Efficient Cross-presentation Depends on Autophagy in Tumor Cells (original) (raw)
. Author manuscript; available in PMC: 2010 Jul 19.
Abstract
Cross-presentation of antigens is critical for the induction of adaptive immunity against tumor cells and infectious pathogens. Currently, it is not known how cross-presentation of tumor antigens is regulated by autophagy. Using both HEK293T cells that expressed the model antigen OVA and melanoma cells as antigen donors, we show that macro-autophagy in tumor cells is essential for cross-presentation by dendritic cells both in vitro and in vivo. Inhibition of autophagy abolished cross-presentation almost completely, whereas induction of autophagy dramatically enhanced cross-presentation of tumor antigens. Moreover, purified autophagosomes were found to be efficient antigen carriers for cross-presentation. Our findings not only identified a novel role for autophagy as an active process in antigen sequestration and delivery to dendritic cells for cross-presentation, but also suggested, for the first time, that isolated autophagosomes may have potential as potent vaccines for immunotherapy against cancer and infectious diseases.
Keywords: autophagy, antigen cross-presentation, DC, autophagosome
Introduction
Antigen cross-presentation is critical for the activation of T cells to viral and tumor antigens that are expressed by parenchymal cells and necessary for the elimination of tumor cells and many pathogens. Cross-presentation is the process by which the antigens from “donor cells” are captured, processed, and then presented to antigen-specific T cells by host professional antigen-presenting cells (APC) (1). Although the mechanisms of antigen capture, processing and presentation by APCs, have been well-appreciated, the forms of the antigen that are actually transferred from the donor cells to the APC are not clear. Furthermore, how the “donor cells” process their antigens and supply them to DCs for cross-presentation has not been well understood. The cellular proteins in the donor cell that are potentially available for cross-presentation are degraded either by the proteasome or in the lysosome after autophagy. It has been suggested that the substrates and/or products of either of these two pathways, e.g. stable intact proteins, proteasome-generated peptides, as well as peptides chaperoned by heat shock proteins, can serve as the relevant form of antigen for cross-presentation (2, 3). Defective ribosomal products (DRiPs) and short-lived proteins are commonly degraded by the proteasome pathway. It has been shown that an active proteasome in virus-infected cells may actually suppress cross-presentation of short-lived proteins, including DRiPs, and that proteasome activity is not required for the cross-priming of CD8 T cells against long-lived proteins (3). Macroautophagy (herein, autophagy) participates in the bulk degradation of long-lived proteins (4) and may play an important role in the digestion of short-lived proteins, such as misfolded proteins or DRiPs (5, 6). During autophagy, a double-membrane structure sequesters misfolded proteins and damaged organelles in the cytosol and forms autophagosomes. Fusion of autophagosomes with the MHC II-containing compartment, such as late endosomes and lysosomes, enhances MHC II presentation of cytosolic proteins and viral antigens (7, 8); however, the role of autophagy in cross-presentation, either in vitro or in vivo, has not been elucidated.
There are three major stages in the autophagy pathway (4). The initiation stage is the de novo formation of an isolation membrane (also called phagophore), which is regulated by mammalian target of rapamycin (mTor), Beclin 1 (Atg6) and type III PI-3 Kinase (hVPS-34). The second stage is elongation during which the isolation membrane expands and damaged organelles or cytosolic materials are captured. A critical step in the second stage is the conjugation of Atg5 to the ubiquitin-like molecule, Atg 12, and to membrane lipid by another ubiquitin-like molecule, Atg8. These two conjugation steps are critical for the expansion of the isolation membrane via tethering and hemifusion of the phagophore membranes (9). The third stage involves formation of autophagosomes, which is followed by rapid transition into autolysosomes upon fusion with lysosomes, and targets the captured materials for degradation.
Because autophagy is the another major cellular pathway mediates the protein degradation, we hypothesized that inhibition of autophagy, like inhibition of the proteasome augmented cross-presentation of short-lived proteins (3), would enhance cross-presentation of long-lived protein antigens. To make this determination, we measured the effects of inhibition or induction of autophagy on cross-presentation by mouse dendritic cells to transgenic T cells using melanoma cells or OVA-expressing human HEK 293T cells as the antigen donor cells. Cross-presentation of antigens derived from whole tumor cells in which autophagy was altered, was assessed by T-cell proliferation both in vitro and in vivo. In addition, autophagosome-enriched fractions and purified autophagosomes were used for the antigen source for the cross-presentation after being loaded onto dendritic cells.
Materials and Methods
Mice
C57BL/6 mice were purchased from the Charles River Laboratories. TCR transgenic OT-I breeders were from the Jackson Laboratory. Pmel-1 breeders transgenic for the TCR that recognize mouse and human gp10025–33 were obtained from Dr. Nicholas P. Restifo (National Cancer Institute, NIH) (10). All mice were maintained and used in accordance with the Earle A. Chiles Research Institute Animal Care and Use Committee.
DNA construction and transfection
Plasmid DNA vector cloning: The Ub-X-GFP (Ub=ubiquitin, X =M, R, or V.) expressing plasmids were kindly provided by Dr. M. G. Masucci (11). The Ub-X-GFP-OVA and Ub-X-GFP-TfR-OVA fusion constructs were made by PCR with Vent polymerase and cloned into the lentiviral vector pWPT (supplementary figure S1). The pWPT vector was kindly provided by Dr. D. Trono and modified to enable the convenient cloning method with the NEB USER Enzyme (New England Biolab, Ipswich, MA). LC3 fusion plamids, pCMV-GFP-LC3, and pCMV-tdTomato-LC3 were kindly provided by Dr. T. Johansen (22) and subcloned into pWPT vector after PCR with Vent polymerase. CFP-LC3 and tdTomato-Ub fusion plasmids were constructed by PCR and cloned into pWPT vector. The second generation lentiviral siRNA vector containing a new design of Mir30 modified short hairpin RNA (shRNAmir) against Beclin 1 (V2MM-9827) and nonspecific control shRNAmir plasmids were purchased from Open Biosystems (Birmingham, AL). For the generation of recombinant lentiviruses, HEK 293 cells were transiently transfected with vector plasmid pWPT or pGIPz, virus packaging plasmid pPAX2, envelope plasmid VSV-G MD2, and helper plasmid pAdv. Viral supernatant was used to infect tumor cells and tumor cells were sorted based on GFP expression by flow cytometry (OVA fusion proteins) or selected with puromycin (Beclin 1).
Fractionation and purification of autophagosomes
HEK 293T cells were transfected with CFP-LC3 and OVA antigen. After 18 hrs of treatment with bortezomib (100nM) and NH4Cl (10 mM), cells were disrupted by mild sonication at 115 V, 56-60HZ using the G112SP1G Special Ultrasonic Cleaner (Laboratory Supplies CO., Inc.). Here bortezomib was used as an inhibitor of protesome activity, and an inducer of autophagy as well (supplementary figure S3B). NH4Cl was used to block lysosomal fusion and degradation of autophagosomes. Cell lysates were precleared by centrifugation at 300g for 10min, and then were separated into the crude autophagosome-containing large vesicles and the supernatant consisting of cytosolic components by a 15-min centrifugation at 10,000 g. A portion of the crude large vesicle preparations was fractionated by self-generated percoll gradient sedimentation. Vesicles from 5-million cells were resuspended in 20mM HEPES, PH7.0, with 0.3M sucrose, 27% percoll, and 1mM EDTA. The gradient was centrifuged 30 min at 36,000g in a NVT65 rotor in a Beckman Optima L-90K Ultracentrifuge (Beckman Coulter, Inc.). Twelve fractions were collected using Pasteur pipettes. For autophagosome purification, GFP-LC3-marked autophagosomes were pulled down using Sheep anti-Mouse IgG Dynabeads (Dynal Biotec) in combination with mouse anti-GFP antibody (Stressgen Bioreagents, Victoria, BC, Canada) according to the manufacturer’s recommendations. Fractions of the flow through, wash, and eluate were collected for further analysis. The distribution of autophagosomes within each fraction was determined by western blot of the GFP-LC3 marker, and the ability of these fractions to activate OTI CD8 T cells in the presence of DCs was analyzed by CFSE dilution.
Western blotting
1×106 HEK293 cells expressing GFP-OVA fusion protein (treated or untreated) were lysed in 100 μl RIPA buffer. The lysates were mixed with 4X NuPAGE LDS sample buffer and samples (10μl each) were resolved by 4–20% SDS-PAGE (Invitrogen). Proteins were transferred to a nitrocellulose membrane, incubated with primary antibodies diluted in blocking buffer (5% dry milk) overnight, and then exposed to HRP-conjugated secondary antibodies for 1 hr. Protein bands were revealed by using chemiluminescent reagents (Pierce). The primary antibodies included rabbit anti-actin (1:2,000, Sigma), biotin-conjugated anti-GFP (1:5000, Rockland, Gilbertsville, PA), anti-Beclin 1 (1:1000, ProSci), and mouse anti-GFP (1:1000, Stressgen). The secondary antibodies were avidin-HRP (1:10000, eBioscience), goat-anti rabbit-HRP (1:10000, Jackson ImmunoResearch), and goat anti-mouse-HRP (1:10000, Jackson ImmunoResearch).
Fluorescent Microscopy
Images of live cells were taken using a Leica inverted microscope capable of digital epifluorescence imaging. A GFP filter (excited at 470/40, dichromatic mirror at 495, and long pass emission filter 500LP) and an Orange filter (excited at 525/50, dichromatic mirror at 555, and band pass emission filter 590/50) were used to capture fluorescent images.
Modulation of autophagy and proteasome activity
Autophagy of antigen donor cells was inhibited by the addition of 3-methyladenine (3-MA, 10 mM, Sigma) or wortmannin (1.0 μM, Calbiochem) for 12–18 hrs. To induce autophagy, donor cells were grown in the presence of rapamycin (20 nM, ALEXIS), or HBSS (amino acid starvation), for 12–18 hrs. NH4Cl (10 mM, Sigma) or Bortezomib (100 nM, Millenium Pharmaceuticals) was used for 18–24 hrs to block lysosome or proteasome activity respectively.
ATG12 or Beclin 1 knock down by siRNA
Synthetic ATG12 siRNAs and control siRNAs were purchased from Invitrogen. HEK 293T cells expressing GFP-OVA fusion protein were transfected with siRNAs together with INTERFERin (Polyplus Transfection) following the manufacturer’s protocol. Knockdown of ATG 12 was verified by RT-PCR 24 hrs later. mRNA was isolated form fresh cells using the RNeasy Mini Kit (Qiagen) 24–40 hrs after siRNA tranfection and reverse transcribed to first strand cDNA using an oligo dT primer (Promega). For knockdown of mouse Beclin 1, B16 F10 melanoma cells were infected with shRNAmir lentiviral vector (pGIPz, Open Biosystems) and selected with puromycin.
CFSE labeling and in vitro/in vivo CFSE dilution assay
Cross presentation of antigens to OT-I or Pmel-1 naive T cells was assessed by measuring dilution of CFSE by flow cytometry (9). DCs were isolated from spleens of C57BL6 mice after sequential intravenous injection of plasmid DNA encoding murine Flt3 ligand and GM-CSF (12). For the in vitro CFSE assay, antigen donor cells (3 ×105), with or without treatment, were irradiated at 10,000cGY and incubated with 2×106 DC for 6 hours, the mixed cells were then washed three times before the addition of 3×106 CFSE-labeled OT-I or Pmel-1 T cells. Splenocytes from OT-I or Pmel-1 mice were labeled with 5 μM CFSE according to the manufacturer’s protocol (Invitrogen). T-cell proliferation was measured after 4 or 5 days of DC and T cell co-incubation. For the in vivo CFSE assay, antigen donor cells (HEK293T, FEMX, or B16F10) treated with various chemicals or siRNAs were collected and washed extensively in PBS, and subcutaneously injected into both flanks of C57BL6 mice. The same day 5×106 Thy1.1+ OT1 or Pmel-1 T cells labeled with CFSE were adoptively transferred into these mice intravenously. Lymph nodes were collected 5 days later and single cell suspensions were prepared. Proliferation of CFSE-labeled T cells was analyzed and the percentage of adoptively transferred T cells to total CD8+ lymphocytes was determined by flow cytometry analysis.
Results and Discussion
The autophagy pathway in antigen donor cells affects cross-presentation
Since lysosomal degradation of proteins in the final stage of autophagy could decrease the supply of antigens for cross-presentation, we examined whether activation or inhibition of autophagy in donor cells would affect cross-presentation of tumor antigens. First, HEK 293T cells that expressed the V-GFP-TFR-OVA fusion protein were used as the antigen donor. The V-GFP-TFR-OVA fusion protein is a membrane-bound, long-lived protein (supplementary figure S1). OVA-expressing HEK 293T cells were treated with rapamycin or subjected to starvation to induce autophagy, or they were treated with the PI3K inhibitors, 3-methyl-adenosine (3-MA), to prevent the initiation of autophagy. The treated HEK293T cells were irradiated and incubated with dendritic cells for 6 hrs and then CFSE-labelled naïve OT-I transgenic cells were added to the culture. T-cell proliferation was measured by flow cytometry analysis of the CFSE profile of gated CD8+ T cells. To our surprise, cross-presentation of OVA antigen was greatly increased after treatment with rampamycin (Figure 1A) or starvation (not shown). Conversely, inhibition of autophagy in antigen donor cells with either 3-MA significantly decreased cross-presentation of OVA antigen to OT-I T cells, close to the level of background T-cell proliferation without antigen (Fig. 1A). Moreover, percentage of cell death, which was judged by staining treated HEK293T cells with annexin and propidium iodide (PI) or 7-AAD, did not appear to be related to the alteration in cross-presentation due to autophagy modulation. No signinigicant apoptosis nor necrosis of tumor cells was identified 18 hours after any of the treatments, indicating that cell viability was not affected by either the induction or inhibition of autophapy before irradiation (Fig. 1A). However, cells underwent progressive death after irradiation and coincubation with DCs. Induction of autophagy prior to irradiation hindered cell death, while inhibition of autophagy accelerated cell death (supplementary figure S2). To ensure that these observations were not an artifact of the OVA model, we also examined cross-presentation of an endogenous tumor antigen, gp100, expressed by the human melanoma cell line, FEMX, after inhibiting and inducing autophagy. By assessing proliferation of adoptively transferred gp100-specific pmel-1 transgenic T cells (10), we found that in vivo cross-presentation of gp100 was also regulated by autophagy. Treatment of FEMX tumor cells with rapamycin before injection into mice significantly increased cross-presentation of gp100 antigen as indicated by both CFSE profile and number of pmel-1 T cells in the draining lymph nodes (Fig. 1B and C). Most importantly, both autophagy inhibitors, 3-MA and wortmannin, almost completely abolished the cross-presentation of gp100 to gp100-specific transgenic T cells. As expected, treatment of either OVA-expressing HEK 293T or FEMX cells with rapamycin led to conversion of autophagy gene Atg8, from a LC3-I form to LC3-II form (Fig. 1D). These results indicate that cross presentation of tumor antigens depends on the early phase of autophagy (i.e. formation of early autophagosomes). Interestingly, when NH4Cl was included to prevent lysosome acidification and fusion and the subsequent degradation of proteins, cross-presentation was augmented. These data suggested that while the early phases of autophagy (initiation and formation of autophagosomes) was required for efficient cross-presentation, the late phase of autophagy during which the encapsulated proteins are degraded in lysosomes hinders cross-presentation. Apoptosis and necrosis of tumor cells have been reported to affect antigen cross-presentation (13, 14); here we showed that autophagy in antigen donor cells also had a dramatic effects on the efficiency of cross-presentation for both model and endogenous tumor antigens.
Figure 1. Cross-presentation of OVA and gp100 to transgenic T cells was greatly affected by the inhibition or induction of autophagy in the antigen donor cells.
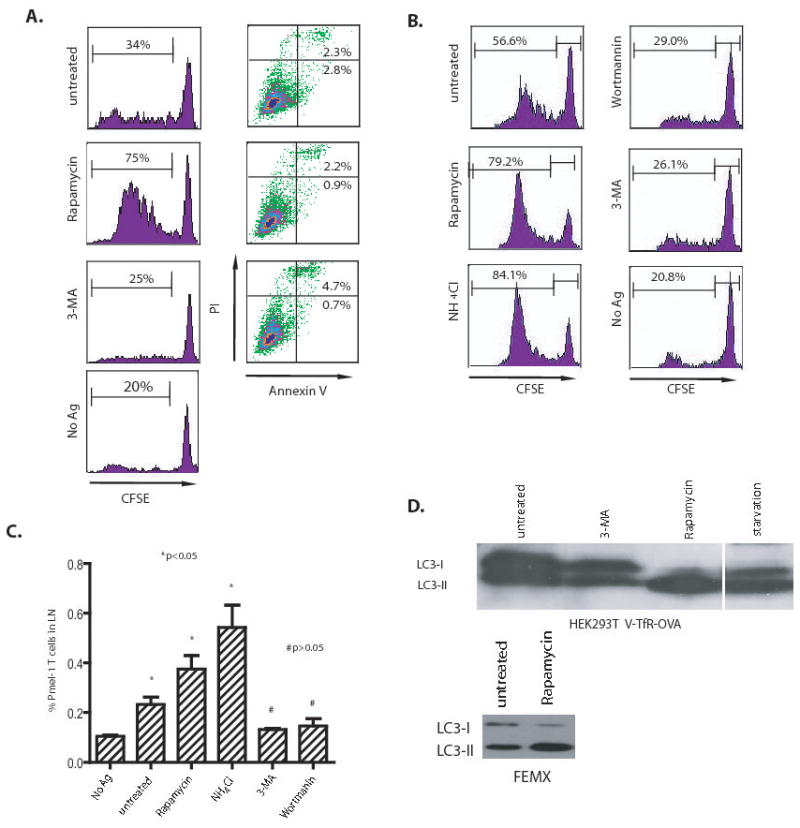
(A) Modulation of cross-presentation of the OVA model antigen in vitro. HEK 293T cells expressing the GFP-OVA fusion protein were treated with the autophagy-inhibitors, 3-MA or wortmanin, or the autophagy-inducer, rapamycin for 18 hours, or subjected to starvation by culturing donor cells in HBSS for 18 hours. PI and FITC-labelled annexin V were used to stain treated cells to measure the levels of apoptosis or necrosis, respectively. To assess cross-presentation, treated tumor cells were irradiated and incubated with DC for 6 hours. CFSE-labeled naive T cells from OT-I TCR transgenic mice were added to the mixture of DC and 293T donor cells and cultured for 4 to 5 days. The dilution of CFSE label in OT-I T cells was determined by flow cytometry. The results are expressed as the percentage of OT-I T cells that had undergone at least one division 4 days after coculture with DC and donor cells. The data represent typical results of three to five independent experiments.
(B) and (C) Modulation of cross-presentation of the melanoma gp100 antigen in vivo. Cells from the human melanoma cell line FEMX were treated with the indicated agents as described above and then injected into the flanks of naïve C57BL/6 mice. CFSE-labelled naïve spleen cells from pmel-1 transgenic mice were adoptively transferred into tumor-bearing mice and lymph nodes draining the tumors were collected at day 6. Both the CFSE profile of pmel-1 CD8 T cells (B) and the percentage of pmel-1 T cells among the lymph node lymphocytes (C) were determined by flow cytometry. Mice that received no tumor or untreated FEMX cells were included as the controls. Each group consisted of 4 mice and the experiment was repeated once with similar results. The difference between treated and untreated groups was significant (*p <0.05); however, the difference between no Ag and 3-MA or wortmannin-treated group was not significant (#p >0.05).
(D) HEK 293T cells expressing OVA antigen or FEMX melanoma cells were treated with 20 nM rapamycin or 10 mM 3-MA for 18 hours. Lysates were prepared from both untreated and treated cells. Lysate from starved HEK 293T cells was included as positive control. Ten microgram of total proteins were loaded and subjected to SDS PAGE and western blot analysis with rabbit anti-LC3 antibody.
Knock down of the autophagy initiation gene, Beclin 1, in antigen donor tumor cells greatly reduced cross-presentation
The above-mentioned inhibitors of autophagy are not specific and may affect other cellular processes. Therefore, we sought to confirm the role of autophagy in cross-presentation using RNA interference to knock down the essential autophagy initiation gene, Atg6/Beclin 1, in B16 F10 murine melanoma cells. Atg6/Beclin 1 regulates autophagy by forming a complex with Class III phosphatidylinositol 3′-kinase (PI3K), a critical lipid kinase involved in membrane trafficking and fusion, and modulating its activity (15). To knock down Beclin 1, a stable B16 F10 cell line that expressed shRNAmir (a short hair pin modified with backbone of mir-30 microRNA) was generated using a lentiviral vector. The Beclin 1 protein level in transduced B16 F10 was reduced to approximately one-half that found in B16 F10 melanoma cells that expressed the control nonspecific NS shRNAmir (Fig. 2A). To show that F10 melanoma cells expressing shRNAmir against Beclin-1 were in fact defective in autophagy induction, the rapamycin induced conversion of LC3 was examined in F10 cells expressing either control shRNA or Beclin-1-specific shRNAmir. As expected, rapamycin increased LC3-II found in F10 cells expressing control shRNAmir but not F10 cells expressing shRNAmir specific to Beclin-1 (Fig. 2B). To examine the effects of Beclin 1 knock down on cross-presentation, each melanoma cell line was injected into mice infused with CFSE-labelled gp100-specific pmel-1 T cells. Melanoma cells with a knock down of Beclin 1 were far less able to cross-present gp100 as evidenced by significantly less T-cell proliferation compared to T cells that were exposed to tumor cells treated with control siRNA (Fig. 2C). Knock down Beclin-1 reduced the percentage of pmel-1 T cells in the draining lymph nodes to the background level of control mice without tumor injection (Fig. 2D). Since Beclin-1 knockdown could have affected direct presentation by B16F10 cells, we performed an additional experiment in TAP-1 deficient mice; endogenous DC from these mice will not be able to cross-present peptides to T cells. The near complete absence of pmel-1 proliferation in TAP-1 deficient mice injected with tumors treated with control or Beclin-1 siRNA essentially ruled out direct presentation. Thus, cross-presentation was significantly impaired in Beclin-1 knockdown mice. This also confirmed an earlier study that showed cross-presentation was the major pathway by which T cells were activated and induced to proliferate in mice inoculated with B16 melanoma cells (16).
Figure 2. Knock-down of Beclin 1 in antigen donor cells decreased cross-presentation of tumor antigens by DC.

(A) B16 F10 melanoma cells were transduced with lentiviral vectors encoding a mir-30 modified short hairpin RNA (shRNAmir) specific for mouse Beclin 1 or a nonspecific control shRNAmir. The specific knockdown of Beclin 1 was confirmed by western blot analysis with anti-beclin 1 antibody. Anti- actin antibody was included as the control.
(B) F10 melanoma cells expressing either control or Beclin-1 shRNAmirs were treated with 20 nM rapamycin for 18 hours. Lysates were prepared from both untreated and treated cells. Ten microgram of total proteins were loaded and subjected to SDS PAGE and western blot analysis with rabbit anti-LC3 antibody.
(C) B16 F10 melanoma cells that were transduced with beclin 1 or control shRNAmir were injected into both flanks of C57BL/6 WT or TAP1 knock-out mice (n=4) that received 5 x106 Thy1.1+ pmel-1 transgenic T cells. Six days after tumor injection, the CFSE profile of transferred pmel-1 T cells from the draining lymph nodes was determined.
(D) The percentage of pmel-1 T cells found among the lymphocytes in the draining LNs of each mouse was determined by flow cytometry. All values indicate the mean with standard deviation (n=4). The difference between control and Beclin-1 knockdown was significant; however, the difference between beclin-1knockdown and no antigen was not significant.
The data represent the results from three independent experiments.
Knock down of Atg12, a critical gene for the formation of autophagosomes, in antigen donor cells significantly reduced cross-presentation
In order to determine what portion of the autophagy pathway was important for cross-presentation, we investigated whether the formation of autophagosomes (the second stage of autophagy) was required for efficient cross-presentation. The elongation phase of autophagy, which leads to the formation of autophagosomes that capture damaged organelles or cytosolic material, requires the initial conjugation of Atg12-Atg5 and the subsequent conjugation with Atg8 (LC3) and membrane phospholipids (4). This aspect of the pathway was inhibited by specific knockdown of Atg12 by siRNA in OVA-expressing HEK 293T cells. Decreased Atg12 expression was documented by RT-PCR (Fig. 3A). The lack of Atg12 protein would prevent incorporation of Atg8 in the lipid membrane and interfere with formation of the autophagosome. Thus we would expect fewer LC3-positive autophagosomes in Atg12 knockdown cells. This can be readily seen in Fig. 3B where there were far fewer LC3-positive punctuates in donor cells transfected with Atg12 siRNA compared to cells transfected with control siRNA. This confirmed that autophagy had been reduced and fewer autophagosomes had been produced. The donor cells’ ability to cross-present antigen in vitro following knockdown of Atg12 was notably reduced (Fig. 3C). In vivo cross-presentation was also decreased when antigen donor cells were transfected with Atg12 siRNA before inoculation into mice (Fig. 3D). Four days after tumor inoculation the percentage of OT-I T cells in the draining lymph nodes of mice immunized with donor tumor cells transfected with Atg12 siRNA was significantly reduced compared to the percentage of T cells in mice immunized with untransfected or control siRNA transfected donor cells. These results further support an important role for autophagy in the efficient cross-presentation of protein antigens from tumor cells.
Figure 3. Cross-presentation was reduced when autophagy was inhibited by knock down of Atg12.

(A) RT-PCR for Atg12 mRNA in HEK 293 T cells expressing the GFP-OVA fusion protein after transient transfection with Atg12 or control siRNA. Actin mRNA was included as the control.
(B) Diminished formation of tdTomato-LC3 punctates after ATG12 knockdown. HEK 293T cells stably expressing tdTomato-LC3 fusion protein following infection with a viral vector were transfected with Atg12 siRNA or control siRNA. Fourty-eight hours later, transfected cells were treated with rapamycin and NH4Cl for 6 hours to induce the formation of punctates.
(C) Reduced cross-presentation of OVA from donor cells after transfection with Atg12 siRNA in vitro. HEK 293T cells that expressed GFP-OVA fusion protein were transfected with Atg 12 or luciferase siRNA 24 hours before they were loaded onto DC. CFSE-labeled OT-I naive T cells were added after 6 hrs of DC and 293T coculture and the dilution of CFSE-label in OT-I T cells at day 4 was determined by flow cytometry. The results are typical of five independent experiments. The numbers indicate the percentage of OT-I T cells that had undergone at least one cell division.
(D) Reduced cross-presentation of OVA from donor cells after transfection with Atg12 siRNA in vivo. HEK 293T cells expressing GFP-OVA fusion protein were transfected with either control siRNA or Atg12 siRNA. Two days later, control or Atg12 siRNA treated cells were injected into both flanks of mice that also received 5 × 106 Thy1.1+ OT-I transgenic T cells. HEK 293T cells expressing GFP protein were used as the negative control. The percentage of OT-I T cells found in the draining LNs of each mouse was determined individually by flow cytometry five days after injection. All values were shown as the mean with s.d. (n=4).
The typical results from three independent experiments are shown.
Autophagosomes are efficient carriers of protein antigens from donor tumor cells
Having showed both that NH4Cl blockade of the fusion between autophagosomes and lysosomes enhanced cross-presentation, and that autophagosome formation was required for cross-presentation, our results strongly suggested that autophagosomes could be the antigen carriers for cross-presentation. Using fluorescent microscopy, we visulaized localization of the protein antigen Ub-V-GFP-OVA in the LC3-positive autophagosome punctuates in HEK293T cells (supplementary figure S3A). To determine whether autophagosomes served as an antigen carrier for efficient cross-presentation, we lysed tumor cells by sonication and separated the whole cell lysate into supernatant and pellet by high-speed centrifugation (10,000g × 15min). Cytosolic proteins and microsomes remained in the supernatant, while large vesicles, which included mitochondria, lysosomes, and autophagosomes, were found in the pellet. Although the supernatant contained more GFP-OVA antigen than the pellet (data not shown), the pelleted vesicles were significantly better in stimulating proliferation of OT-I T cells when used to pulse DCs (Fig. 4A). Using a percoll gradient and high-speed centrifugation, we further fractionated the vesicles from the pellet according to their density. Autophagosomes are light vesicles and band at lower density than mitochnodria and lysosomes (15). Individual fractions were recovered and incubated with DCs and then used to stimulate CFSE-labelled naïve OT-I T cells. The ability of each individual fraction to cross-present OVA correlated directly with the amount of LC3, a specific marker of autophagosomes, in that fraction (Fig. 4B). While LC3, and therefore presumably autophagosomes, were found in many fractions, the three fractions in which LC3 could not be detected were devoid of cross-presentation ability. Using melanoma cells, we also found that fractions enriched for autophagosomes mediated the cross-presentation of gp100 antigen to naïve pmel-1 T cells (unpublished data).
Fig. 4. Autophagosomes are the source of antigen for cross-presentation.

(A) Cross-presentation activity was found primarily in the large vesicles obtained from centrifugation of cell lysates. HEK 293T cells expressing OVA and CFP-LC3 were treated with bortezomib and NH4Cl for 24 hrs and then lysed by mild sonication. The lysate was fractionated into supernatant and a pellet containing large vesicles by high-speed centrifugation (10,000g for 15 min). The pellet was resuspended in the original volume. DCs were incubated with an equal volume of supernatant or resuspended pellet and used to stimulate CFSE-labelled OT-I T cells in vitro. The dilution of CFSE-label in OT-I T cells at day 4 was determined by flow cytometry. Data represent the mean and standard deviation from four independent experiments.
(B) The highest cross-presentation activity and the greatest amount of LC3 was found in the lighter density fraction of large vesicles. The pellet was resuspended in 27% Percoll solution and subjected to ultra-speed centrifugation (36,000g for 30 min). One ml fractions were collected from the top to the bottom of the gradient. The distribution of CFP-LC3 in the gradients was analyzed by western blot with anti-GFP antibody (above the bar graph). DCs were loaded with each fraction and used to stimulate CFSE-labelled OT-I T cells in vitro. The dilution of CFSE-label in OT-I T cells at day 4 was determined by flow cytometry (shown in the bar graph). Data represent a typical result from three independent experiments.
(C) Purified autophagosomes delivered OVA very efficiently for cross-presentation. HEK 293T cells were transiently transfected with plasmids encoding GFP-LC3 and OVA. Forty-eight hours after transfection, cells were treated and lysed as above, and the large vesicles were prepared from lysates. The resuspended pellet was purified using a mouse anti-GFP antibody and magnetic beads conjugated with anti-mouse IgG. The control was performed in the absence of the mouse anti-GFP antibody. Different fractions, including the crude vesicles before purification, flow through (FT), three washing steps (W1, W2, W3), and the bead fractions (B30 and B120) were obtained and kept in their original volume. Thirty ls of each fraction were loaded onto DC, which were used to stimulate OT-1 T cells as above (FT, W1, W2, W3, B30). An additional tube with 120 ls of bead fraction was included (B120). The figure above the bar graph shows the western blot analysis of each fraction with anti-GFP antibody. Data represent a typical result from two independent experiments.
To confirm that autophagosomes labeled with LC3 were the carriers of antigen for efficient cross-presentation, autophagosomes were purified from lysates of HEK 293T cells that expressed both GFP-LC3 and OVA proteins by using a mouse anti-GFP antibody and anti-mouse IgG conjugated magnetic beads (Fig. 4C). Mock purification in the absence of anti-GFP antibody was also performed to ensure that purification was specific to GFP-LC3. Western blot analysis confirmed the specificity of purification. GFP-LC3 positive materials were obtained only when the anti-GFP antibody was used. After purification, each fraction was loaded onto DCs which were used to stimulate naïve OT-1 T cells as described above. The bead fraction collected in the presence of antibody to GFP conferred strong cross-presentation ability to DCs in a dose-dependent fashion; DCs pulsed with the same fraction isolated in the absence of GFP antibody failed to stimulate OT-1 T cells even if more material was used for loading.
Cross-presentation of tumor antigens has been shown to involve both intact whole proteins and chaperoned protein products that were generated either during protein synthesis or degradation (16, 17). Additional factors that may influence cross-presentation include the role of particulate versus soluble antigens and the role of apoptotic versus necrotic cells as antigen donors for professional antigen presenting cells. The half-life of proteins and the quantity available also are important factors in cross-presentation. Our results identified another novel, critical regulatory element of cross presentation and strongly suggest that autophagy in the antigen donor cells is a central process by which antigens are sequestrated, accumulated, and degraded prior to cross-presentation. Proteins targeted to the proteasome for degradation (short-lived proteins and DRiPs) are not efficiently cross-presented; however, proteins that accumulate in autophagosomes e.g. long-lived proteins, or accumulated DRiPs and short-lived proteins after inhibition of proteasome function, could subsequently be delivered to DC for cross-presentation if fusion with lysosomes was blocked.
Autophagy is a highly conserved, global cellular process in many cell lineages and tumor cell lines. A number of pathogens, including bacteria, viruses and parasites, induce autophagy in infected cells (18). Our findings also suggest that autophagy represents a physiologically relevant mechanism for cross-presentation of antigens derived from either tumor cells or pathogens. A deficiency of autophagy has been associated with tumorigenesis and persistence of viral infection (4, 19). Our results imply that defective autophagy could promote tumorigenesis or chronic infections by diminishing intrinsic cellular immunity (autophagy mediated degradation of intracellular pathogens), reducing innate immunity (reduced activation of innate immune cells), and by inhibiting adaptive immunity (less efficient cross-presentation). Our results suggest a new strategy by which the efficacy of vaccines that depend on cross-presentation may be increased, namely by induction of autophagy in vivo or using isolated autophagosomes as the antigen vehicles to cross-prime antitumor immune response. Our preliminary data (Supplementary Figure S4) from both mouse melanoma and lung cancer models supports the later possibility and we are planning new clinical trials using autophagosomes isolated from tumor cells as the novel cancer vaccine.
Supplementary Material
S1
S2
S3
S4
Acknowledgments
We thank Drs. B. A. Fox, S Aung, M. Crittenden, M. Fang, and C. Twitty for their input. We thank D. Healy for cell sorting and N. Handy, A. James, and Y. Trinh for their technical assistance. We are grateful to Drs. M. G. Masucci, D. Trono, and T. Johansen for reagents. This work was supported by the Providence Portland Medical Foundation, the M. J. Murdock Charitable Trust, the American Cancer Society research scholar grant LIB-106810 (to H.-M. Hu), and Human Services Public Health Service grant R01-CA107243 (to H.-M. Hu) from NIH.
References
- 1.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 2.Serna A, Ramirez MC, Soukhanova A, Sigal LJ. Cutting edge: efficient MHC class I cross-presentation during early vaccinia infection requires the transfer of proteasomal intermediates between antigen donor and presenting cells. J Immunol. 2003;171:5668–5672. doi: 10.4049/jimmunol.171.11.5668. [DOI] [PubMed] [Google Scholar]
- 3.Norbury CC, Basta S, Donohue KB, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. doi: 10.1126/science.1096378. [DOI] [PubMed] [Google Scholar]
- 4.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 6.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 7.Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 8.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a Ubiquitin-like Protein Required for Autophagosome Formation, Mediates Membrane Tethering and Hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 12.He Y, Pimenov AA, Nayak JV, Plowey J, Falo LDJ, Huang L. Intravenous injection of naked DNA encoding secreted flt3 ligand dramatically increases the number of dendritic cells and natural killer cells in vivo. Hum Gene Ther. 2000;11:547–554. doi: 10.1089/10430340050015734. [DOI] [PubMed] [Google Scholar]
- 13.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 14.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stromhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen L, Rock KL. Cellular protein is the source of cross-priming antigen in vivo. Proc Natl Acad Sci U S A. 2004;101:3035–3040. doi: 10.1073/pnas.0308345101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–599. doi: 10.1038/ni1201. [DOI] [PubMed] [Google Scholar]
- 18.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host & Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1
S2
S3
S4