animals | − (−) | 0.34 (0.56) | 0.37 (0.53) | − (−) | 0.41 (0.57) | − (−) | − (−) | 0.18 (0.36) | 0.24 (0.27) |
To show that users can benefit from the integrated confidence score, we analyzed the performance enrichment for high confidence scores. We reevaluated the performance of the YLoc predictors on the BaCelLo animals IDS by considering only proteins that could be predicted with a minimum confidence score. For statistical reasons, we excluded classes with less than five instances. The performance of YLoc for different minimum confidence scores is shown in Table 2. For the subset of proteins that can be predicted with high confidence, YLoc shows increased prediction performance. Consequently, predictions made with high confidence scores can be rated as more reliable.
Table 2.
Performance of YLoc on the BaCelLo animal IDS (27) for different minimum confidence scores
| Predictor | Measure | 0.0 | 0.2 | 0.4 | 0.6 | 0.8 | 0.9 |
|---|---|---|---|---|---|---|---|
| YLoc-LowRes | _F_1 | 0.75 | 0.76 | 0.78 | 0.80 | 0.84 | 0.95 |
| ACC | 0.79 | 0.79 | 0.81 | 0.86 | 0.91 | 0.93 | |
| No. of instances | 576 | 467 | 395 | 299 | 189 | 118 | |
| YLoc-HighRes | _F_1 | 0.69 | 0.74 | 0.76 | 0.76 | 0.77 | 0.77 |
| ACC | 0.74 | 0.78 | 0.80 | 0.82 | 0.83 | 0.84 | |
| No. of instances | 576 | 507 | 470 | 428 | 391 | 354 | |
| YLoc+ | _F_1 | 0.67 | 0.69 | 0.72 | 0.77 | 0.76 | 0.81 |
| ACC | 0.58 | 0.60 | 0.62 | 0.65 | 0.65 | 0.69 | |
| No. of instances | 576 | 494 | 423 | 324 | 219 | 142 |
We tested YLoc+’s ability to predict multiple localization sites in a nested 5-fold cross-validation scheme on the DBMLoc data set (22). We found that YLoc yields an ACC of 0.64 and an _F_1 of 0.68 using multilabel measures. YLoc+ correctly identifies half of the proteins as multiple targeted and predicts both locations correctly in about one-third of the cases.
WEB SERVER
The YLoc web server requires protein sequences in FASTA format as input. It allows users to predict the location of at most 20 proteins. For large-scale predictions, users can access YLoc via SOAP or HTTP using the Python-based client scripts provided on the YLoc web site. Users can choose between three YLoc predictors, YLoc-LowRes, YLoc-HighRes and YLoc+, and three protein origins, animals, fungi and plants. In addition, they can switch off the use of GO term-based features. In this case, YLoc uses models in which the GO terms from close homologs are replaced by sequence-based features. Consequently, these YLoc models rely less on the presence of close homologous proteins. Every prediction will be assigned with a prediction ID that can be used to retrieve results later on. Alternatively, users can simply bookmark the waiting page or result page to obtain results later. Currently, predictions are saved for 2 weeks. The location prediction of a single protein takes 10–20 s, depending on the protein length.
Prediction results are displayed in three levels of details. The prediction summary presents the predicted location(s), the probability of those and the confidence score for every query protein. The probability of a location is simply how likely the protein is located in this compartment. In contrast, the confidence score is a measure of reliability. A low confidence score implies the possibility that the real probability can differ considerably from the predicted probability. However, a high confidence score signifies that the predicted probability is close to the real probability for being located in the predicted location. Consequently, higher confidence scores imply a higher reliability of the prediction for the individual sequence. In addition, an explanation in natural language clarifies why the prediction has been made. This explanation includes the two most likely reasons for this localization, for example: ‘The most important reason for making this prediction is the strong SP sorting signal’ or ‘Moreover, it is a barely charged protein.’ This information can be very important since it might already give a hint of the underlying mechanism for this protein localization.
The detailed prediction page provides more information on a particular protein prediction. For example, the probability distribution of the locations is provided. It is important to know the runner-up locations, especially for low confidence predictions, since rather ambiguous predictions should be inspected manually. YLoc also provides the most similar protein from Swiss-Prot 42.0 and associated GO terms. More details of how protein attributes influence the prediction are given in a large attribute table (Figure 1). The attributes are expressed in biological terms and ordered according to their absolute discrimination score, which corresponds to its influence on the prediction. A positive discrimination score implies that the attribute value is very typical for the predicted location, but atypical for some other location. In contrast, a negative discrimination score implies that the attribute value is more typical for some other location than the predicted one. A simple +/− encoding shows whether an attribute is typical for a location or not. By simply inspecting only the first lines of the table, it is sometimes already obvious which biological property lead to the prediction outcome and is likely to be responsible for the real localization of the protein. In addition, it gives hints of which parts of the protein should be considered for protein engineering.
Figure 1.

The attribute table of the YLoc web service lists all attributes in order of their influence on the prediction outcome. All attributes are expressed in biological terms. The +(+) or −(−) indicates whether that attribute value is (very) typical or (very) untypical for a location.
How a particular biological attribute is calculated can be found on a detailed attribute page (Figure 2). For example, YLoc-LowRes (animal version) calculates the strength of the SP sorting signal using the ‘autocorrelation of every third hydrophobic amino acid within the first 20 amino acids in the N-terminus’. Knowing how the attribute value is calculated is essential to understand which particular amino acids and properties encode for possible sorting signal. Furthermore, the attribute is visualized. Embedded Javascript code displays the distribution of proteins from the different locations regarding this feature. The provided protein distributions are very helpful for understanding how proteins from different locations behave with respect to a biological property or sorting signal.
Figure 2.

The detailed attribute page of the feature ‘secretory pathway sorting signal’ in YLoc-LowRes (animal version). The distribution of proteins from the cy, mi, nu and SP over the different attribute intervals is shown.
APPLICATION
The interpretable YLoc web service can be applied to numerous tasks that range from large-scale predictions to the identification of sorting signals. A very interesting application example is supervised protein engineering. YLoc can identify biological properties, e.g. example sorting signals that might be responsible for the localization. For example, human fumerate hydratase (FH, SwissProt AC P07954) is primarily located in the mi. The three YLoc predictors (animal version) detect the correct location and identify a mitochondrial targeting peptide (mTP). After truncating the leading 43 residues, FH lacks an mTP and shows a negatively charged N-terminus which is unfavorable for mitochondrial localization. Consequently, YLoc predicts FH to be cytoplasmic. In fact, the truncated FH protein is a known cytoplasmic isoform of FH encoded by the same gene (29). This example shows that YLoc can by valuable in location engineering of proteins.
YLoc VIA SOAP
For large-scale predictions, YLoc can be accessed via SOAP. The corresponding WSDL can be downloaded from the YLoc web site. In addition, we provide a Python-based script. Alternatively, YLoc can be accessed via an HTTP-based client that is also available for download.
CONCLUSION
As an interpretable web server for predicting subcellular localization of proteins, YLoc explains why a prediction was made and what features are likely to be responsible for the protein localization. This information can be very helpful to understand the localization of a protein and thus can assist in location engineering of proteins. Furthermore, a confidence score rates the reliability of a prediction. At the same time, it performs comparably with other state-of-the-art predictors. We believe that YLoc is a valuable alternative to experimental methods and current state-of-the-art predictors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
LGFG Promotionsverbund ‘Pflanzliche Sensorhistidinkinasen’ of the Universtity of Tübingen (S.B.). Funding for open access charge: LGFG Promotionsverbund ‘Pflanzliche Sensorhistidinkinasen’ of the Universtity of Tübingen.
Conflict of interest statement. None declared.
Supplementary Material
[Supplementary Data]
ACKNOWLEDGEMENTS
We thank Jan Schulze for technical support and Nora Toussaint for comments on the manuscript.
REFERENCES
- 1.Nakai K, Kanehisa M. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horton P, Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbor classifier. Intell. Syst. Mol. Biol. 1997;5:147–152. [PubMed] [Google Scholar]
- 3.Chou K, Cai Y. Prediction and classification of protein subcellular location-sequence-order effect and pseudo amino acid composition. J. Cell. Biochem. 2003;90:1250–1260. doi: 10.1002/jcb.10719. [DOI] [PubMed] [Google Scholar]
- 4.Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J. Mol. Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 5.Petsalaki E, Bagos P, Litou Z, Hamodrakas S. PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Genomics Proteomics Bioinformatics. 2006;4:48–55. doi: 10.1016/S1672-0229(06)60016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pierleoni A, Martelli P, Fariselli P, Casadio R. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22:e408–e416. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- 7.Höglund A, Dönnes P, Blum T, Adolph H, Kohlbacher O. MultiLoc: prediction of protein subcellular localization using N-terminal targeting sequences, sequence motifs and amino acid composition. Bioinformatics. 2006;22:1158–1165. doi: 10.1093/bioinformatics/btl002. [DOI] [PubMed] [Google Scholar]
- 8.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 9.Horton P, Park K, Obayashi T, Fujita N, Harada H, Adams-Collier C, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou K, Cai Y. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 2002;277:45765–45769. doi: 10.1074/jbc.M204161200. [DOI] [PubMed] [Google Scholar]
- 11.Scott M, Thomas D, Hallett M. Predicting subcellular localization via protein motif co-occurrence. Genome Res. 2004;14:1957–1966. doi: 10.1101/gr.2650004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg A, Raghava G. ESLpred 2: improved method for predicting subcellular localization of eukaryotic proteins. BMC Bioinformatics. 2008;9:503. doi: 10.1186/1471-2105-9-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin HN, Chen CT, Sung TY, Ho SY, Hsu WL. Protein subcellular localization prediction of eukaryotes using a knowledge-based approach. BMC Bioinformatics. 2009;10:S8. doi: 10.1186/1471-2105-10-S15-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Tung C, Ho S, Hwang S, Ho S. ProLoc-GO: utilizing informative Gene Ontology terms for sequence-based prediction of protein subcellular localization. BMC Bioinformatics. 2008;9:80. doi: 10.1186/1471-2105-9-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady S, Shatkay H. Pacific Symposium on Biocomputing. World Scientific; 2008. EpiLoc: a (working) text-based system for predicting protein subcellular location; pp. 604–615. [PubMed] [Google Scholar]
- 16.Fyshe A, Liu Y, Szafron D, Greiner R, Lu P. Improving subcellular localization prediction using text classification and the Gene Ontology. Bioinformatics. 2008;24:2512–2517. doi: 10.1093/bioinformatics/btn463. [DOI] [PubMed] [Google Scholar]
- 17.Chou K, Cai Y. A new hybrid approach to predict subcellular localization of proteins by incorporating Gene Ontology. Biochem. Biophys. Res. Commun. 2003;311:743–747. doi: 10.1016/j.bbrc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 18.Scott M, Calafell S, Thomas D, Hallett M. Refining protein subcellular localization. PLoS Comput. Biol. 2005;1:e66. doi: 10.1371/journal.pcbi.0010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou K, Shen H. Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J. Proteome Res. 2007;6:1728–1734. doi: 10.1021/pr060635i. [DOI] [PubMed] [Google Scholar]
- 20.Blum T, Briesemeister S, Kohlbacher O. MultiLoc2: integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinformatics. 2009;10:274. doi: 10.1186/1471-2105-10-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briesemeister S, Blum T, Brady S, Lam Y, Kohlbacher O, Shatkay H. SherLoc2: a high-accuracy hybrid method for predicting protein subcellular localization. J. Proteome Res. 2009;8:5363–5366. doi: 10.1021/pr900665y. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S, Xia X, Shen J, Zhou Y, Sun Z. DBMLoc: a database of proteins with multiple subcellular localizations. BMC Bioinformatics. 2008;9:127. doi: 10.1186/1471-2105-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall M. Proceedings of the Seventeenth International Conference on Machine Learning. Morgan Kaufman Publishers; 2000. Correlation-based feature selection for discrete and numeric class machine learning; pp. 359–366. [Google Scholar]
- 24.Whitten I, Frank E. Data Mining: Practical Machine Learning Tools and Techniques. San Fransisco, CA: Morgan Kaufman Publishers; 2005. [Google Scholar]
- 25.Fayyad UM, Irani K. Proceedings of the 13th International Joint Conference on Artificial Intelligence. Morgan Kaufmann Publishers; 1993. Multi-interval discretization of continuous-valued attributes for classification learning; pp. 1022–1027. [Google Scholar]
- 26.Briesemeister S, Rahnenführer J, Kohlbacher O. Going from where to why – interpretable prediction of protein subcellular localization. Bioinformatics. 2010;26:1232–1238. doi: 10.1093/bioinformatics/btq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casadio R, Martelli P, Pierleoni A. The prediction of protein subcellular localization from sequence: a shortcut to functional genome annotation. Brief. Funct. Genomic Proteomic. 2008;7:63–67. doi: 10.1093/bfgp/eln003. [DOI] [PubMed] [Google Scholar]
- 28.Tsoumakas G, Katakis I. Multi-label classification: an overview. Int. J. Data Warehousing Min. 2007;3:1–13. [Google Scholar]
- 29.Tolley E, Craig I. Presence of two forms of fumarase (fumarate hydratase E.C. 4.2.1.2) in mammalian cells: immunological characterization and genetic analysis in somatic cell hybrids. Confirmation of the assignment of a gene necessary for the enzyme expression to human chromosome 1. Biochem. Genet. 1975;13:867–883. doi: 10.1007/BF00484417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Data]