Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis (original) (raw)
Abstract
Polκ and Rev1 are members of the Y family of DNA polymerases involved in tolerance to DNA damage by replicative bypass [translesion DNA synthesis (TLS)]. We demonstrate that mouse Rev1 protein physically associates with Polκ. We show too that Rev1 interacts independently with Rev7 (a subunit of a TLS polymerase, Polζ) and with two other Y-family polymerases, Polι and Polη. Mouse Polκ, Rev7, Polι and Polη each bind to the same ∼100 amino acid C-terminal region of Rev1. Furthermore, Rev7 competes directly with Polκ for binding to the Rev1 C-terminus. Notwith standing the physical interaction between Rev1 and Polκ, the DNA polymerase activity of each measured by primer extension in vitro is unaffected by the complex, either when extending normal primer-termini, when bypassing a single thymine glycol lesion, or when extending certain mismatched primer termini. Our observations suggest that Rev1 plays a role(s) in mediating protein–protein interactions among DNA polymerases required for TLS. The precise function(s) of these interactions during TLS remains to be determined.
Keywords: DNA polymerases/mutagenesis/Polκ/Rev1/translesion DNA synthesis
Introduction
Recent years have witnessed the discovery of multiple specialized DNA polymerases in prokaryotic and eukaryotic cells (Friedberg et al., 2002; Goodman, 2002). Most of these enzymes belong to an evolutionarily related protein superfamily, the polymerase Y family (Ohmori et al., 2001), members of which are devoid of 3′→5′ proofreading exonuclease activity and replicate undamaged DNA in vitro with low fidelity and weak processivity. The Y family of polymerases can replicate past a spectrum of template DNA damage by a process known as translesion synthesis (TLS). These features are shared by several other specialized polymerases from the A, B and X families.
We and others previously reported features of the mouse and human PolK (DinB)/POLK (DINB) genes and their polypeptide products, DNA polymerase κ (Polκ) (Gerlach et al., 1999; Ohashi et al., 2000; Zhang et al., 2000). Primer extension assays have shown that human Polκ can support TLS across sites of base loss, acetylaminofluorene-G adducts, benzo[_a_]pyrene-G adducts (Hubscher et al., 2002) and thymine glycol (Fischhaber et al., 2002). However, the enzyme does not support primer extension past thymine–thymine (T<>T) dimers or [6-4] pyrimidine–pyrimidone photoproducts (Hubscher et al., 2002).
Similar to another TLS DNA polymerase, Polζ, Polκ can extend terminal mismatches on undamaged templates in vitro (Haracska et al., 2002a; Prakash and Prakash, 2002; Washington et al., 2002). In addition, Polκ can extend primer-terminal nucleotides inserted opposite damaged bases by other specialized DNA polymerases (Frank et al., 2001; Haracska et al., 2002a; Zhang et al., 2002).
Rev1 is also a member of the Y family of polymerases (Ohmori et al., 2001). In contrast to its relatives, Rev1 has limited catalytic activity in vitro, which is mainly reflected in the preferential and limited incorporation of dCMP in a template-directed manner regardless of the template nucleotide (Nelson et al., 1996). Rev1 is required for error-prone TLS by polζ, but its dCMP transferase activity is not obligatory for this function (Baynton et al., 1999; Nelson et al., 2000; Lawrence, 2002). Rev1 is unable to support TLS across pyrimidine dimers or [6-4] photoproducts. Nonetheless, the REV1 gene is required for UV radiation-induced mutagenesis in yeast and human cells (Lawrence, 2002). Collectively, these observations suggest that Rev1 plays an as yet unidentified role(s) in TLS that is unrelated to the dCMP transferase activity. This suggestion is supported by recent studies showing that chicken DT40 cells in which the nucleotidyl transferase domain and C-terminal domain of Rev1 protein have been inactivated are abnormally sensitive to a variety of DNA-damaging agents (Simpson and Sale, 2003).
To further our understanding of the role of Polκ in TLS and mutagenesis, we have searched for proteins that interact with mouse Polκ (mPolκ). Here we show that mPolκ specifically interacts with mouse Rev1 protein (mRev1). We have mapped a limited C-terminal domain of mRev1 that is necessary and sufficient for this interaction. Importantly, we observed that mRev1 interacts with several other specialized DNA polymerases, notably mPolι, mPolη and the Rev7 subunit of the heterodimeric specialized polymerase mPolζ. In each case, the limited C-terminal domain of mRev1 is required for these interactions.
We also show that the catalytic activities of mRev1 and mPolκ acting in concert are not detectably altered when copying undamaged, normally base-paired DNA in vitro. However, in particular template DNA sequence contexts, mRev1 and mPolκ together exhibit primer extension activity that is greater than the additive activity of the individual polymerases when synthesizing DNA past a thymine glycol base or extending certain mismatched primer-termini. We show that this enhanced activity does not derive from Polκ/Rev1 protein complex formation.
Results
Mouse Polκ interacts with mouse Rev1 protein
To identify proteins that interact with mPolκ, a mouse testis cDNA library constructed in the two-hybrid vector pACT2 was screened using mPolκ (amino acids 100–616) as bait. Prior to the screen we determined that the bait alone did not yield transactivation in the assay. We screened ∼6 × 106 clones on quadruple drop-out (QDO) plates depleted for adenine (ade), histidine (his), leucine (leu) and tryptophan (trp). Four resulting positive colonies represented fragments of the Y-family protein mRev1 (amino acids 239–1249, 632–1249, 767–1249 and 871–1249). Yeast containing both mPolκ- and Rev1-expressing plasmids were able to grow on QDO plates (Figure 1A), whereas if either of the plasmids contained no insert, there was no growth on QDO plates (Figure 1A). Interaction between mPolκ and mRev1 in the yeast two-hybrid system was further confirmed by measuring β-galactosidase activity from a lacZ reporter gene in extracts of cells transformed with relevant plasmid pairs (Figure 1B).

Fig. 1. Interaction between mPolκ and mRev1. (A) AH109 was co-transformed with plasmid combinations as indicated and plated on QDO medium. The combinations tested were: 1, mDinB-pGBT9 + Rev1-pGADT7; 2, mDinB-pGBT9 + pGADT7; 3, mRev1-pGADT7 + pGBT9; 4, pGBT9 + pGADT7. Only the mDinB-pGBT9 + Rev1-pGADT7 combination was viable. The presence of ‘bait’ and ‘prey’ plasmids in co-transformed cells was controlled by growth on DDO media. (B) Extracts prepared from yeast transformed with plasmid combinations described above were assayed for β-galactosidase activity. Values are in Miller units. Data represent the average of three independent experiments with error bars representing standard deviations. (C) Association between mouse Polκ and Rev1 in cos7 cells. Lysates from HA-mPolκ and Myc-mRev1 co-transfected cos7 cells were analyzed by immunoprecipitation and western blotting, as indicated. A mock antibody (normal rabbit serum) was used in controls. Input lanes contained 1/25 the lysates used in the experiments. Top panel, Myc-mRev1 co-immunoprecipitates with HA-mPolκ. Bottom panel, HA-mPolκ co-immunoprecipitates with Myc-mRev1. (D) Immuno precipitation with a mixture of 0.3 µM each of purified mRev1 and mPolκ. Upper panel, the blot was probed with anti-mPolκ antibody. Lane 1 contains 1/35 the amount of purified mPolκ used in the reactions. Lanes 2–5 show immunoprecipitation of the mRev1/mPolκ mixture with the following: lane 2, normal rabbit serum; lane 3, anti-Rev1 serum with mRev1 omitted; lane 4, anti-Rev1 serum with mPolκ omitted; lane 5, anti-Rev1 serum. Lower panel, the blot was stripped and probed with anti-Rev1 antibody. IP and IB indicate immunoprecipitate and immunoblot, respectively.
Interaction between mRev1 and mPolκ was also demonstrated by immunoprecipitation. Mouse Polκ and mouse Rev1 proteins tagged with HA (HA-mPolκ) and Myc (Myc-mRev1) epitopes at their N-termini were expressed from mammalian expression vectors. Western analysis using antibodies specific to the HA or Myc epitopes confirmed co-expression in cos7 cells (Figure 1C). Cell lysates were immunoprecipitated with either anti-HA or anti-Myc polyclonal antibodies using normal rabbit serum as a mock control (Figure 1C). HA-mPolκ co-precipitated with Myc-mRev1 regardless of which antibody was used for immunoprecipitation or western analysis. However, neither protein was detected when rabbit serum was used as an immunoprecipitation control (Figure 1C). A mixture of stoichiometric equivalents of purified mRev1 and mPolκ was also co-precipitated with anti-Rev1 antibody (Figure 1D, lane 5), while anti-Rev1 antibody did not precipitate mPolκ alone (Figure 1D, lane 3).
Interaction between mPolκ and mRev1 requires the C-terminal 100 amino acids of mRev1
The shortest of the four polypeptides that interacted with mPolκ (amino acids 100–616) in the two-hybrid screen contains the C-terminal 378 amino acids, suggesting that a limited C-terminal region of mRev1 is necessary and sufficient for interaction with mPolκ. To map this region more precisely we constructed mRev1 cDNAs carrying various deletions and tested these in two-hybrid assays. Clones containing the C-terminal 100 amino acid residues (amino acids 1150–1249) yielded positive interactions, whereas clones expressing smaller mRev1 polypeptides did not (Figure 2A).

Fig. 2. Deletion mapping of mRev1 region required for interaction with mPolκ (A and B). (A) Deletion mutants of mRev1 were tested for their ability to interact with full-length mPolκ in the yeast two-hybrid system. On auxotroph selective plates (QDO + x-α-gal), yeast co-transformed with full-length or truncated mRev1 constructs 1–5 plus the mDinB plasmid are viable, showing blue colonies within 3 days. In contrast, yeast co-transformed with truncated mRev1 constructs 6–7 plus the mPolκ plasmid are not viable. (B) _In vitro_-translated Myc-mPolκ was added to glutathione beads coupled with either GST (lane 2), GST–Rev1-3 (lane 3), GST–Rev1-4 (lane 4), GST–Rev1-5 (lane 5) or GST–Rev1-6 (lane 6) fusion proteins. The input lane (lane 1) contains 1/20 of the IVTT product used in the experiment. Interactions were examined by western analysis using monoclonal antibody against Myc. (C) Deletion mapping of the mPolκ region required for interaction with mRev1. Deletion mutants of mPolκ were tested for their ability to interact with full-length mRev1 in the yeast two-hybrid system as described above.
To confirm these results, mPolκ was incubated with GST-tagged mRev1 proteins expressed from various deletion constructs and coupled to glutathione–agarose beads. The washed beads were resuspended in SDS loading buffer and bound proteins were detected by western analysis using monoclonal antibody against the Myc epitope. Once again mPolκ only bound GST–mRev1 polypeptides that included the C-terminal amino acids 1150–1249 (Figure 2B). The reciprocal experiment was performed in which proteins expressed from different deletion constructs of mouse PolK cDNA were examined for interaction with full-length mRev1 protein with the two-hybrid assay. Only truncated mPolκ polypeptides that included amino acid residues 230–616 (present in the bait protein for the initial two-hybrid screen) yielded interactions (Figure 2C). This region of mPolκ includes two conserved helix–hairpin–helix (HhH) domains as well as an undefined domain conserved in all members of the DinB subfamily of the Y superfamily (Gerlach et al., 1999).
Mouse Rev1 interacts with other novel DNA polymerases through its C-terminal domain
We examined the interaction of Rev1 with other specialized DNA polymerases known to support TLS in vitro or in vivo. Using the yeast two-hybrid system we observed interactions between mRev1 and mPolι (Figure 3A) and between mRev1 and mPolη (Figure 3B). To confirm these results we co-transfected Flag and Myc epitope-tagged constructs (Flag-mPolι and Myc-mRev1; Flag-mPolη and Myc-mRev1) into cos7 cells and incubated cell lysates with anti-Flag affinity beads. Precipitated proteins were detected by western analysis using antibodies against the Myc or Flag epitopes. Mouse Rev1 protein was shown to interact with mPolι (Figure 3C) and mPolη (Figure 3D). The specificity of these interactions was demonstrated with appropriate controls (Figure 3C and D). As observed with the mRev1/mPolκ interaction the C-terminal 100 amino acids of Rev1 are sufficient for interaction with mPolι or mPolη (Figure 3). Neither of these proteins interacted with mPolκ in yeast two-hybrid and immunoprecipitation experiments (data not shown).

Fig. 3. Deletion mapping of mRev1 to determine the minimal region required for interaction with mPolι (A) or mPolη (B) by the yeast two-hybrid assay. (C) Association between mRev1 and mPolι in cos7 cells. Anti-Flag M2 agarose affinity gel was incubated with the cos7 cell lysates expressing Myc-mRev1 and Flag-mPolι or Myc-mRev1 (control). Top panel, immunoblotting to detect Myc-mRev1. Lanes 1 and 2, input containing 1/50 the lysate used for immunoprecipitation. Lanes 3 and 5, immunoprecipitation of lysates with anti-Flag M2 antibody. The lysates express Myc-mRev1 (lane 3) or Myc-mRev1 and Flag-mPolι (lane 5), respectively. Lane 4, Myc-mRev1 and Flag-mPolι lysates were precipitated with mock antibody (HA). Bottom panel, the blot was stripped and probed with anti-Flag monoclonal antibody. (D) Interaction between mRev1 and mPolη in cos7 cells. Lysates expressing Myc-mRev1 and Flag-mPolη were precipitated and detected analogously to (C).
It has been previously shown that human Rev1 protein interacts with human Rev7 protein, a subunit of the heterodimeric TLS polymerase Polζ, and that the Rev7-binding domain resides in the Rev1 C-terminus (amino acids 1130–1251) (Murakumo et al., 2001; Masuda et al., 2003). Since this region of Rev1 is highly conserved between the mouse and human proteins, we examined mRev1 and mRev7 proteins for this interaction. Cell lysates expressing Myc-mRev1 and Flag-mRev7 were precipitated with anti-Flag affinity beads. Western analysis of the precipitates using an anti-Myc monoclonal antibody demonstrated co-precipitation of mRev1 with Flag-mRev7 (Figure 4A). No mRev1 protein was detected in control experiments in which only the Myc-mRev1 lysate was precipitated (Figure 4A). This interaction was confirmed with in vitro GST pull-down experiments. Recombinant mRev1 was incubated with equal amounts of either GST or GST–mRev7 proteins and bound to glutathione–agarose beads. After extensive washes bound proteins were analyzed by western analysis using polyclonal antibody against human Rev1 (amino acids 245–847). GST–mRev7 protein bound specifically to mRev1 (Figure 4B, lane 4). Diminished amounts of mRev1 were recovered with the GST–mRev7 beads in the presence of increasing amounts of purified mPolκ (Figure 4B, lanes 5 and 6), suggesting that mRev7 and mPolκ compete directly for binding to mRev1. Mouse Polκ and mRev7 did not interact in the yeast two-hybrid system or in GST pull-down assays (data not shown).
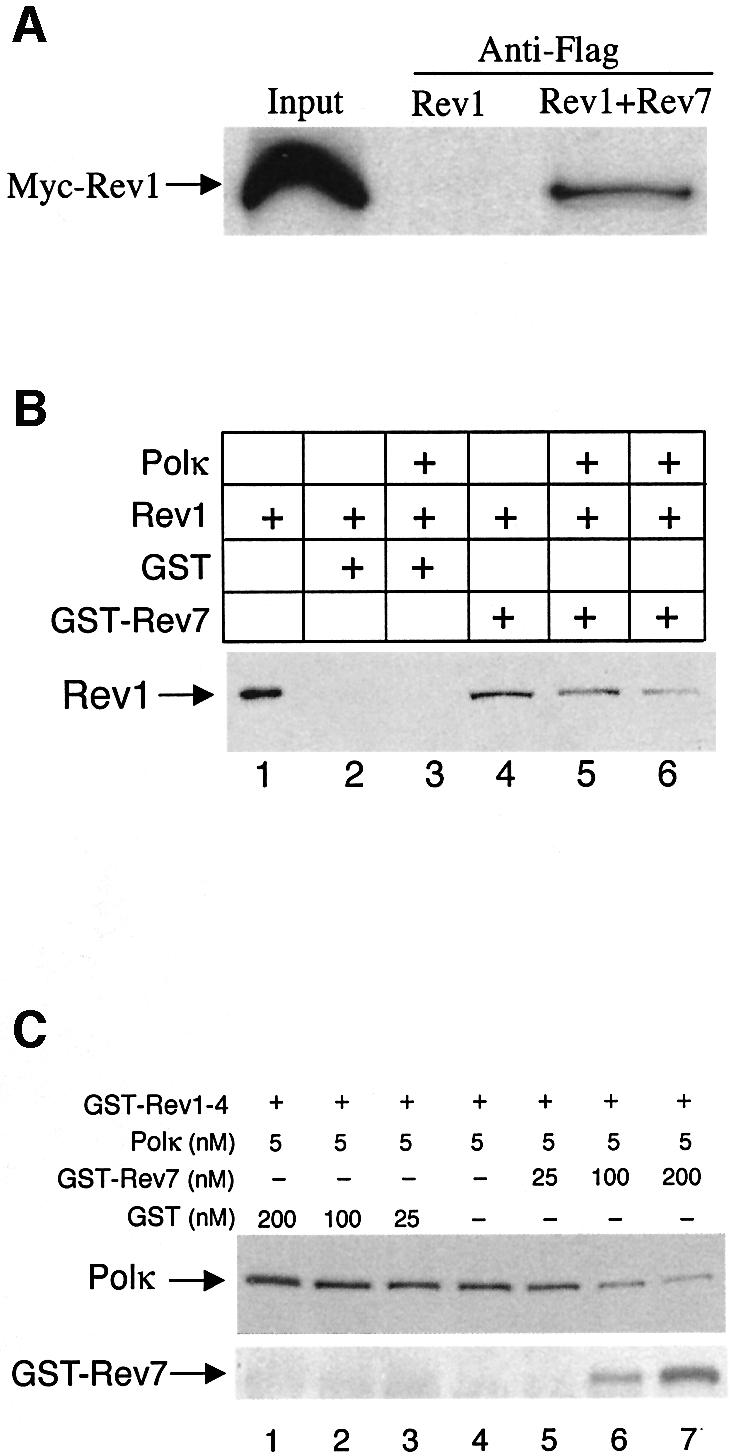
Fig. 4. Association between mRev1 and mRev7. (A) Extracts of cos7 cells expressing Flag-mRev7 and Myc-mRev1 were incubated with anti-Flag M2 agarose affinity gel. Retained proteins were detected by immunoblotting with monoclonal antibody against Myc. Input lanes contain 1/40 of the lysates used in the experiments. (B) GST pull-down of mRev1 with GST–Rev7. Recombinant mRev1 (45 nM) was incubated with 40 µg GST or GST–Rev7 coupled to glutathione beads in the absence or presence of purified mPolκ. Bound proteins were resolved by 8% SDS–PAGE followed by immunoblot analysis with anti-Rev1 antibody. Lane 1 contains 1/10 of the mRev1 used in the experiments. Lane 2, GST+mRev1; lane 3, GST + mRev1 + 450 nM mPolκ; lane 4, GST–Rev7 + mRev1; lane 5, GST–Rev7 + mRev1 + 45 nM mPolκ; lane 6, GST–Rev7 + mRev1 + 450 nM mPolκ. (C) Rev7 competes with Polκ for binding to the mRev1 C-terminus. Immobilized GST–Rev1-4 (amino acids 1124–1249) fusion protein (5 µg) was incubated with a fixed amount of recombinant mPolκ (5 nM) in the presence of increasing concentrations (0–200 nM) of GST–Rev7 or GST, as indicated. Bound proteins were resolved by 8% SDS–PAGE followed by immunoblot analysis with anti-mPolκ antibody (top panel). The blot was stripped and probed with anti-Rev7 antibody (bottom panel). As more GST–Rev7 protein is added, there is an increase in GST–Rev7 binding and a decrease in mPolκ binding to GST–Rev1-4. Data are representative of three independent experiments.
Since the C-terminal 100 amino acids of mRev1 protein are required for binding to both mPolκ and mRev7, and in light of the observation that mPolκ and mRev7 may compete directly for binding to mRev1, we examined interactions between the three proteins GST–Rev1-4 (amino acids 1124–1249), mPolκ and mRev7. The C-terminal region of mRev1 was bound to glutathione–agarose beads and washed extensively with GST protein until saturated. Bound beads were incubated with a fixed concentration of mPolκ followed by increasing amounts of GST–mRev7 protein eluted from glutathione beads. Following extensive washing the association between Polκ and GST–Rev1-4 was evaluated by immunoblot analysis with a polyclonal antibody against mPolκ. Increasing amounts of GST–mRev7 protein in the incubation (but not GST alone) decreased the amount of Polκ bound to GST–Rev1-4 and increased the amount of GST–Rev7 bound (Figure 4C).
In summary, our results demonstrate that mouse Polκ, Polι, Polη and Rev7 each interact with mRev1. In the cases of mPolκ, mPolι and mPolη, the C-terminal 100 amino acids of mRev1 are sufficient for the interaction. Additionally, mPolκ and mRev7 compete directly for binding to mRev1 protein.
Catalytic activity of the mouse Polκ/Rev1 complex
In view of the observation that purified mPolκ and mRev1 form a stable complex in solution (Figure 1D) we compared the polymerase activity of each alone and when incubated together. The two proteins were introduced into reaction mixtures by mixing droplets of each on the side of incubation tubes and gently pushing the mixed droplets into reactions containing the remaining components pre-warmed to 37°C. When individual protein droplets were mixed for a minute prior to addition to the primer extension reaction no differences were noted compared to shorter mixing times, indicating that 1–2 s was sufficient for mPolκ/mRev1 to attain binding equilibrium. We also performed experiments in which each of the four dNTPs were introduced individually to demonstrate that the fidelity of mPolκ or mRev1 was unaltered when replicating normally base-paired DNA together.
Experiments with a native and correctly base-paired primer-template. Visual examination and quantitation (shown below each lane in Figure 5A) of individual radiolabeled bands generated by primer extension of undamaged template DNA normally base-paired with a primer demonstrated that, in the presence of both mPolκ and mRev1, the relative quantities of total nucleotides incorporated were essentially the sum of that observed with each polymerase alone (Figure 5A). Additionally, replication fidelity was unaltered (data not shown). We conclude that neither DNA polymerase activity nor fidelity is significantly altered when a mixture of the two polymerases extends a normally base-paired primer annealed to undamaged template DNA.
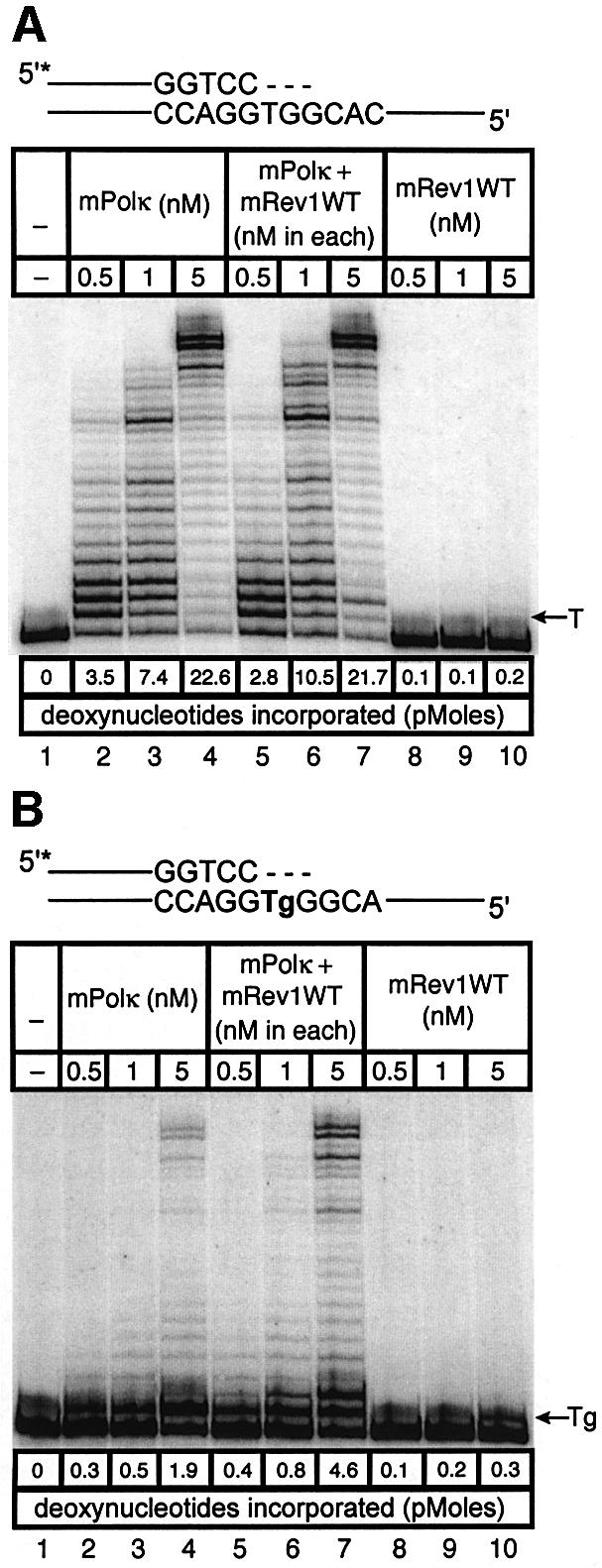
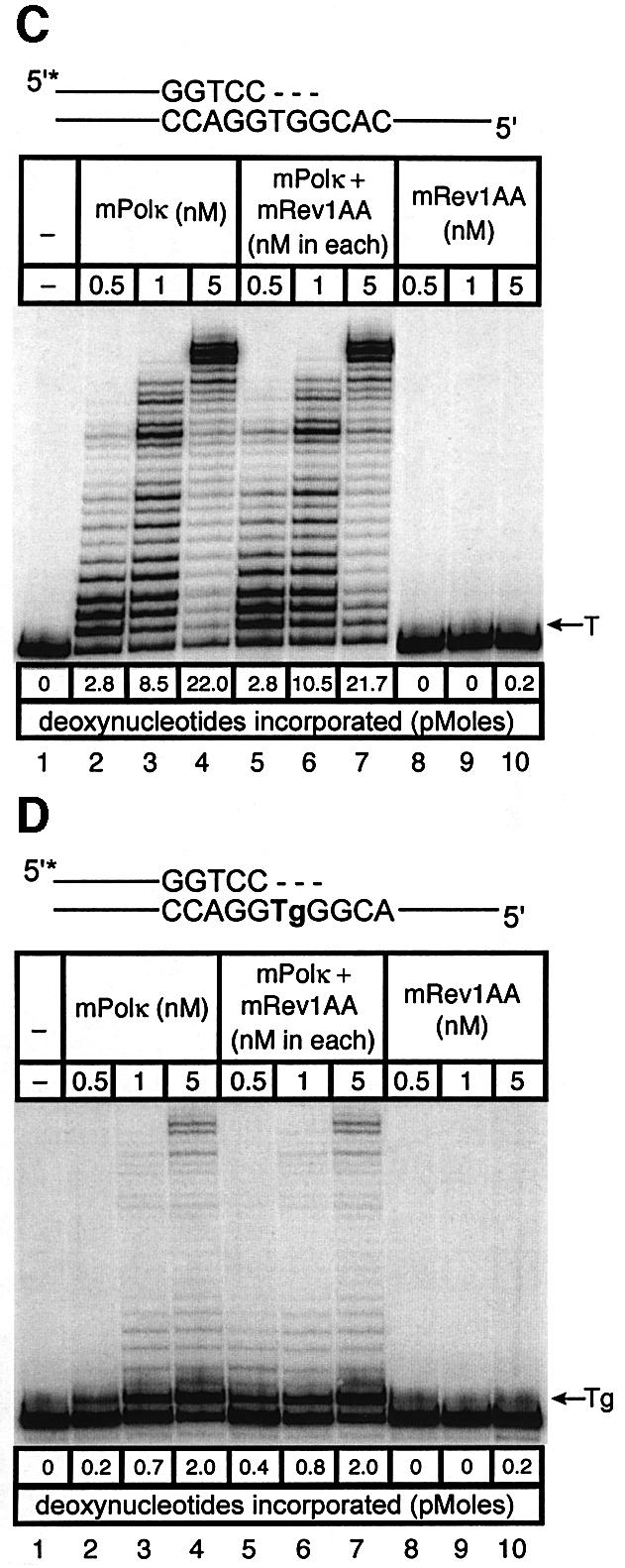
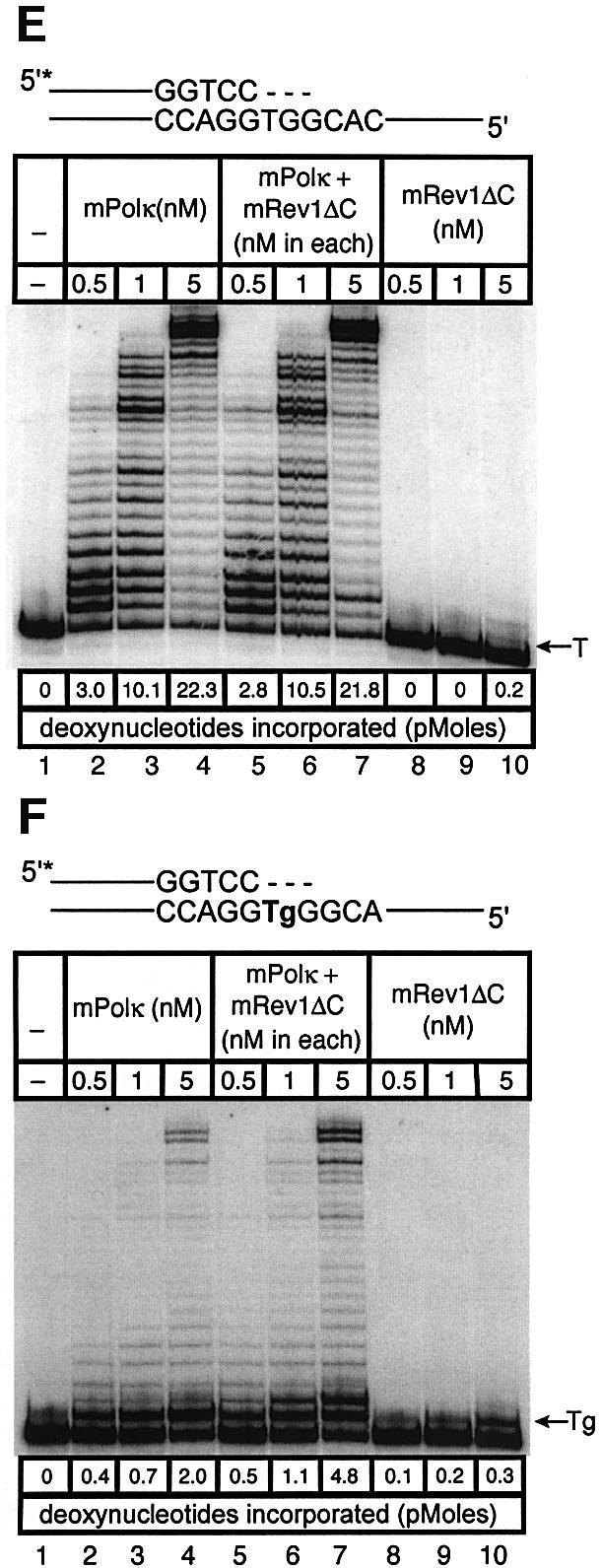
Fig. 5. Direct interaction does not influence the polymerase activities of mRev1 and mPolκ in vitro on undamaged base-paired primer-templates or opposite a thymine glycol template base. Radiolabeled DNA primer-templates and the four dNTPs were incubated with mPolκ, mRev1 or both proteins. Reaction products were resolved by DPAGE. For each panel: lane 1, control with no enzyme; lanes 2, 3 and 4, mPolκ alone at 0.5, 1 and 5 nM, respectively; lanes 5, 6 and 7, mPolκ and mRev1 at 0.5, 1 and 5 nM each, respectively; lanes 8, 9 and 10, mRev1 alone at 0.5, 1 and 5 nM respectively. (A) Undamaged base-paired primer-template substrate (local sequence context indicated in the scheme above the gel). (B) Primer-template substrate containing a single thymine glycol base (local sequence context indicated in the scheme above the gel); thymine glycol is represented as ‘Tg’. (C and D) Analogous to (A) and (B) except that the nucleotidyl transferase-defective mRev1AA protein was used instead of wild-type mRev1. (E and F) Analogous to (A) and (B) except that the C-terminal deletion mRev1ΔC protein lacking the domain for binding to mPolκ was used instead of wild-type mRev1. The position on the gels corresponding to the primer extended by a single nucleotide (opposite template T or template Tg) is indicated by an arrow to the right of the gel. The total quantity of deoxynucleotides incorporated/reaction is indicated below each lane of each gel as pmol.
Experiments with template DNA containing base damage. Human Polκ can efficiently bypass thymine glycol bases during primer extension in vitro (Fischhaber et al., 2002). In the present studies we asked whether mPolκ is also endowed with this property and, if so, whether this property is altered in the presence of mRev1. Experiments were performed using a DNA oligonucleotide containing thymine glycol as the next template base. The polymerase activity in each primer extension reaction was determined quantitatively and expressed as total deoxynucleotide incorporated per reaction (see results below each lane in Figure 5B). Like the human protein, mPolκ is able to support efficient TLS across thymine glycol (Figure 5B). Additionally, mRev1 bypasses thymine glycol (Figure 5B). In this case the polymerase activity when the two proteins were incubated together was greater than the additive activities of each alone (Figure 5B). For example, 1.9 pmol of dNMP was incorporated by mPolκ (Figure 5B, lane 4) and 0.3 pmol was incorporated by mRev1 (Figure 5B, lane 10). However, when the two enzymes were incubated together 4.6 pmol rather than the expected 2.2 pmol of dNMP was incorporated (Figure 5B, lane 7).
This result suggests that physical interaction between mPolκ and mRev1 during primer extension stimulates nucleotide incorporation opposite thymine glycol. However, the two template bases immediately following thymine glycol are both G. Thus, an alternative explanation for the stimulation derived from known properties of Rev1 protein. Mouse Rev1 may realign the template by skipping the thymine glycol and incorporate C opposite the two template G residues. If the resulting partially extended primer-template provides a better substrate for mPolκ, more robust extension beyond the lesion may result, a scenario in accord with the two-step, two-polymerase model for TLS proposed by others (Bridges and Woodgate, 1985; Johnson et al., 2000; Pages and Fuchs, 2002).
To distinguish between these two possibilities we generated mutant forms of mRev1 protein. In one case the nucleotidyl transferase domain of mRev1 was inactivated by changing conserved aspartate and glutamate residues to alanine (D568A, E569A, designated mRev1AA). This form of mRev1 is expected to retain a stimulatory effect promoted by protein–protein interaction, even though it is unable to support nucleotide incorporation. In the other case mRev1 was deleted of the C-terminal region required for interaction with mPolκ (designated mRev1ΔC). This form of mRev1 is expected to retain catalytic activity but to be inactive for stimulation mediated by interaction between the two proteins.
Mouse Rev1AA protein was devoid of polymerase activity, while mRev1ΔC retained the ability to support incorporation of C (Figure 5C and E). However, the amount of primer extension with mPolκ and mRev1AA was identical to that observed with mPolκ alone, using either an undamaged template (Figure 5C) or the thymine glycol template (Figure 5D). Similarly the extent of primer extension with mPolκ and mRev1ΔC (Figure 5E and F) was identical to that supported by mPolκ and mRev1 (Figure 5A and B). Collectively, these results support the two-step, two-polymerase model for TLS and indicate that mixing mPolκ and mRev1 does not significantly alter the polymerase function of either polymerase when bypassing thymine glycol in vitro.
Experiments with mispaired primer-termini. Polκ can extend mispaired primer termini (Washington et al., 2002). We examined such primer extension by a mixture of mPolκ and mRev1 proteins using annealed primers terminating either in a correct base pair (A:T) or a mismatched base pair (C:T). Once again the correctly paired A:T substrate showed little difference in primer extension when mPolκ was incubated alone or with mRev1 (Figure 6A). However, the C:T mismatched substrate supported slightly enhanced activity when incubated with mRev1 (Figure 6B). In experiments using the mRev1AA and mRev1ΔC mutant proteins the results were completely analogous to those obtained with thymine glycol (Figure 6C–F). We performed an additional primer extension experiment in which the template G immediately 5′ to the primer terminus was changed to C to alter the sequence of the next template base from that preferred by mRev1. No enhanced activity was observed with the altered template (Figure 6G and H).
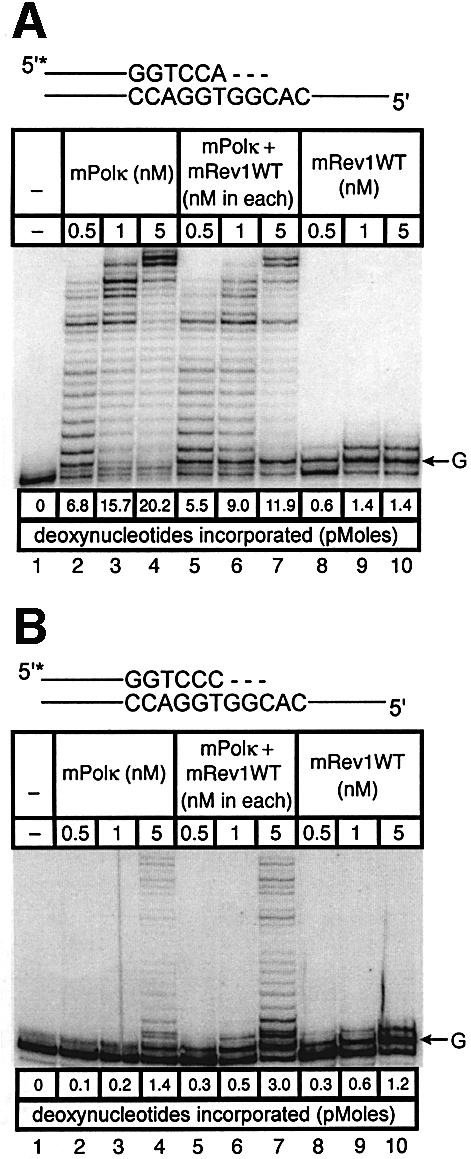
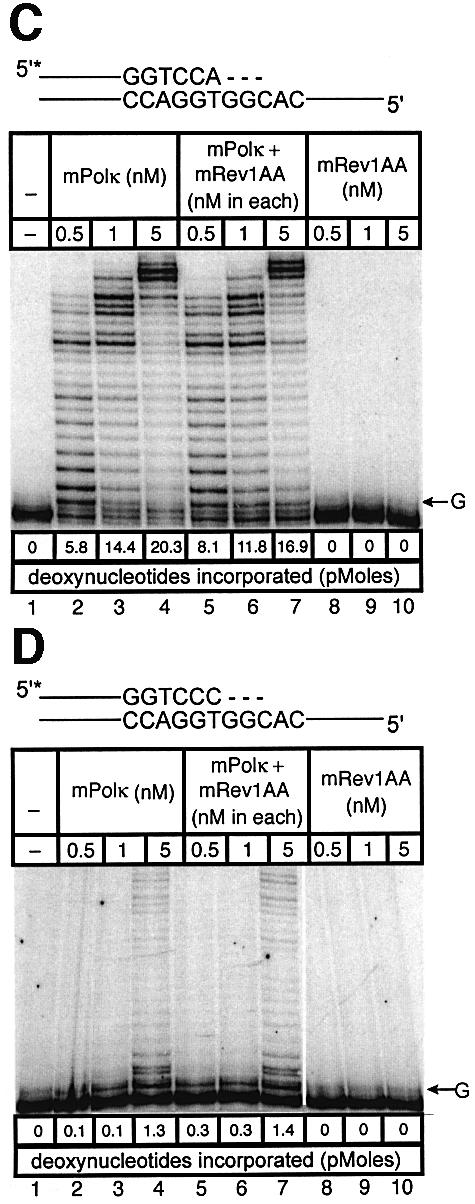
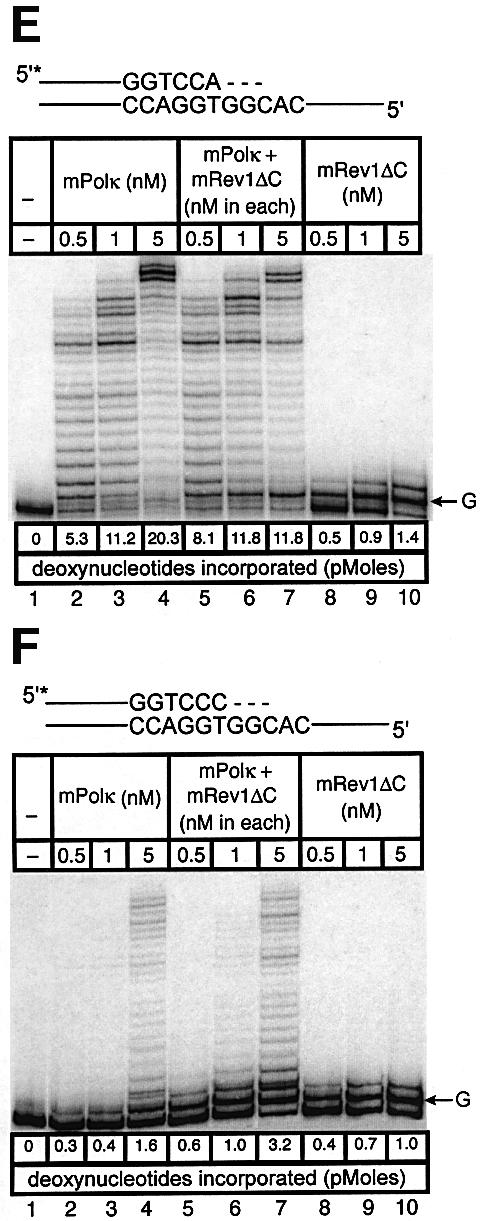
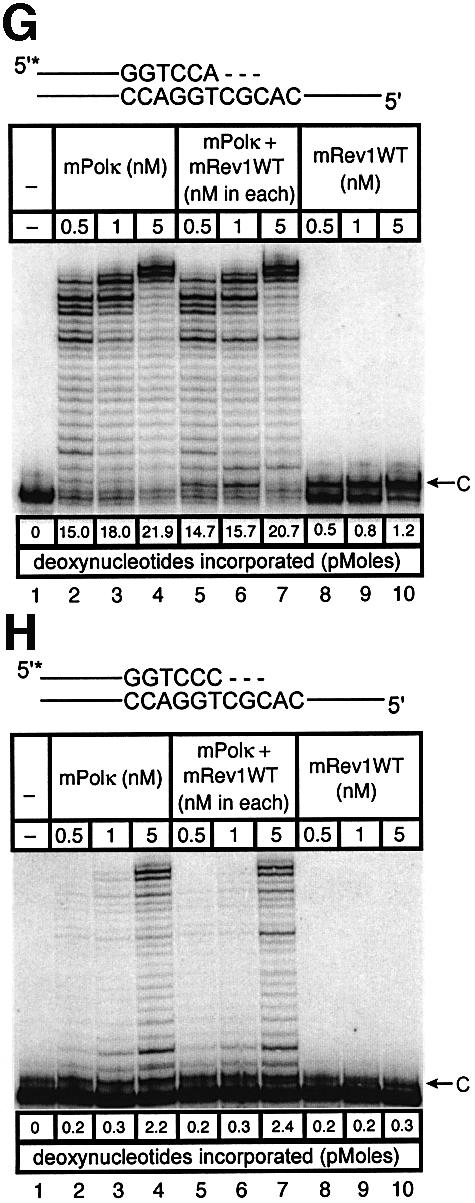

Fig. 6. Direct interaction does not influence the polymerase activities of mRev1 and mPolκ in vitro on a terminally mismatched primer-template. Radiolabeled DNA primer-templates and the four dNTPs were incubated with mPolκ, mRev1 or both proteins. Reaction products were resolved by DPAGE. For each panel: lane 1, control with no enzyme added; lanes 2, 3 and 4, mPolκ alone at 0.5, 1 and 5 nM, respectively; lanes 5, 6 and 7, both mPolκ and mRev1 at 0.5, 1 and 5 nM each, respectively; lanes 8, 9 and 10, mRev1 alone at 0.5, 1 and 5 nM respectively. (A) Undamaged base-paired primer-template substrate (local sequence context indicated in scheme above the gel). (B) C:T terminally-mismatched substrate as indicated in scheme above the gel. (C and D) Analogous to (A) and (B) except that the nucleotidyl transferase-defective Rev1AA protein was used instead of wild-type Rev1. (E and F) Analogous to (A) and (B) except that the C-terminal deletion Rev1ΔC protein lacking the domain for binding to mPolκ was used instead of wild-type Rev1. (G and H) Analogous to (A) and (B) except that the next 5′ template base was C instead of G. (I) Time-course experiment in which primer extension of 5 nM of each enzyme was monitored at 1, 5 and 10 min. Plots of quantitated data are shown (inset) indicating that polymerase activity is in the linear range under these conditions. The position on the gels corresponding to the primer extended by a single nucleotide (opposite template G or template C) is indicated by an arrow to the right of the gel. The total quantity of deoxyribonucleotides incorporated/reaction is indicated below each lane of each gel as pmol.
As an added control we performed a time-course experiment with the sequence context used in the mismatch experiments, during which the activity of the highest enzyme concentration was monitored at intervals (Figure 6I). Each enzyme supported a linear response for nucleotide incorporation (Figure 6I, inset). These experiments clearly indicate that the observed enhancement of polymerase activity when mPolκ and mRev1 are incubated together does not derive from direct protein–protein interaction, but is the result of the template sequence context immediately 5′ to the mismatched primer-terminus.
Discussion
Unrelieved arrested DNA replication threatens the viability of dividing cells. Not surprisingly, both prokaryotic and eukaryotic cells have evolved strategies for coping with this threat. The discovery that many eukaryotic cells, in particular higher eukaryotes, are endowed with multiple low fidelity DNA polymerases (mammalian cells contain at least eight such enzymes) that can catalyze DNA synthesis past sites of base damage in vitro has yielded insights about DNA damage tolerance by the process of TLS. Regardless of the specific types of base damage in DNA handled by TLS, a question of considerable interest is how switching is effected at sites of arrested replication between high fidelity polymerases in the replicative machinery and one or more specialized enzymes that support TLS. Nor is it known how a particular specialized polymerase(s) is selected for bypass of a particular type(s) of base damage.
Recent studies have demonstrated that some TLS polymerases can bind to accessory proteins in the replicative machinery, in particular PCNA (Haracska et al., 2001a,b,c, 2002b). Additionally, it has been shown that Polη and Polι can physically associate (Kannouche et al., 2002). These observations are open to multiple interpretations, but they suggest that (some) specialized polymerases may associate with one another and with the replicative machinery during TLS.
A number of in vivo studies suggest that Polκ is required for TLS across oxidative base damage in DNA (Schenten et al., 2002; Velasco-Miguel et al., 2003). Consistent with this notion, purified human (Fischhaber et al., 2002) and mouse (present study) Polκ support accurate TLS past thymine glycol lesions in vitro. There is also convincing evidence that mPolκ is required for TLS across benzo[_a_]pyrene adducts in vivo (Ogi et al., 2002). To further our understanding of the biological role of Polκ during TLS in mammalian cells, we searched for proteins with which it interacts and observed that mRev1, another Y-family polymerase, consistently interacts with mPolκ. Rev1 is essential for error-prone bypass of a variety of lesions, a function that is independent of its dCMP transferase activity (Baynton et al., 1999; Nelson et al., 2000; Lawrence, 2002). Additionally, Rev1 protein is unable to support TLS across T<>T dimers or [6-4] photoproducts, but is nonetheless required for UV radiation-induced mutagenesis in yeast and in mammalian cells. The C-terminal 100 amino acids of mRev1 are required for its interaction with mPolκ. Remarkably, the same region is required for its interaction with all the other specialized polymerases thus far tested (Polι, Polη and the non-catalytic Rev7 subunit of Polζ).
The present studies may cast further light on the central issue of the mechanism of polymerase switching and regulation during TLS. According to the two-step, two-polymerase model, the first enzyme supports the incorporation of just 1 or 2 nucleotides opposite the lesion, while the second polymerase extends the primer beyond the lesion site to a position downstream of the damage where the high fidelity replicative machinery can again operate effectively, and where its associated 3′→5′ proof-reading exonuclease activities cannot remove nucleotides incorporated during TLS (Goodman, 2002). Rev1 may conceivably function as a scaffold for mediating polymerase switching at the arrested lesion site. In this role Rev1 may bind the first (‘lesion’) polymerase. Following nucleotide incorporation opposite the lesion it may release this polymerase and bind the second (‘extension’) polymerase. Different polymerases may be selected by competition (Pages and Fuchs, 2002). This general model of polymerase hand-off offers a central role of Rev1 in the efficiency and possibly also the fidelity of nucleotide incorporation during TLS. The observation that different polymerases bind the same region of Rev1 protein implies significant structural similarities in the Rev1-binding domains of each. Studies are in progress to verify this.
Alternatively, Rev1 may function as a molecular docking site that delivers different TLS polymerases to the primer-template terminus at sites of arrested DNA replication, perhaps through a specific interaction between Rev1 and one or more proteins of the arrested replication machinery. It is not obvious how selection of a particular specialized polymerase for TLS across a particular type of base damage is then accomplished. Perhaps different Rev1/polymerase complexes have varying affinity for different sites of base damage.
In summary, the present studies hint at an important role(s) for Rev1 protein in polymerase selection during the process of TLS. The models proposed here raise a number of cogent questions, the answers to which will likely provide further insights into the mechanism of TLS and the avoidance of such synthesis on undamaged DNA, except perhaps in specific circumstances where mutagenesis is physiological, e.g. somatic hypermutation.
Materials and methods
Plasmids
For yeast two-hybrid assays, full-length and truncated fragments of mDinB cDNA were cloned in pGBKT7 or pGBT9 (Clontech). Full-length and truncated mutants of mRev1 cDNA were cloned in pGADT7 (Clontech). For in vivo binding assays, full-length mRev1 and mDinB cDNAs were cloned in pCMV-Myc or pCMV-HA (Clontech) to produce Myc or HA fusion proteins. Mouse Rev7 and Polη cDNAs were amplified by RT–PCR using mouse testis total RNA as template (Murakumo et al., 2000; Yamada et al., 2000). Mouse Polι cDNA was amplified by PCR. Each of these cDNAs was cloned in pCMV5-Flag vector to generate Flag fusion proteins. For GST pull-down experiments, Rev7 cDNA and Rev1 C-terminal fragments were cloned in pGEX4T-2 (Amersham) to produce GST fusion proteins.
For preparation of mPolκ expression vector, the mouse DinB cDNA was subcloned into the _Nde_I site of the pET-16b vector (Novagen) or _Nde_I and _Sap_I sites of the IMPACT-CN system vector pTXB1 (New England Biolabs). The pET-16b-mDinB plasmid yields an N-terminally His10-tagged mPolκ protein. The pTXB1-mDinB plasmid yields a tagless mPolκ protein. To prepare the expression construct for mRev1AA, mutations D568A and E569A were generated in Rev1-pBAD22A using the QuikChange Mutagenesis Kit (Stratagene). To construct the truncated Rev1-pBAD22A-expressing vector, an _Aat_II/_Spe_I fragment that includes the mRev1 C-terminus (amino acids 574–1249) was cut off from Rev1-pBAD22A (Masuda et al., 2002); the _Aat_II/_Apa_LI fragment from Rev1-pGBKT7 that includes mRev1 (amino acids 574–1135) was inserted. The resulting plasmid, Rev1ΔC-pBAD22A, expresses a truncated mRev1 protein lacking the C-terminal 114 amino acids.
Yeast two-hybrid assay
The pGBKT7/mDinB100–616 plasmid, containing the mouse Polκ nucleotidyl transferase domain and the tandem HhH domain, was used to screen a mouse testis cDNA library. This bait construct was transformed into AH109 and then crossed with strain Y187 pretransformed with a mouse testis cDNA library in pACT2 according to the manufacturer’s instructions (Clontech). Library plasmids were scored positive by their ability to confer growth on QDO plates. Positive clones were isolated and confirmed by back-transformation into AH109 with the bait construct. Empty vectors served as negative controls. The presence of ‘bait’ and ‘prey’ plasmids in co-transformed cells was controlled by growth on double drop-out (DDO) plates deleted of leu and trp. A liquid culture assay for β-galactosidase activity was performed quantitatively according to the manufacturer’s instructions (Clontech). β-galactosidase activity was determined by averaging the results of three independent experiments.
Cell culture and reagents
Cos7 cells were grown in DMEM supplemented with 10% fetal bovine serum. For transient transfection experiments, cos7 cells were grown in 10 cm culture dishes and transfected with pCMV-HA-mDinB, pCMV-Myc-mRev1, pCMV5-Flag-Rev7, pCMV5-Flag-Polι or pCMV5-Flag-Polη, as indicated, using lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were harvested for further analysis 68 h after transfection.
Antibodies
Rabbit polyclonal anti-Myc, anti-HA, mouse monoclonal anti-HA and anti-Myc and anti-Flag M2 were purchased from Covance. Anti-Flag M2 Agarose affinity gel was purchased from Sigma. Polyclonal antiserum against mPolκ was obtained by immunizing a hamster with purified his10-mPolκ.
Lysate preparation, co-immunoprecipitation and western blotting
Cos7 cells were transiently transfected with pCMV-Myc-mRev1, pCMV-HA-mDinB, pCMV5-Flag-Rev7, pCMV5-Flag-Polι or pCMV5-Flag-Polη for 68 h to express Myc-mRev1, HA-mPolκ, Flag-Rev7, Flag-Polι and Flag-Polη fusion proteins. Harvested cells were disrupted in cos cell lysis buffer [1% Triton X-100, 50 mM Tris–HCl, 300 mM NaCl, 5 mM Na2EDTA, 1× protease inhibitor cocktail (Roche), pH 7.4]. Cell lysates were clarified by centrifugation (15 000 g, 20 min, 4°C) and incubated with either anti-Flag M2 agarose affinity gel or the protein A/G plus agarose beads (Santa Cruz Biotechnology) conjugated with polyclonal antibody against c-Myc or HA (3 h, 4°C), with gentle inversion mixing. Beads were pelleted by centrifugation, washed with buffer (0.1% Triton X-100, 50 mM Tris–HCl, 300 mM NaCl, 5 mM Na2EDTA, 1× protease inhibitor cocktail, pH 7.4). Bound proteins were analyzed by SDS–PAGE and transferred to PVDF membranes. Proteins were detected by immunoblotting with mouse monoclonal antibodies against Myc (9E10), HA (16B12) or Flag (M2), as indicated.
Co-immunoprecipitation of purified mRev1 and mPolκ was in the presence of binding buffer I (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5 mM Na2EDTA, 10% glycerol, 0.1% Tween-20, 0.75 mg/ml BSA, 1 mM PMSF, 1× protease inhibitor cocktail) or binding buffer II (25 mM Tris–HCl, 25 mM HEPES–NaOH, 250 mM NaCl, 1 mM DTT, 10% glycerol, 0.1 mg/ml BSA, 0.01% NP-40, 1 mM PMSF, 1× protease inhibitor cocktail, pH 7.4). The mixture [0.3 µM each of purified mRev1 and tagless mPolκ (50 µl)] was incubated with rotation (2 h, 4°C). The protein complex was adsorbed onto protein A/G plus agarose beads coupled with anti-Rev1 by incubating overnight (4°C). Beads were washed with binding buffer containing protease inhibitors. Bound proteins were detected by immunoblotting with hamster antibody against mPolκ or rabbit antibody against human Rev1245–847 (Masuda et al., 2003).
In vitro transcription and translation
In vitro transcription and translation (IVTT) of full-length mPolκ was performed using the TNT T7 quick-coupled transcription/translation system (Promega) according to the manufacturer’s instructions. The expression vector encoding full-length mPolκ (pGBKT7-DinB) was added to reaction mixtures and incubated (90 min, 30°C). Reaction products were used in GST pull-down assays.
GST pull-down assay
GST fusion proteins were expressed in Escherichia coli BL21 transformed with pGEX-4T-2 with the induction of isopropyl β-d-thiogalactopyranoside (1 mM, 4 h, 30°C). Cells were harvested and disrupted in bacterial lysis buffer (50 mM imidazole, 100 mM NaCl, 10 mM Na2EDTA, 1 mM DTT, 0.2 mg/ml lysozyme, 1 mM PMSF, 1× protease inhibitors, pH 6.8) with sonication. Resulting GST fusion proteins were purified on glutathione–agarose (Sigma), eluted with 10 mM reduced glutathione (Sigma) and dialyzed against 50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10% glycerol, 1 mM DTT. For assessment of in vitro interaction, equal amounts of GST or GST fusion proteins (∼80 µg) coupled to glutathione–agarose beads were incubated with equal amounts of IVTT proteins (30 µl) in 150 µl binding buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5 mM Na2EDTA, 10% glycerol, 0.1% Tween-20, 0.75 mg/ml BSA, 1 mM PMSF, 1× protease inhibitor cocktail). For interaction between mRev1 and mRev7, equal amounts of GST or GST fusion proteins (∼40 µg) coupled to glutathione–agarose beads were incubated with equal amounts of recombinant mRev1 (45 nM) in 200 µl binding buffer. Tubes were incubated (2 h, 4°C) on a rocker. Beads were washed with binding buffer and resuspended in 30 µl SDS loading buffer. Samples were separated by SDS–PAGE, transferred to PVDF membranes and detected by immuno-chemiluminescence using monoclonal antibody against Myc (9E10), or polyclonal antibody against human Rev1245–847. To test whether mRev7 and mPolκ compete in binding with the mRev1 C-terminus we measured binding of Polκ to immobilized GST–Rev1-4 beads in the presence of increasing concentrations of GST–Rev7. Increasing concentrations of GST were used as controls. GST–Rev1-4 (amino acids 1124–1249) beads were saturated with GST (10 µg/µl) prior to the binding assays. After incubating (3 h, 4°C) beads were washed with binding buffer and resuspended in 20 µl SDS loading buffer. Samples were separated by SDS–PAGE and detected by immunoblotting using a hamster polyclonal antibody against mPolκ or rabbit polyclonal antibody against human Rev7 (Murakumo et al., 2001).
Expression and purification of mPolκ
mPolκ was expressed in E.coli BL21-codonplus (DE3)-RP cells harboring pET-16b-mDinB or pTXB1-mDinB with the induction of IPTG (0.25 mM, 4 h). A cell pellet harboring pET-16b-mDinB was resuspended in chilled lysis buffer I (50 mM sodium phosphate buffer pH 8.0, 300 mM NaCl, 2% glycerol, 1× protease inhibitors). After adding lysozyme, DNase I and incubating (1 h, 4°C) the lysate was sonicated and clarified by centrifugation (12 000 g, 4°C, 20 min). The supernatant was applied to a Ni–NTA agarose column (Qiagen) pre-equilibrated with lysis buffer I. The resin was washed with 10 column vol (CV) of lysis buffer I and 10 CV buffer II (Lysis buffer I + 1 M NaCl), followed by 2 CV of Tris-equilibration buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl and 2% glycerol). Protein was eluted with increasing concentrations of buffered imidazole pH 8.0 in Tris-equilibration buffer. For tagless mPolκ, the cell pellet was resuspended in chitin column buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 mM Na2EDTA) in the presence of DNase I and protease inhibitors. Cells were sonicated and purified as described by the IMPACT-CN System manual (New England Biolabs), whereby mPolκ was >95% cleaved from the chitin-binding domain after 18 h at 4°C. Protein fractions containing mPolκ were pooled and dialyzed against 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1% glycerol and 3 mM DTT overnight. The sample was further purified by anion exchange chromatography using a Source 30Q column (Amersham Biosciences). A step wash was performed with 120 mM NaCl before the NaCl gradient was applied, eluting mPolκ at 150 mM NaCl. Fractions containing mPolκ were pooled and vacuum dialyzed against gel filtration buffer (20 mM Tris–HCl pH 8.0, 400 mM NaCl, 1% glycerol and 3 mM DTT) to a volume of 4 ml, and then applied to a pre-equilibrated HiLoad 26/60 Superdex 200 prep grade column. Fractions containing mPolκ were pooled and subjected to vacuum dialysis followed by flash freezing.
Expression and purification of mRev1
mRev1, mRev1AA and mRev1ΔC were expressed and purified from E.coli BL21(DE3) as described (Masuda et al., 2002).
Preparation of thymine glycol DNA template
A template DNA containing a single thymine glycol DNA base was prepared by oxidation of DNA with osmium tetroxide as described (Fischhaber et al., 2002).
In vitro primer extension assay
The primer for in vitro primer-extension experiments (Figure 5) was P5-OX-SS 5′-d(GAATTCCTGCAGCCCAGGATCGACTGGTCC). The thymine glycol-containing template was identical to that reported (Fischhaber et al., 2002). In control experiments a template of identical sequence was used but with thymine rather than thymine glycol at the appropriate position. In experiments presented in Figure 6 primers P5-OX-SS-A, 5′-d(GAATTCCTGCAGCCCAGGATCGACTGGTCCA) or P5-OX-SS-C, 5′-d(GAATTCCTGCAGCCCAGGATCGACTGGTCCC) were annealed to either 5′-d(ATTCCAGACTGTCAATAACACGGTG GACCAGTCGATCCTGGGCTGCAGGAATTC) or the same oligo in which the underlined base was changed to C (Figure 6G and H). DNA oligonucleotides were purified, 5′-end-labeled with [γ-32P]ATP and T4 polynucleotide kinase (Invitrogen) and desalted. Primers were annealed to template strands in a stoichiometric ratio of 1:1.5 (primer:template, 50 mM NaCl, 5 mM Tris–HCl, 0.5 mM Na2EDTA, pH 8.0) by heating (90°C, 5 min) and then cooling to room temperature.
Enzyme samples were diluted from concentrated stocks immediately prior to use in primer extension assays. 1× dilution buffer for mPolκ enzyme: 50 mM Tris–HCl, 5 mM MgCl2, 1 mM DTT, 10% glycerol, 0.1 mg/ml BSA, pH 7.0; 1× dilution buffer for mRev1: 50 mM HEPES–NaOH, 500 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol, 0.1 mg/ml BSA, pH 7.5. Primer extension experiments were performed as follows. Radiolabeled primer-templates (5 nM) were incubated with mPolκ (0.5, 1 or 5 nM), mRev1 (0.5, 1 or 5 nM) or both enzymes (0.5, 1 or 5 nM in each) in 1× reaction buffer (25 mM potassium phosphate, 2.5 mM MgCl2, 0.1 mg/ml BSA, 10% glycerol, 5 mM DTT, 25 µM dATP, 25 µM dCTP, 25 µM dGTP, 25 µM TTP, pH 7.4, 10 min, 37°C). Enzymes were introduced as droplets on the sides of Eppendorf tubes containing the rest of the reaction mixture. In reactions containing just one of the two enzymes, a droplet containing dilution buffer was added instead. Protein droplets were pushed together, mixed by pipette and then pushed into the reaction mixture (already prewarmed to 37°C) to initiate primer extension. In kinetic experiments all conditions were identical to those described above except that 5 nM enzyme(s) was used and incubations were for varying amounts of time prior to quenching (1, 5 or 10 min).
Reactions were quenched by adding formamide gel loading solution (95% formamide, 5% 1× TBE, 0.025% xylene cyanol, 50 µl), heated (90°C, 5 min), and resolved by DPAGE. Gel images were recorded on a storage phosphor screen and analyzed using Molecular Dynamics software. Each primer-extension experiment was performed at least twice. Data quantification was performed by determining the fraction of the total radioactive signal in individual lanes (representing the fraction of 1.25 pmol primer-template extended) in a given gel band, subtracting an appropriate background, weighting the band according to the number of nucleotides incorporated beyond the N-mer primer (i.e. N + 1, N + 2, etc.) and then summing the weighted gel band results to determine the total number of nucleotides incorporated over the entire lane.
Acknowledgments
Acknowledgements
The authors acknowledge Dr Roger Woodgate for Polι cDNA, Dr Murakumo for Rev7 antibody, Dr Wen Lai for hamster antiserum against mPolκ and Annie Wood and Liqun Wang for technical assistance. These studies were funded by grants ES11344 (USPHS) (E.C.F.), DOE grant DE-FG02-01ER63073 (C.K.), Pew Scholars Program in the Biomedical Sciences (C.K.) and a Grant-in-Aid from the Ministry of Education, Science, Sports, Culture and Technology of Japan (K.Y.).
References
- Baynton K., Bresson-Roy,A. and Fuchs,R.P. (1999) Distinct roles for Rev1p and Rev7p during translesion synthesis in Saccharomyces cerevisiae. Mol. Microbiol., 34, 124–133. [DOI] [PubMed] [Google Scholar]
- Bridges B.A. and Woodgate,R. (1985) The two-step model of bacterial UV mutagenesis. Mutat. Res., 150, 133–139. [DOI] [PubMed] [Google Scholar]
- Fischhaber P.L., Gerlach,V.L., Feaver,W.J., Hatahet,Z., Wallace,S.S. and Friedberg,E.C. (2002) Human DNA polymerase κ bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J. Biol. Chem., 277, 37604–37611. [DOI] [PubMed] [Google Scholar]
- Frank E.G., Tissier,A., McDonald,J.P., Rapic-Otrin,V., Zeng,X., Gearhart,P.J. and Woodgate,R. (2001) Altered nucleotide misinsertion fidelity associated with poliota-dependent replication at the end of a DNA template. EMBO J., 20, 2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E.C., Wagner,R. and Radman,M. (2002) Specialized DNA polymerases, cellular survival and the genesis of mutations. Science, 296, 1627–1630. [DOI] [PubMed] [Google Scholar]
- Gerlach V.L., Aravind,L., Gotway,G., Schultz,R.A., Koonin,E.V. and Friedberg,E.C. (1999) Human and mouse homologs of Escherichia coli DinB (DNA polymerase IV), members of the UmuC/DinB superfamily. Proc. Natl Acad. Sci. USA, 96, 11922–11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M.F. (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem., 71, 17–50. [DOI] [PubMed] [Google Scholar]
- Haracska L., Johnson,R.E., Unk,I., Phillips,B., Hurwitz,J., Prakash,L. and Prakash,S. (2001a) Physical and functional interactions of human DNA polymerase η with PCNA. Mol. Cell. Biol., 21, 7199–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L., Johnson,R.E., Unk,I., Phillips,B.B., Hurwitz,J., Prakash,L. and Prakash,S. (2001b) Targeting of human DNA polymerase ι to the replication machinery via interaction with PCNA. Proc. Natl Acad. Sci. USA, 98, 14256–14261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L., Kondratick,C.M., Unk,I., Prakash,S. and Prakash,L. (2001c) Interaction with PCNA is essential for yeast DNA polymerase η function. Mol. Cell, 8, 407–415. [DOI] [PubMed] [Google Scholar]
- Haracska L., Prakash,L. and Prakash,S. (2002a) Role of human DNA polymerase κ as an extender in translesion synthesis. Proc. Natl Acad. Sci. USA, 99, 16000–16005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L., Unk,I., Johnson,R.E., Phillips,B.B., Hurwitz,J., Prakash,L. and Prakash,S. (2002b) Stimulation of DNA synthesis activity of human DNA polymerase κ by PCNA. Mol. Cell. Biol., 22, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubscher U., Maga,G. and Spadari,S. (2002) Eukaryotic DNA polymerases. Annu. Rev. Biochem., 71, 133–163. [DOI] [PubMed] [Google Scholar]
- Johnson R.E., Washington,M.T., Haracska,L., Prakash,S. and Prakash,L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature, 406, 1015–1019. [DOI] [PubMed] [Google Scholar]
- Kannouche P., Fernandez de Henestrosa,A.R., Coull,B., Vidal,A.E., Gray,C., Zicha,D., Woodgate,R. and Lehmann,A.R. (2002) Localization of DNA polymerases η and ι to the replication machinery is tightly co-ordinated in human cells. EMBO J., 21, 6246–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C.W. (2002) Cellular roles of DNA polymerase ζ and Rev1 protein. DNA Repair (Amst), 1, 425–435. [DOI] [PubMed] [Google Scholar]
- Masuda Y., Takahashi,M., Fukuda,S., Sumii,M. and Kamiya,K. (2002) Mechanisms of dCMP transferase reactions catalyzed by mouse Rev1 protein. J. Biol. Chem., 277, 3040–3046. [DOI] [PubMed] [Google Scholar]
- Masuda Y., Ohmae,M., Masuda,K. and Kamiya,K. (2003) Structure and enzymatic properties of a stable complex of the Human REV1 and REV7 proteins. J. Biol. Chem., 278, 12356–12360. [DOI] [PubMed] [Google Scholar]
- Murakumo Y., Roth,T., Ishii,H., Rasio,D., Numata,S., Croce,C.M. and Fishel,R. (2000) A human REV7 homolog that interacts with the polymerase ζ catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J. Biol. Chem., 275, 4391–4397. [DOI] [PubMed] [Google Scholar]
- Murakumo Y., Ogura,Y., Ishii,H., Numata,S., Ichihara,M., Croce,C.M., Fishel,R. and Takahashi,M. (2001) Interactions in the error-prone postreplication repair proteins hREV1, hREV3 and hREV7. J. Biol. Chem., 276, 35644–35651. [DOI] [PubMed] [Google Scholar]
- Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature, 382, 729–731. [DOI] [PubMed] [Google Scholar]
- Nelson J.R., Gibbs,P.E., Nowicka,A.M., Hinkle,D.C. and Lawrence,C.W. (2000) Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol., 37, 549–554. [DOI] [PubMed] [Google Scholar]
- Ogi T., Shinkai,Y., Tanaka,K. and Ohmori,H. (2002) Polκ protects mammalian cells against the lethal and mutagenic effects of benzo[_a_]pyrene. Proc. Natl Acad. Sci. USA, 99, 15548–15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi E., Ogi,T., Kusumoto,R., Iwai,S., Masutani,C., Hanaoka,F. and Ohmori,H. (2000) Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev., 14, 1589–1594. [PMC free article] [PubMed] [Google Scholar]
- Ohmori H. et al. (2001) The Y-family of DNA polymerases. Mol. Cell, 8, 7–8. [DOI] [PubMed] [Google Scholar]
- Pages V. and Fuchs,R.P. (2002) How DNA lesions are turned into mutations within cells? Oncogene, 21, 8957–8966. [DOI] [PubMed] [Google Scholar]
- Prakash S. and Prakash,L. (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev., 16, 1872–1883. [DOI] [PubMed] [Google Scholar]
- Schenten D., Gerlach,V.L., Guo,C., Velasco-Miguel,S., Hladik,C.L., White,C.L., Friedberg,E.C., Rajewsky,K. and Esposito,G. (2002) DNA polymerase κ deficiency does not affect somatic hypermutation in mice. Eur. J. Immunol., 32, 3152–3160. [DOI] [PubMed] [Google Scholar]
- Simpson L.J. and Sale,J.E. (2003) Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J., 22, 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Miguel S., Richardson,J.A., Gerlach,V.L., Lai,W.C., Gao,T., Russell,L.D., Hladik,C.L., White,C.L. and Friedberg,E.C. (2003) Constitutive and regulated expression of the mouse Dinb (Polκ) gene encoding DNA polymerase κ. DNA Repair (Amst), 2, 91–106. [DOI] [PubMed] [Google Scholar]
- Washington M.T., Johnson,R.E., Prakash,L. and Prakash,S. (2002) Human DINB1-encoded DNA polymerase κ is a promiscuous extender of mispaired primer termini. Proc. Natl Acad. Sci. USA, 99, 1910–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A., Masutani,C., Iwai,S. and Hanaoka,F. (2000) Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase η. Nucleic Acids Res., 28, 2473–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Yuan,F., Xin,H., Wu,X., Rajpal,D.K., Yang,D. and Wang,Z. (2000) Human DNA polymerase κ synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res., 28, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wu,X., Guo,D., Rechkoblit,O. and Wang,Z. (2002) Activities of human DNA polymerase κ in response to the major benzo[_a_]pyrene DNA adduct: error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair (Amst), 1, 559–569. [DOI] [PubMed] [Google Scholar]