Catalysis of lysine 48-specific ubiquitin chain assembly by residues in E2 and ubiquitin (original) (raw)
. Author manuscript; available in PMC: 2010 Nov 1.
Summary
Protein ubiquitination is catalyzed by ubiquitin-conjugating enzymes (E2s) in collaboration with ubiquitin-protein ligases (E3s). This process depends on nucleophilic attack by a substrate lysine on a thioester bond linking the C-terminus of ubiquitin to a cysteine in the E2 active site. Different E2 family members display specificity for lysines in distinct contexts. We addressed the mechanistic basis for this lysine selectivity in Ubc1, an E2 that catalyzes the ubiquitination of lysine 48 (K48) in ubiquitin, leading to the formation of K48-linked polyubiquitin chains. We identified a cluster of polar residues near the Ubc1 active site, as well as a residue in ubiquitin itself, that are required for catalysis of K48-specific ubiquitin ligation but not for general activity toward other lysines. Our results suggest that the active site of Ubc1, as well as the surface of ubiquitin, contain specificity determinants that channel specific lysines to the central residues involved directly in catalysis.
Introduction
The attachment of ubiquitin to proteins is used as a regulatory mechanism to control many aspects of protein function, including localization, binding partners, and rate of degradation (Deshaies and Joazeiro, 2009; Hochstrasser, 2009; Pickart and Eddins, 2004). Protein ubiquitination is carried out by a series of three enzymes: an E1 or ubiquitin-activating enzyme, an E2 or ubiquitin-conjugating enzyme, and an E3 or ubiquitin-protein ligase. The E1 employs ATP hydrolysis to generate a high-energy thioester bond between the C-terminus of ubiquitin and a cysteine in the E1. Ubiquitin-charged E1 interacts with one of many different E2 proteins, resulting in the transfer of the ubiquitin C-terminus to a cysteine on the E2. A specific E3 then binds both the ubiquitin-charged E2 and a protein substrate, promoting transfer of the ubiquitin C-terminus to a lysine side chain on the substrate.
The initial attachment of ubiquitin to substrate results in monoubiquitination. In many cases, a lysine on the attached ubiquitin is then ubiquitinated, and sequential cycles of this process generate a polyubiquitin chain (Deshaies and Joazeiro, 2009; Pierce et al., 2009; Ye and Rape, 2009). Although all lysines in ubiquitin can be used for chain formation, many polyubiquitin chains are linked through the same lysine in each monomer (Pickart and Fushman, 2004; Xu et al., 2009). Lysine 48 (K48) of ubiquitin is a common site of attachment, resulting in K48-linked chains that target the modified protein to the proteasome for destruction (Thrower et al., 2000).
For members of the RING-domain family of E3s, the final chemical step in protein-ubiquitin ligation involves nucleophilic attack by the lysine ε-amino group on the thioester bond of the E2-ubiquitin conjugate, leading to isopeptide bond formation through an oxyanion intermediate (Capili and Lima, 2007; Dye and Schulman, 2007; Matyskiela et al., 2009; Pickart and Eddins, 2004; Ye and Rape, 2009). Residues in the E2 active site catalyze this reaction. A conserved asparagine near the active-site cysteine might stabilize the oxyanion reaction intermediate (Wu et al., 2003), while other E2 residues are thought to orient and deprotonate the attacking lysine (Capili and Lima, 2007; Yunus and Lima, 2006).
Important insights into the catalysis of ubiquitin transfer come from studies of sumoylation, the process by which the ubiquitin-like protein SUMO is linked to a substrate lysine by mechanisms analogous to those used in ubiquitin attachment. SUMO attachment involves a specific E2 called Ubc9, which interacts directly with some substrates in the absence of an E3. Three residues (N85, Y87, D127) near the active-site cysteine of Ubc9 are particularly important for catalysis (Capili and Lima, 2007; Yunus and Lima, 2006). The side chains of these residues position the attacking lysine and also provide a hydrophobic microenvironment that desolvates the lysine and thereby lowers its effective p_Ka_, enhancing deprotonation (and thus nucleophilic attack) at physiological pH. Two of these residues (N85 and D127) are well conserved in other E2s. The third, Y87, is absent in several other E2s; in these cases, however, a nearby leucine might fulfill the function of the missing tyrosine.
Further insights come from structural studies of the mechanisms by which lysine 63 of ubiquitin attacks a ubiquitin conjugate of the E2 Ubc13 (Eddins et al., 2006). In this case, the attacking lysine is positioned in the E2 active site by residues that fulfill the same functions as the key catalytic residues of Ubc9. N85 and D127 of Ubc9 are conserved in Ubc13. Y87 is replaced by aspartate, but the space occupied by Y87 in Ubc9 is occupied by a leucine, L121, in Ubc13.
Thus, catalysis of the chemical step in protein ubiquitination depends primarily on conserved residues near the active-site cysteine of the E2. Clearly, however, the rate of ubiquitination is also influenced by the occupancy of the E2 active site by the attacking substrate, which is determined by substrate affinity and concentration. In the case of protein ubiquitination, substrate affinity for the E2 alone is very low (Petroski and Deshaies, 2005), and it is unlikely that free E2-ubiquitin conjugates are attacked productively at the low substrate concentrations inside the cell. Efficient ubiquitination generally requires an E3, which carries a specific substrate-docking site adjacent to its E2 binding site. This increases substrate affinity, which drives E2 active-site occupation and thereby promotes significant rates of ubiquitination. E3 binding might also cause a conformational change in the E2 active site that enhances catalytic rate (Ozkan et al., 2005; Petroski and Deshaies, 2005).
Given that residues in the E2 help position the attacking lysine, it follows that different E2s exhibit specificity for lysines in specific contexts (Ye and Rape, 2009). As mentioned above, the E2 Ubc13, together with its cofactor Mms2, is highly specific for K63 of ubiquitin, and structural analysis has revealed how a specific face of ubiquitin, termed the hydrophobic patch, interacts with the Ubc13-Mms2 complex to orient K63 toward the active-site cysteine (Eddins et al., 2006). The E2 Cdc34, which works with the E3 SCF, displays specificity for K48 of ubiquitin, and catalysis of K48 ubiquitination depends in part on an ‘acidic loop’ inserted near the active-site cysteine (Petroski and Deshaies, 2005).
The importance of lysine specificity is illustrated by studies of the anaphase-promoting complex or cyclosome (APC), a multisubunit E3 that assembles polyubiquitin chains on numerous mitotic regulators, thereby targeting them to the proteasome for destruction (Matyskiela et al., 2009; Peters, 2006; Sullivan and Morgan, 2007; Thornton and Toczyski, 2006). Chain assembly on yeast APC targets occurs in two steps: ubiquitin is attached to a lysine on the substrate protein, after which K48 on attached ubiquitins is modified to generate K48-linked polyubiquitin chains (Rodrigo-Brenni and Morgan, 2007). These two steps involve lysines in different structural contexts. The first ubiquitin is attached to an APC substrate at one of several lysines that seem randomly distributed, without a clear sequence context, in a substrate region that is likely to be disordered. In contrast, the second ubiquitin is attached to a single lysine, K48, that lies within a specific sequence context on the surface of a tightly folded globular protein.
The two steps in chain assembly by the yeast APC depend on two different E2s (Rodrigo-Brenni and Morgan, 2007). The initial ubiquitination of the substrate lysine is catalyzed by Ubc4, an E2 that collaborates with numerous E3s to ubiquitinate lysines in many contexts. Ubc4 has minimal activity toward any of the lysines on ubiquitin. The second step in chain assembly – ubiquitination of K48 of the first ubiquitin – depends on another E2, Ubc1, which is clearly specialized for catalysis of K48 modification and has very low activity toward lysines on the substrate itself.
To gain a better understanding of the mechanisms underlying the lysine specificity of different E2s, we set out to identify the features of the Ubc1 active site that are required for its K48 specificity. We found that K48-specific ubiquitination depends on multiple active-site residues, including one residue that is also important for reducing the rate of ubiquitination of lysines on the protein substrate. We also identified a residue in ubiquitin that is required for K48-specific chain assembly, suggesting that the substrate itself also contributes to catalysis.
Results
The catalytic core of Ubc1 interacts specifically with K48 of ubiquitin
We showed previously that Ubc1 assembles K48-linked ubiquitin chains on APC substrates (Rodrigo-Brenni and Morgan, 2007). To study this reaction in more detail, we used diubiquitin synthesis assays (Petroski and Deshaies, 2005) that allow careful dissection of the K48-specific polyubiquitination reaction in the absence of an E3. Radiolabeled ubiquitin conjugated to Ubc1 is the donor in these assays, and successful attack of this donor by acceptor ubiquitin in solution results in the formation of radiolabeled diubiquitin.
We charged Ubc1 with a radiolabeled ubiquitin moiety in which K48 was mutated to arginine, thereby preventing it from acting as the attacking ubiquitin. Following the conjugation reaction, we added EDTA and NEM to inactivate all unoccupied active-site cysteines on E1 and E2. These conditions allowed only a single round of attack by free ubiquitin (Petroski and Deshaies, 2005).
In pilot experiments, saturating amounts of unlabeled wild-type ubiquitin were added to radiolabeled ubiquitin-Ubc1 conjugate, and the appearance of diubiquitin, as well as the disappearance of conjugated E2, were measured by gel electrophoresis under nonreducing conditions. The formation of diubiquitin was linear with time up to 5 minutes, after which it leveled off due to depletion of the conjugated species (Figures S1A, B). Diubiquitin synthesis was negligible when the attacking ubiquitin carried the K48R mutation, confirming the K48 specificity of the reaction (Figure S1C).
This single-turnover assay allowed us to measure initial rates of diubiquitin synthesis over a range of ubiquitin concentrations (Figure 1A, left), resulting in estimates of ubiquitin-E2 affinity (apparent Kd) and catalytic rate (apparent _k_2) (Yunus and Lima, 2006). We estimated the catalytic turnover rate by dividing the rate of diubiquitin synthesis by the initial amount of E2-ubiquitin conjugate. The results revealed an apparent Kd of 356 μM and an apparent _k_2 of 0.0015 s-1 (Figure 1B; Table S1).
Figure 1. The catalytic core of Ubc1 binds ubiquitin.
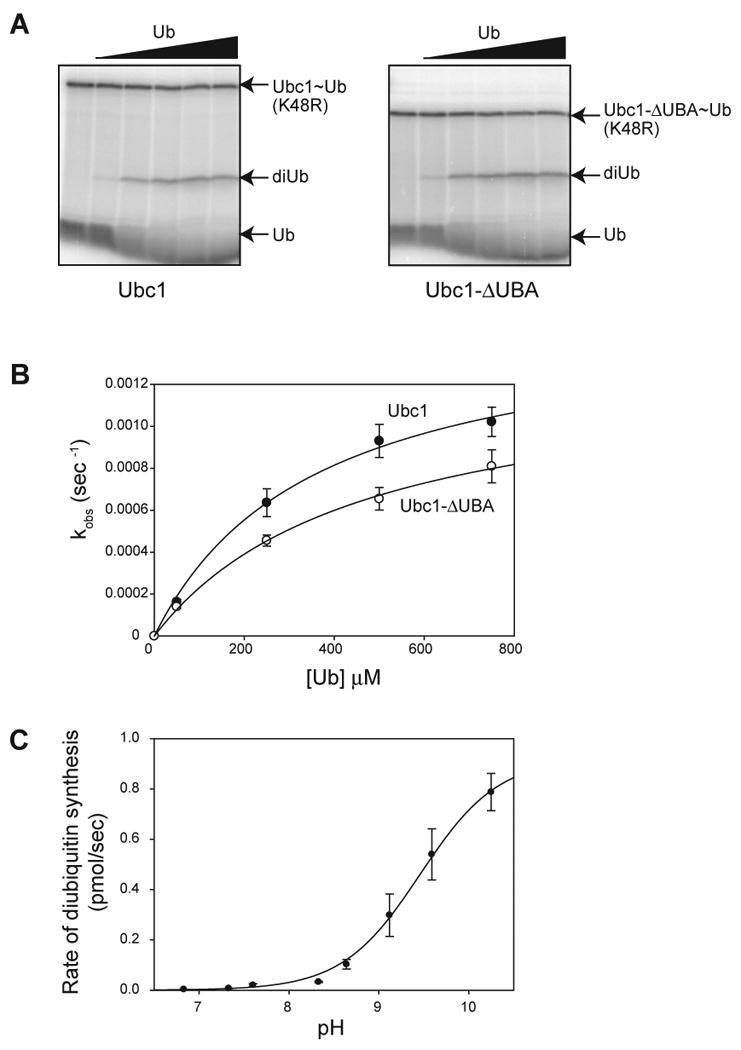
(A) Purified Ubc1 (left) or Ubc1-ΔUBA (right) was conjugated with 32P-labeled K48R-ubiquitin and treated with NEM and EDTA to prevent recharging of Ubc1 and E1. Increasing amounts of unlabeled ubiquitin (50 μM to 750 μM) were added. After 2 min, reactions were stopped by addition of nonreducing sample buffer and analyzed by SDS-PAGE. Free ubiquitin (Ub), diubiquitin (diUb) and charged Ubc1 (Ubc1∼Ub) are indicated. See Figure S1 for a time course of similar reactions.
(B) Initial rates of diubiquitin synthesis were measured over a range of ubiquitin concentrations in reactions like those in panel A (n=5 for Ubc1; n=3 for Ubc1-ΔUBA). The rate of diubiquitin synthesis was divided by total Ubc1-Ub to estimate turnover rate (kobs). Data were fit to a rectangular hyperbola using the ligand-binding module of SigmaPlot. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
(C) Initial rates of diubiquitin synthesis were determined at saturating ubiquitin concentration (1 mM) over a range of pH values (6.86-10.26). At each pH, diubiquitin formation was measured over a time course. The appearance of diubiquitin was plotted as a function of time and fit to a linear function using Excel. The rate of diubiquitin synthesis (the slope of the linear function) was plotted as a function of pH and fit to a sigmoidal function using SigmaPlot. Experiments were done in triplicate and the error bars represent SEM.
Ubc1 contains a C-terminal UBA domain that is linked by a flexible tether to the core UBC domain (Merkley and Shaw, 2004). In other proteins, UBA domains bind ubiquitin and various diubiquitin species (Dikic et al., 2009; Raasi et al., 2005). We showed previously that the UBA domain of Ubc1 is not required for K48 specificity (Rodrigo-Brenni and Morgan, 2007), but it remained possible that it contributes in some way to recognition or orientation of the attacking ubiquitin. We found, however, that removal of the UBA domain had little effect on ubiquitin affinity or catalytic rate (Figure 1A, B; Table S1). We conclude that the core UBC domain of Ubc1 is the primary site of binding and catalysis for the attacking ubiquitin.
We next explored the pH dependency of the diubiquitin synthesis reaction. These experiments were motivated by previous studies of protein sumoylation by the SUMO E2, Ubc9, in which the effect of pH on the reaction was used to estimate the p_Ka_ of the attacking lysine (Yunus and Lima, 2006). As mentioned in the Introduction, these studies suggested that hydrophobic residues near the active-site cysteine of Ubc9 suppress the attacking lysine p_Ka_ to a value of about 8.5. We therefore measured the maximal rate of diubiquitin synthesis with saturating amounts of ubiquitin (1 mM) over a range of pH values (6.83-10.26) (Figure 1C). Reaction rate increased dramatically at higher pH, and the inflection point on the sigmoidal pH-dependency curve was at pH 9.4. A plot of log(rate) versus pH revealed a slope of about one (not shown), consistent with a single titratable group in the active site. The most plausible titratable base is the attacking lysine, as in the case of Ubc9. The predicted p_Ka_ of K48 in ubiquitin is 10.4. We therefore speculate that the effective p_Ka_ of the attacking lysine is suppressed in Ubc1 reactions by about 1 pH unit. This degree of p_Ka_ suppression is not as great as that seen with Ubc9 (Yunus and Lima, 2006), but could nevertheless indicate that catalytic residues in Ubc1 alter the local microenvironment as in Ubc9.
Residues in the active site of Ubc1 are involved in K48-specific chain assembly
We next sought to identify residues in the active site of Ubc1 that are required for its K48 specificity. To identify candidate residues, we compared the sequences of E2s that are known to build K48-linked chains (Ubc1 and its human ortholog E2-25K) with sequences of Ubc4-related E2 proteins, which don't exhibit specificity for any lysine of ubiquitin. We focused on residues that are different between Ubc1 and Ubc4 but identical in E2-25K and Ubc1 (Figure S2). We further narrowed our search using previously determined tertiary structures of Ubc1 (Hamilton et al., 2001; Merkley and Shaw, 2004) and Ubc4 (Cook et al., 1993) to focus on residues near the active-site cysteine that are solvent-exposed, and therefore more likely to participate in catalysis or ubiquitin binding.
Two clusters of amino acids satisfied these criteria (Figure S2). Cluster I contains V83, T84 and A86; cluster II contains Q122 and A124. Although the two clusters are far apart in the primary sequence of Ubc1, they lie close to each other in the tertiary structure, on the face of Ubc1 near the active-site cysteine (Figure 2, top). In a structural model of the Ubc1-ubiquitin thioester conjugate (Hamilton et al., 2001), these residues lie on the opposite side of the cysteine from the conjugated ubiquitin. A comparison of Ubc1 with Ubc9 (Figure 2, bottom) also suggests that these residues are well suited for positioning ubiquitin for attack of the thioester.
Figure 2. Conserved residues in the active site of Ubc1.
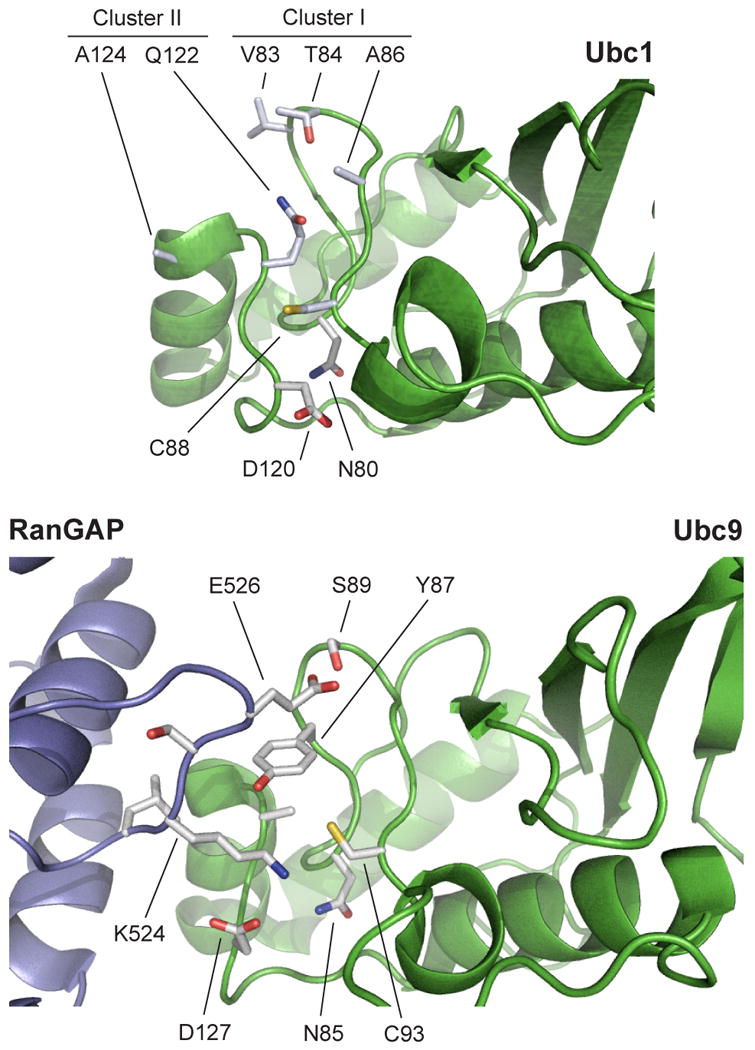
(top) The Ubc1 active site contains five surface-exposed side chains, in two clusters, that are conserved in human E2-25K but are not present in Ubc4 (see Figure S2 for E2 sequence alignments). In the Ubc1-ubiquitin conjugate (Hamilton et al., 2001; Merkley and Shaw, 2004), ubiquitin is positioned to the lower right of the active-site cysteine (C88) in this orientation (not shown). Additional residues known to be important for catalysis in other E2s (D120, N80; see Figure S2) are also labeled.
(bottom) The structure of Ubc9 in complex with its substrate RanGAP1 (Bernier-Villamor et al., 2002) illustrates the orientation of the attacking lysine (K524). The active-site cysteine of Ubc9 (C93) is surrounded by three residues (Y87, D127, and N85) that are important in catalysis (Yunus and Lima, 2006). Note that the position of Y87 is occupied by the side chain of Q122 in Ubc1 (top). RanGAP1 contains a glutamate (E526) that is a key part of the SUMO consensus sequence and interacts with a serine (S89) in Ubc9. In Ubc1 (top), T84 is found in a similar position, consistent with a role in substrate orientation. Images constructed with PyMOL (DeLano, 2008).
We first focused on the residues of cluster I, which we changed to the amino acids found at the same positions in Ubc4 (V83N, T84Δ, A86N). This mutant could be charged with ubiquitin by E1 to the same extent as wild-type Ubc1 (data not shown), indicating that it interacts normally with E1 and that the mutations did not cause gross conformational defects.
We tested the activity of the Ubc1-cluster I mutant in an APC-dependent reaction. As in our previous work, wild-type Ubc1 catalyzed the assembly of K48-linked chains on a radiolabeled cyclin substrate (Figure S3A). When chain formation was blocked with methyl-ubiquitin or a K48R ubiquitin mutant, Ubc1 catalyzed only the slow attachment of a single ubiquitin to a lysine on the substrate. The Ubc1-cluster I mutant also catalyzed the initial ubiquitin attachment to the substrate but did not promote assembly of K48-linked chains (Figure S3A).
Rates of initial ubiquitin attachment to the substrate were further assessed using reactions with methyl-ubiquitin. These rates were similar with Ubc1 and the Ubc1-cluster I mutant (Figure S3B). Thus, the mutant displays normal (low) activity in the attachment of the initial ubiquitin to the substrate, but has no detectable activity toward K48 of attached ubiquitins.
Threonine 84 in Ubc1 is required for ubiquitin chain formation
To identify the residues in cluster I that are responsible for K48 specificity, we tested the activity of single point mutants. All mutants were able to conjugate ubiquitin, and thus interact with E1, to the same degree as wild-type Ubc1 (data not shown). In APC-dependent reactions, Ubc1-V83N and Ubc1-A86N showed moderate decreases in K48-linked chain assembly, while Ubc1-T84Δ recapitulated the complete loss of K48-specific chain assembly that was seen with the Ubc1-cluster I mutant (Figure 3A). Again, the first step of protein target ubiquitination was not greatly affected in any of the mutants. The rate of methyl ubiquitin incorporation was 4-fold lower with the Ubc1-T84Δ mutant (Figure 3D).
Figure 3. The hydroxyl group of T84 is required for K48-specific polyubiquitination.
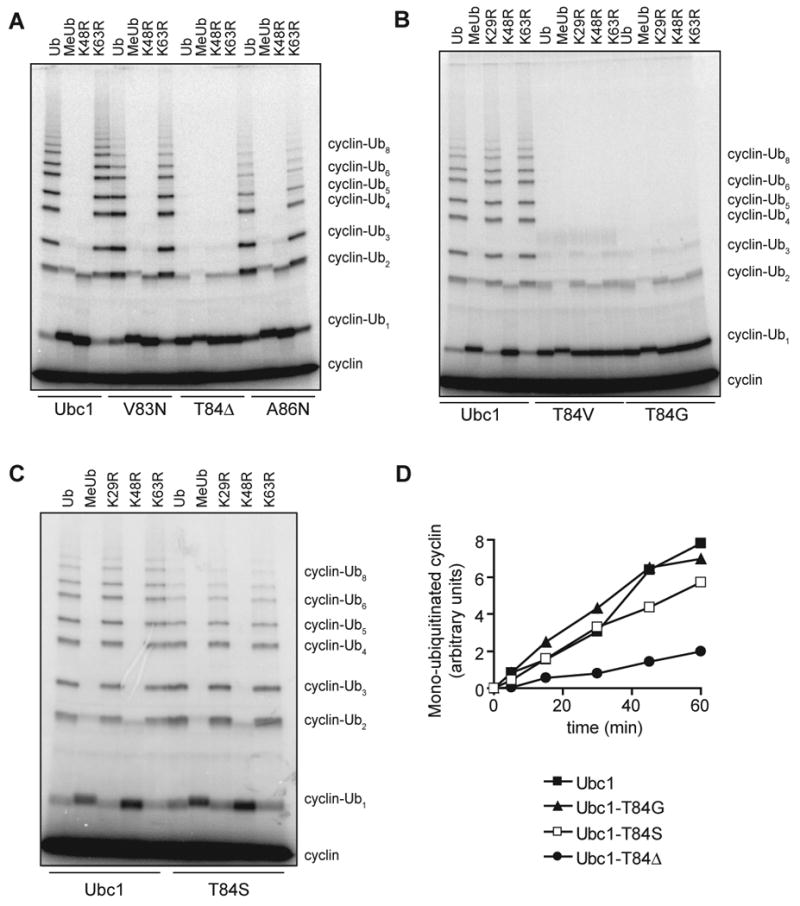
(A-C) Purified Ubc1 or the indicated mutant was incubated for 15 min with E1, ATP and the indicated ubiquitin species (MeUb indicates methyl-ubiquitin). E1/E2 mixes were added to APC, Cdh1 and 125I-cyclin B and incubated for 45 min at room temperature. Reaction products were analyzed by SDS-PAGE and PhosphorImager. See Figure S3 for analysis of the cluster I mutant containing all three mutations.
(D) Methyl-ubiquitin incorporation was measured in APC reactions with Ubc1 or the indicated mutant. Each E2 was incubated for 15 min with E1, ATP and methyl-ubiquitin, and then added to an APCCdh1 mix containing 125I-cyclin B at time zero. Reaction products over time were analyzed by SDS-PAGE and PhosphorImager, and quantified with ImageQuant.
Mutation of T84 to valine or glycine caused severe chain-assembly defects like those seen with Ubc1-T84Δ (Figure 3B), but the rate of methyl-ubiquitin incorporation was unaffected (Figure 3D). We also changed T84 to a serine, which contains a hydroxyl group but is one methyl group shorter than threonine. Ubc1-T84S was able to build K48-linked chains at a rate only slightly lower than that of wild-type Ubc1 (Figure 3C). We observed the same effects of T84Δ and T84S mutations in APC reactions with an N-terminal fragment of yeast securin (amino acids 1-110) (data not shown). We conclude that the hydroxyl group of threonine 84 is required for efficient K48-specific chain formation.
Threonine 84 promotes catalysis of diubiquitin synthesis
We next attempted to use the diubiquitin synthesis assay (Figure 1) to identify the step in the catalytic cycle that is defective in the T84G mutant. However, Ubc1-T84G displayed negligible activity under our normal reaction conditions (data not shown), consistent with its weak chain-assembly activity in APC reactions. We reasoned, however, that we might obtain greater activity at higher pH, as is the case with wild-type Ubc1. We measured the pH dependency of diubiquitin formation by Ubc1-T84G with saturating amounts of ubiquitin. Activity was greatly increased at higher pH, and the resulting sigmoidal curve (Figure 4A) had an inflection point at pH 9.4, about the same as that seen with wild-type Ubc1 (Figure 1C).
Figure 4. T84 is required for K48-specific catalysis.

(A) Rates of diubiquitin synthesis were measured with the Ubc1-T84G mutant and saturating ubiquitin (1 mM) over a range of pH values (6.86-10.26), as in Figure 1C. Experiments were done in triplicate and the error bars represent SEM.
(B) Diubiquitin formation by Ubc1 and Ubc1-T84G was measured over a range of ubiquitin concentrations (50 μM to 750 μM) in reactions like those in Figure 1A, except that the pH of the reaction was 10.26 and the reaction time was 10 s for Ubc1 and 2 min for Ubc1-T84G. Reaction products were analyzed by SDS-PAGE and PhosphorImager. Data from two experiments were quantified with ImageQuant, and the resulting plots were fit to a rectangular hyperbola using SigmaPlot. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
(C) Diubiquitin formation by Ubc1-T84G was measured with wild-type ubiquitin or ubiquitin-K48R in reactions like those in Figure 1A, except at pH 10.15 for 2 min. Reaction products were analyzed by SDS-PAGE and PhosphorImager. Data from four experiments were quantified with ImageQuant, and the resulting plots were fit to a rectangular hyperbola using Prism. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
We measured initial rates of diubiquitin synthesis with increasing amounts of ubiquitin for both Ubc1 and Ubc1-T84G at pH 10.26 (Figure 4B; Table S1). Apparent Kd values for Ubc1 and Ubc1-T84G were similar at pH 10.26, and were similar to the value obtained with wild-type Ubc1 at pH 7.4 (Figure 1B). The apparent _k_2 for wild-type Ubc1 at pH 10.26 was almost 50-fold higher than the value at pH 7.4. Most importantly, _k_2 with Ubc1-T84G at pH 10.26 was 100-fold lower than the value with wild-type Ubc1 at the same pH (Table S1). Thus, T84 is not essential for binding of ubiquitin but enhances the catalytic rate. Given that T84 is too distant from the active-site cysteine to contribute directly to catalysis in the traditional sense, its catalytic role might be to help position ubiquitin for attack and thereby reduce the formation of non-productive enzyme-substrate complexes (see Discussion).
The low catalytic rate of the Ubc1-T84G mutant was reduced only slightly by mutation of K48 in ubiquitin (Figure 4C). Thus, mutation of T84 almost completely abolishes K48-specific activity, and much of the low activity of the Ubc1-T84G mutant is directed toward non-K48 amino groups on ubiquitin.
Glutamine 122 and alanine 124 contribute to K48 specificity
We next assessed the function of the second cluster of Ubc1 residues that appeared to be good candidates for determinants of K48 specificity (see above). We constructed a mutant in which the two residues of cluster II were changed to the residues at those positions in Ubc4 (Q122L, A124P). This mutant was able to conjugate ubiquitin at rates similar to wild-type Ubc1 (data not shown). In APC-dependent reactions, the cluster II mutant displayed a major defect in K48-specific chain assembly (Figure S3C), as well as an unexpected defect not seen with the cluster I mutant: an increased rate of monoubiquitination on lysines in the cyclin substrate (Figure S3C, D).
To determine the effects of individual residues in cluster II, we tested single point mutants in APC-dependent reactions. The A124P mutation greatly reduced K48-specific chain formation without changing cyclin monoubiquitination; thus, a proline at this position disrupts K48 specificity (Figure 5A). The Q122L mutant displayed a blend of two defects like those seen with the cluster II mutant. First, the Q122L mutation caused a reduced rate of K48-linked chain formation (Figure 5A). In addition, the chains were more heterogeneous than those produced by wild-type Ubc1, apparently as a result of an increase in the number of lysines modified on the cyclin substrate (as revealed by the increased amount of diubiquitinated product in reactions with methyl- or K48R-ubiquitin; Figure 5A).
Figure 5. Q122 and A124 contribute to lysine specificity.
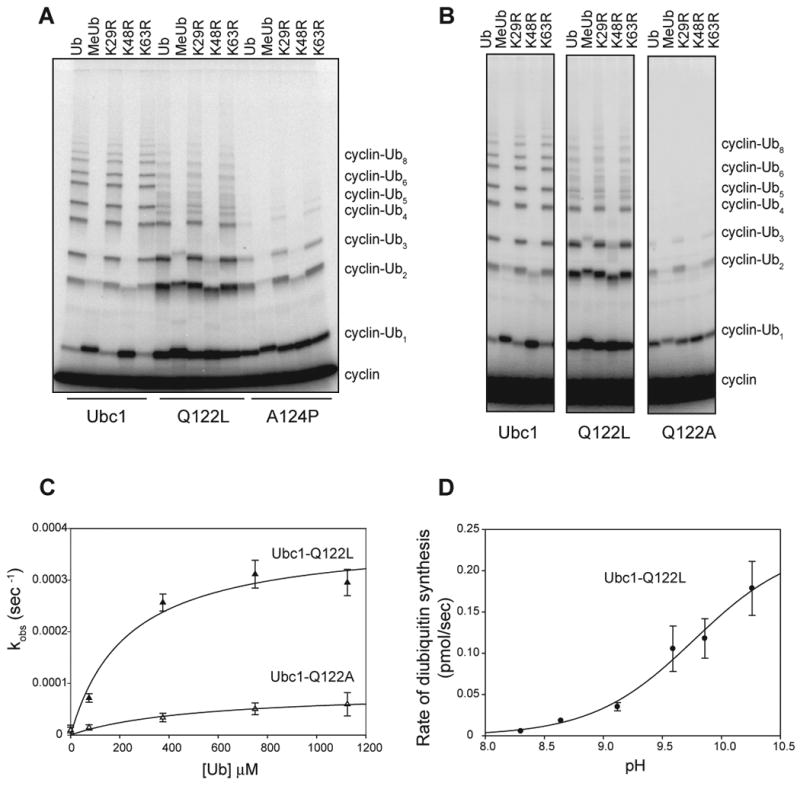
(A, B) Purified Ubc1 or the indicated mutant was incubated for 15 min with E1, ATP and the indicated ubiquitin species. E1/E2 mixes were added to APC, Cdh1 and 125I-cyclin B and incubated for 45 min at room temperature. Reaction products were analyzed by SDS-PAGE and PhosphorImager. See Figure S3 for analysis of the cluster II mutant containing both Q122L and A124P mutations. See Figure S4 for analysis of mutants in which Q122 is replaced with other amino acids.
(C) Initial rates of diubiquitin synthesis were measured in 2-min reactions with Ubc1-Q122L or Ubc1-Q122A over a range of ubiquitin concentrations, as in Figure 1A. Data (n=3) were fit to a rectangular hyperbola using the ligand-binding module of SigmaPlot. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
(D) Rates of diubiquitin synthesis were measured with the Ubc1-Q122L mutant and saturating ubiquitin (1 mM) over a range of pH values (6.86-10.26), as in Figure 1C. Experiments were done in duplicate and the error bars represent SEM.
Further insights were obtained by replacing Q122 with other amino acids. Replacement with alanine resulted in a severe defect in chain assembly but no gain in cyclin monoubiquitination (Figure 5B). Similar results were obtained when Q122 was changed to asparagine or glutamate (Figure S4). We speculate that the side chain at position 122 can serve two functions that are both lost in the alanine mutant. With a glutamine at this position, the result is a gain of one function: efficient K48-specific chain assembly. When a leucine is placed at this position, the enzyme displays some K48-specific activity but also gains a distinct second function: more efficient monoubiquitination of cyclin.
We explored the K48-specificity function of Q122 in more detail with the diubiquitin synthesis assay. Ubc1-Q122L was similar to Ubc1 in its affinity for ubiquitin, but apparent _k_2 decreased 4-fold, suggesting a catalytic defect in this mutant (Figure 5C; Table S1). Ubc1-Q122A displayed an even more severe (17-fold) catalytic defect. Analysis of Ubc1-Q122L activity over a range of pH values (Figure 5D) resulted in a sigmoidal curve with an approximate inflection point of at least 9.7, slightly higher than that seen with wild-type Ubc1 (pH 9.4, see Figure 1). At the highest pH tested, the activity of Ubc1-Q122L was 5-fold lower than that of wild-type Ubc1. Thus, Q122 (like T84) is not essential for ubiquitin binding but promotes catalytic rate. Given that the amount of deprotonated K48 is no longer limiting at high pH, the reduced activity of the T84G and Q122L mutants at high pH is likely due to a defect in K48 orientation, and not deprotonation.
Leucine at position 122 increases the rate of cyclin monoubiquitination
Our results suggest that Ubc1-Q122L has a defect in catalytic interactions with K48 of ubiquitin, while also displaying enhanced interactions with lysines on the cyclin substrate. We explored this issue further with direct measurements of the rate of methyl-ubiquitin incorporation (Figure 6A), which showed that the Q122L mutant modified substrate lysines about 5-fold more rapidly than wild-type Ubc1 and 13-fold more rapidly than Ubc1-Q122A. Ubc1-Q122L was not as efficient as Ubc4, which was 10-fold faster in these reactions than wild-type Ubc1.
Figure 6. Q122 limits the lysine specificity of Ubc1.
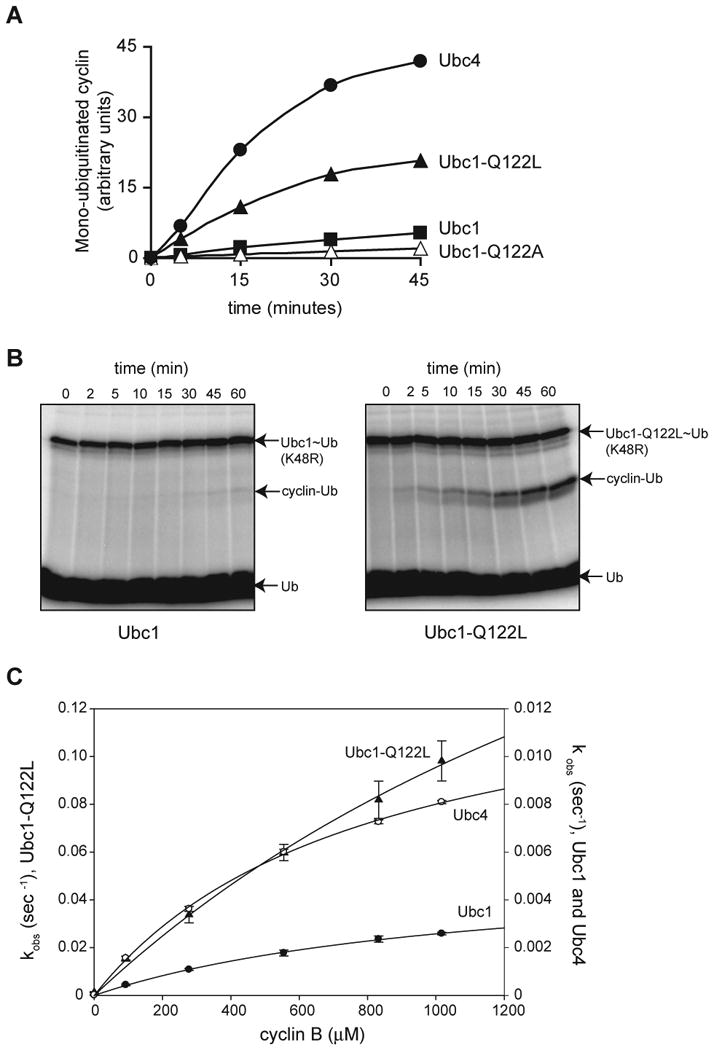
(A) Methyl-ubiquitin incorporation was measured in APC reactions with Ubc1, Ubc1-Q122L, Ubc1-Q122A or Ubc4, as in Figure 3D.
(B) Purified Ubc1 or Ubc1-Q122L was conjugated with 32P-labeled K48R-ubiquitin and treated with NEM and EDTA to prevent recharging of Ubc1 and E1. 12 mg of purified sea urchin cyclin B fragment was added, and samples were removed at the indicated times and analyzed by SDS-PAGE and PhosphorImager.
(C) Ubc1, Ubc1-Q122L or Ubc4 was conjugated with 32P-labeled K48R-ubiquitin and treated with NEM and EDTA to prevent recharging of Ubc1 and E1. E1/E2 was added to reactions containing increasing amounts of purified sea urchin cyclin B (90 μM to 1000 μM) at pH 10.26, for different times (2 min for Ubc1, 10 s for Ubc1-Q122L, and 1 min for Ubc4). Reaction products were analyzed by SDS-PAGE and PhosphorImager. Data from three experiments were quantified using ImageQuant, and the resulting plots were fit to a rectangular hyperbola using SigmaPlot. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
We characterized this change in lysine specificity by modifying the diubiquitin synthesis assay: instead of using unlabeled ubiquitin as the attacking substrate, we used unlabeled cyclin. APC was not present in these assays. When the rate of ubiquitin incorporation into cyclin was measured at pH 7.4, Ubc1 displayed very low activity (0.0027 pmol/min), whereas Ubc1-Q122L had a 30-fold higher rate (0.089 pmol/min) (Figure 6B).
We carried out more detailed measurements at pH 10.26, where we suspected the rate might be increased as in the diubiquitin synthesis assays. We measured the appearance of cyclin-ubiquitin over a range of unlabeled cyclin substrate concentrations (Figure 6C; Table S1). Wild-type Ubc1 displayed a low rate of cyclin ubiquitination (apparent _k_2 = 0.0055 s-1, about 12-fold lower than the value for Ubc1 with ubiquitin). In reactions with Ubc4, which is known to rapidly ubiquitinate lysines on cyclin, apparent _k_2 was 2.5-fold higher. In experiments with Ubc1-Q122L, we were unable to reach saturating cyclin concentrations, but a reasonable hyperbolic fit of the data suggested an approximate _k_2 of 0.33 s-1, about 60-fold higher than that with Ubc1. Thus, placing a leucine at this position in Ubc1 results in far more efficient attack by lysines in cyclin.
Ubc1-Q122L is more efficient than Ubc4 in cyclin ubiquitination reactions lacking the APC (Figure 6C), whereas the converse is true (Ubc4 is more active than Ubc1-Q122L) in APC-dependent reactions (Figure 6A). We cannot explain this difference. As stated in the Introduction, there is evidence that E3 binding promotes E2 catalytic function, and perhaps Ubc4 is more strongly stimulated than Ubc1 by APC binding.
We also characterized a Ubc1-T84Δ, Q122L double mutant, reasoning that the combination of these two mutations would change Ubc1 into an E2 that behaves like Ubc4, which has high activity toward cyclin lysines (like Ubc1-Q122L) and poor K48-specific chain-forming activity (like Ubc1-T84Δ). Indeed, we found that the double Ubc1 mutant generated ubiquitinated cyclin products similar to those seen with Ubc4 (Figure S5A), although the double mutant did not achieve the same level of cyclin monoubiquitination: it displayed a 3-fold higher activity than Ubc1 in methyl-ubiquitin incorporation, whereas Ubc4 exhibited 16-fold higher activity (data not shown).
We also attempted to determine if the critical amino acids we identified in Ubc1 are sufficient to allow K48-specific chain assembly when transferred to Ubc4. We constructed a version of Ubc4 in which the five residues of clusters I and II were all changed to the corresponding residues from Ubc1. This mutant (Ubc4-N82V, Δ83T, N84A, L120Q, P122A) was able to ubiquitinate multiple lysines on cyclin in the presence of APC (Figure S5B), but at a rate slightly lower than that with wild-type Ubc4. K48-specific chains were not observed. Thus, although T84, Q122, and A124 all contribute to the K48-specific activity of Ubc1, they are not sufficient to drive chain assembly when placed on the Ubc4 scaffold.
K48 specificity is required for Ubc1 function in the cell
To address the importance of K48 specificity for Ubc1 function in vivo, we tested the ability of Ubc1 mutants to suppress the mitotic arrest that occurs in cells lacking Ubc1. We used a strain in which endogenous UBC1 is under the control of the GAL1 promoter, allowing its expression to be shut off in glucose-containing medium (Rodrigo-Brenni and Morgan, 2007). We expressed selected UBC1 mutants at an ectopic locus in this strain and measured proliferation on glucose plates (Figure S6). Normal proliferation was observed in cells expressing wild-type UBC1 or UBC1-Q122L, and to a lesser extent in cells expressing UBC1-T84S. However, severe growth defects were seen in mutants lacking K48-specific activity (cluster I and II mutants, as well as Ubc1-T84Δ and Ubc1-T84G mutants) (Figure S6). Further analysis revealed that the Ubc1-T84G mutant had a mitotic-arrest phenotype like that seen in the absence of Ubc1 (data not shown). These results suggest that the K48 specificity of Ubc1 is required for its mitotic function.
Tyrosine 59 of ubiquitin is required for the K48 specificity of Ubc1
We next sought to identify residues in the attacking ubiquitin that are important for K48-specific chain formation by Ubc1. To develop an approach for rapidly generating and testing ubiquitin mutations in APC-dependent polyubiquitin assembly, we constructed a linear fusion linking the C-terminus of ubiquitin to the N-terminus of our model substrate, cyclin B. This provided a substrate that is essentially equivalent to monoubiquitinated cyclin, allowing us to bypass the first step in cyclin ubiquitination and focus on the formation of the K48-specific diubiquitin bond in standard APC assays.
We first characterized radiolabeled Ub-cyclin fusion protein in APC-dependent and -independent assays with Ubc1 or Ubc4 conjugated to unlabeled wild-type ubiquitin (Figure S7). In reactions with Ubc1, a small amount of Ub-cyclin was monoubiquitinated in reactions lacking APC, which presumably resulted from a low level of attack by the fusion protein on E2-ubiquitin conjugates in solution. Addition of APC-Cdh1 complex resulted in extensive chain assembly, which was largely abolished by a K48R mutation in the Ub-cyclin fusion. A background level of ubiquitinated products in K48R reactions presumably resulted from a low level of chain assembly on lysines in the cyclin substrate, as in typical Ubc1-dependent APC assays. In control reactions with Ubc4 as the E2, the typical di- and tri-ubiquitinated reaction products were similar with wild-type or K48R-cyclin fusion (Figure S7, right panel), indicating that Ubc4 was catalyzing ubiquitination of lysines on the substrate but not on K48 of ubiquitin, as in typical Ubc4 assays.
We used the Ub-cyclin fusion to test the effects of mutations in ubiquitin residues with solvent-exposed side chains near K48, reasoning that these residues might be involved in K48-specific attack of Ubc1. Our mutations included the hydrophobic patch mutation, I44A, which is known to disrupt K48-specific chain formation by the E2, Cdc34 (Petroski and Deshaies, 2005). Surprisingly, however, this mutation did not affect the assembly of ubiquitin chains by Ubc1 (Figure 7A). In fact, of 8 mutations tested, only the Y59A mutation resulted in a major chain-assembly defect like that seen with the K48R mutation. None of the mutations had any effect in reactions with Ubc4, which does not build chains under the conditions tested (data not shown).
Figure 7. Tyrosine 59 of ubiquitin is required for K48-specific polyubiquitination.
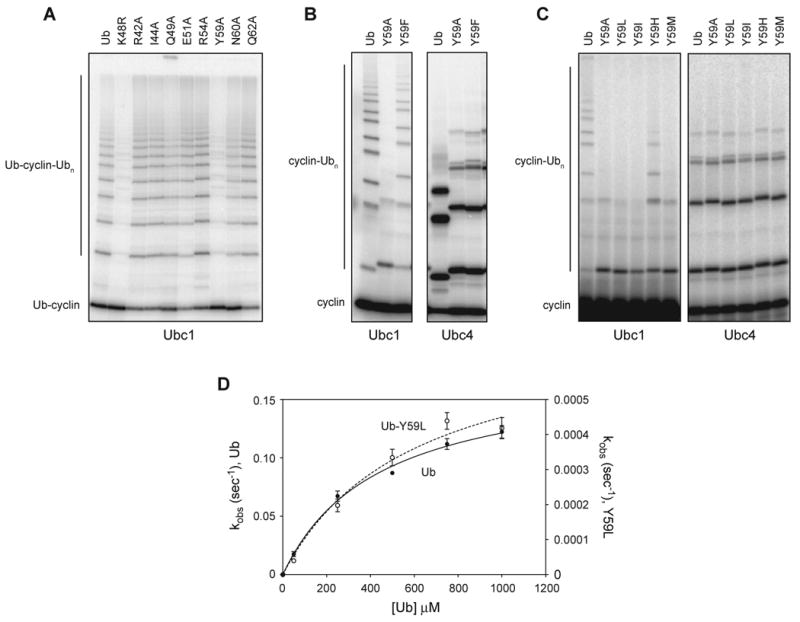
(A) Purified Ubc1 was conjugated to ubiquitin and added to APCCdh1 and a 32P-labeled ubiquitin-cyclin fusion protein carrying the indicated ubiquitin mutation. After 15 min, reaction products were analyzed by SDS-PAGE and PhosphorImager. See Figure S7 for characterization of reactions with the ubiquitin-cyclin fusion protein.
(B, C) Purified Ubc1 or Ubc4 was incubated with E1, ATP and the indicated ubiquitin species for 15 min, then mixed with APCCdh1 and 125I-cyclin B and incubated for 45 min (B) or 60 min (C) at room temperature. Reaction products were analyzed by SDS-PAGE and PhosphorImager. Wild-type ubiquitin in panel B is native protein that migrates more rapidly than the other ubiquitin species, which carry extra sequence derived from an excised tag at the N-terminus (see Experimental Procedures). In panel C, the left autoradiograph was exposed 20-fold longer than the one on the right.
(D) Ubc1 was incubated with E1, ATP and 32P-labeled K48R-ubiquitin for 15 min, and NEM and EDTA were added. E1/E2 mix was added to reactions with increasing amounts (50 μM to 1000 μM) of wild-type ubiquitin or ubiquitin-Y59L at pH 10.15, for different reaction times (5 s for wild-type ubiquitin; 10 min for ubiquitin-Y59L). Reaction products were analyzed by SDS-PAGE and PhosphorImager. Data from three experiments were quantified using ImageQuant, and the resulting plots were fit to a rectangular hyperbola using Prism. Error bars represent SEM. See Table S1 for apparent Kd and _k_2 values.
To address the importance of Y59 in the context of free ubiquitin, we next produced ubiquitin (without the cyclin fusion) containing the Y59A mutation and tested it in standard APC assays, in which the ubiquitin was used for both charging the E2 (donor ubiquitin) and as the attacking ubiquitin. As in the Ub-cyclin fusion, the Y59A mutation essentially abolished polyubiquitin chain assembly on cyclin (Figure 7B). This defect was specific for chain assembly: the first step in the reaction (cyclin monoubiquitination) was unaffected, indicating that the mutation had little effect on conjugation of ubiquitin to the E2 by E1, and little effect on the low level of nonspecific lysine ubiquitination by Ubc1. Similarly, the Y59A mutation did not affect the ubiquitination of lysines on cyclin in Ubc4-dependent reactions (Figure 7B, right). Thus, the Y59A mutation did not cause a general disruption of ubiquitin function but caused a specific defect in K48-specific chain assembly.
Replacement of Y59 with phenylalanine had a relatively minor effect on chain assembly (Figure 7B), indicating that the phenyl ring and not the hydroxyl moiety of Y59 is important for K48-specific diubiquitin bond formation.
Attempts to prepare large quantities of Y59A ubiquitin for detailed analysis were limited by instability of the protein during a heating step in the purification (see Experimental Procedures). Hoping to find a more stable variant, we constructed six additional Y59 mutations. Y59T and Y59N, like Y59A, were unstable at high temperature, but Y59I, Y59H, Y59M, and particularly Y59L were all near wild-type in stability (data not shown). Three of these mutants (Y59L, Y59I, Y59M) displayed severe defects in K48-linked chain assembly with Ubc1 but had no effect on cyclin ubiquitination by Ubc4 (Figure 7C). Mutation to histidine had a relatively mild effect on Ubc1 activity, further suggesting that the ring of Y59 is important for its function.
We analyzed the effects of the Y59L mutation in the diubiquitin synthesis assay, performed at pH 10.15 to enhance activity. The Y59L mutation had little effect on ubiquitin affinity for Ubc1 but caused a 200-fold decrease in catalytic rate (Figure 7D, Table S1). This rate decrease is similar to that seen with the Ubc1-T84G mutant using wild-type or K48R ubiquitin (Figure 4B, C), suggesting that there is little K48-specific activity remaining in reactions with the ubiquitin-Y59L mutant. Consistent with this interpretation, we found that reactions combining ubiquitin-Y59L and Ubc1-T84G displayed the same catalytic rate defect as either mutant alone (data not shown). We conclude that Y59 of ubiquitin, like T84 of Ubc1, is essential for K48-specific catalysis.
Discussion
Polyubiquitin chain assembly occurs in two steps. First, one of many lysines on a substrate attacks an E2-ubiquitin thioester, resulting in transfer of ubiquitin to the lysine (monoubiquitination). In the case of the yeast APC, this first reaction is catalyzed most efficiently by Ubc4, suggesting that the Ubc4 active site is specialized for recognition of lysines that are typically found in a disordered peptide context (Rodrigo-Brenni and Morgan, 2007). Second, a specific lysine in the attached ubiquitin attacks another E2-ubiquitin thioester to initiate assembly of a polyubiquitin chain. In yeast APC reactions, this second step is catalyzed by Ubc1, the active site of which is specialized for recognition of K48 in the globular ubiquitin protein.
Our results demonstrate that the K48 specificity of Ubc1 depends on a group of residues on two loops lying next to each other near the active-site cysteine (Figures 2, S2). Single mutations of key residues in this group (T84, Q122, and A124) result in major defects in K48-specific ubiquitin attack. We propose that the side chains of these residues form a landing pad that globally orients the incoming ubiquitin for efficient K48-specific attack of the Ubc1-ubiquitin thioester.
The mutations we analyzed in detail were found to reduce K48-specific catalytic rate but not ubiquitin binding affinity, as judged by the similar Kd values obtained in diubiquitin synthesis assays. Thus, we did not identify a mutation that significantly disrupts ubiquitin binding, perhaps indicating that the interacting surface depends on multiple low affinity interactions involving several residues.
The high Kd values we observed in our diubiquitin synthesis reactions are similar to those observed with other E2s (Petroski and Deshaies, 2005), suggesting that the affinity of the attacking ubiquitin for an E2-ubiquitin conjugate is very low. This interaction, in the 350 μM range, would not be expected to result in significant reaction rates at the low concentrations of ubiquitin (∼10 μM) inside the cell. Productive attack of Ubc1 occurs only at significant rates in the context of the E3, where binding of the substrate near the E2 would greatly increase its effective concentration and thereby drive the reaction. In addition, the binding of the E3 is thought to enhance the catalytic activity of the E2 (Ozkan et al., 2005; Petroski and Deshaies, 2005), which would increase _k_cat/KM even at low substrate occupancy.
Our results with the diubiquitin synthesis assay also indicate that the binding of ubiquitin to the Ubc1 active site, as reflected in apparent Kd, does not depend on the C-terminal UBA domain. This domain has ubiquitin-binding activity, but its function remains unclear. As deletion of the UBA domain reduces the processivity of chain assembly by the APC (Rodrigo-Brenni and Morgan, 2007), one possibility is that the UBA domain increases the affinity of Ubc1 for APC-bound substrates carrying multiple ubiquitins.
Much of our work focused on T84, which we found is required specifically for K48-dependent catalysis. Mutation of this residue abolished K48-linked chain formation without affecting the slow rate of ubiquitination of non-K48 lysines. The T84G mutant appears to display normal affinity for ubiquitin but has a major catalytic defect. Based on the normal pH dependency of the reaction with T84G, we conclude that T84 does not help suppress the p_Ka_ of the attacking lysine. In the tertiary structure of Ubc1, the T84 side chain is exposed on a surface loop and appears disordered in the NMR ensemble (Merkley and Shaw, 2004) (Figure 2). Mutation of T84 is not likely to change the conformation of this loop or nearby side chains in Ubc1. In addition, the T84 side chain is about 12 Å from the active-site cysteine of Ubc1, which is too distant for it to be directly involved in positioning K48 during the reaction. Interestingly, T84 is located in a position similar to that of S89 in Ubc9, which helps orient the substrate by interacting with a glutamate near the attacking lysine (Figure 2, bottom). We therefore suspect that T84 influences catalytic rate through indirect or global effects on K48 positioning, perhaps through interactions between the T84 hydroxyl and residues in ubiquitin other than K48.
We found that mutation of A124 to a proline (as in Ubc4) also resulted in a significant defect in K48-specific ubiquitination. The placement of a proline at this position, at the junction of a loop and an alpha helix (Figure 2), might be expected to generate local conformational changes that influence the positions of nearby residues, such as T84 or Q122.
Q122 is another important residue in K48-specific ubiquitination. Mutation of this residue to leucine, as in Ubc4, resulted in a 4-fold drop in the rate of K48-specific diubiquitin synthesis, and mutation to alanine or other amino acids greatly reduced K48-specific chain formation. As in the case of T84G, the Q122L and Q122A mutations affected catalytic rate and not ubiquitin binding affinity. Like the side chain of T84, that of Q122 is exposed and flexible on an ordered surface loop (Figure 2), and mutations at this site are not expected to cause local conformational changes in the protein backbone. A comparison of Ubc1 structure with that of Ubc9 (Figure 2) suggests that Q122 is well positioned to influence the orientation of the attacking lysine, either directly or indirectly through interactions with nearby residues on ubiquitin.
Interestingly, the decreased K48-specific activity of the Q122L mutant was accompanied by an increase in its activity toward non-K48 lysines on the cyclin substrate. This change in behavior was particularly dramatic in studies of the attack of Ubc1 by cyclin substrate in the absence of the APC. Wild-type Ubc1 ubiquitinated lysines on the cyclin substrate very poorly, whereas Ubc1-Q122L displayed 60-fold higher activity toward cyclin. Thus, a leucine at this position greatly enhances the ubiquitination of lysines in the cyclin substrate. The glutamine that is normally at this position in Ubc1 limits its activity toward these lysines.
Our results with the Q122L mutant are reminiscent of previous studies of the SUMO E2, Ubc9 (Yunus and Lima, 2006). Ubc9 contains a tyrosine, Y87, that contributes to a hydrophobic microenvironment that helps position the attacking lysine and suppress its p_Ka_ (Figure 2, bottom). A tyrosine is found at this position in several E2s (Yunus and Lima, 2006). Many other E2s, however, do not have this tyrosine but instead contain a leucine, at a different position, that seems to fulfil the tyrosine's role. Ubc4 is a member of this latter group: indeed, it is this leucine of Ubc4 (L120) that is changed to a glutamine (Q122) in Ubc1 (Figure S2). Thus, it seems likely that this leucine in Ubc4 helps catalyze non-K48 lysine ubiquitination. Ubc1, however, is unique among yeast E2s in not containing either the tyrosine (which is S81 in Ubc1) or the leucine (Q122). Furthermore, our pH dependency studies suggest that the lysine p_Ka_ is not suppressed by Ubc1 as much as it is in Ubc9 reactions. We therefore speculate that deprotonation and positioning of the attacking K48 side chain involves mechanisms that are at least partly distinct from those employed by Ubc9.
We also explored the residues in ubiquitin itself that are important for K48-linked chain formation. Mutation of numerous residues adjacent to K48 of ubiquitin had little effect, but K48-specific activity was severely inhibited by mutation of Y59 to alanine or small hydrophobic residues. Activity was not greatly affected by mutation to phenylalanine and only partly inhibited by mutation to histidine, arguing that the ring structure of Y59, and not simply its hydrophobicity, is critical for activity. Interestingly, iodination of Y59 reduces ubiquitin chain assembly by E2-25K, the human ortholog of Ubc1 (Pickart et al., 1992), indicating that the function of the Y59 ring is disrupted by iodination.
Detailed analysis of the Y59L ubiquitin mutant revealed a severe reduction of catalytic rate, with little effect on affinity for Ubc1. Y59 is therefore required for the catalysis of K48-specific ubiquitination, providing a potential example of substrate-assisted catalysis. In the tertiary structure of ubiquitin, Y59 does not contact the K48 side chain but could partly limit its movement. The Y59 side chain is partially buried and interacts with the hydrophobic protein core (notably L50), and the reduced heat stability we observed with some Y59 mutants might have resulted from local packing defects. The Y59L mutant, on the other hand, is nearly as heat-stable as wild-type ubiquitin but still displays a major catalytic defect. We suspect that the ring of Y59 in ubiquitin, like the hydroxyl of T84 in Ubc1, helps orient ubiquitin for productive attack of the E2. Indeed, Y59 and T84 might depend on each other for a common orientation function, which would explain our observation that mutation of either residue (or the two mutations in combination) abolishes almost all K48-specific activity. Another intriguing possibility arises from the fact, mentioned above, that Ubc1 is unusual among E2s because it does not contain the tyrosine or leucine that helps form a hydrophobic microenvironment around the attacking lysine. Perhaps Y59 on the ubiquitin substrate helps fulfil this role.
Numerous lines of evidence suggest that the K48 specificity of Ubc1 depends on mechanisms that are distinct from those used by another K48-specific E2, Cdc34. First, our work shows that Ubc1 activity toward K48 depends on a polar cluster of residues that are found near the cysteine of the Ubc1 active site but are not found in Cdc34; on the other hand, the K48 specificity of Cdc34 depends on an acidic loop, not found in Ubc1, that is inserted in the active site (Figure S2) (Petroski and Deshaies, 2005). Second, we found that Ubc1 activity was unaffected by mutation of I44 in the ubiquitin hydrophobic patch, whereas this mutation greatly reduces Cdc34 activity. Finally, iodination of Y59 was shown previously to inhibit activity with E2-25K, as mentioned above, but did not affect activity with the human Cdc34 ortholog (Pickart et al., 1992). We are left with the remarkable conclusion that two enzymes have evolved distinct mechanisms to catalyze K48-specific polyubiquitin assembly.
It is becoming clear that the E2 active site contains residues that use varied and poorly understood mechanisms to catalyze lysine attack. Moreover, the active sites of different E2 proteins contain unique combinations of residues that confer specificity for lysines in distinct contexts. General E2s, such as Ubc4, carry residues that promote effective attack by a lysine in a polypeptide context that appears nonspecific and disordered. Other E2s (e.g. Ubc1, Ubc13, and Cdc34) carry active-site residues that direct enzyme activity toward specific lysines, such as K48 or K63, within the globular context of ubiquitin. In addition, the ubiquitin substrate itself contributes residues that help determine lysine specificity.
Experimental Procedures
Plasmids, expression and purification of recombinant proteins
Expression and purification of Ubc1, Ubc4 and Ubc1-ΔUBA were described previously (Rodrigo-Brenni and Morgan, 2007). Ubc1 and Ubc4 mutations were created by site-directed mutagenesis.
A plasmid encoding GST-K48R-ubiquitin (Petroski and Deshaies, 2005) was transformed into BL21 cells. A single colony was incubated overnight in 100 ml LB/Amp media at 37°C, diluted into 1 liter of fresh media and grown at 37°C to an OD600 of 0.60, after which IPTG was added to 1 mM for 4 h at 37°C. Cells were harvested by centrifugation, washed in cold water and frozen in liquid nitrogen. The frozen pellet was melted rapidly and incubated with 4 vol breakage buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10% glycerol, protease inhibitor cocktail, 1 mM DTT, 1.25 mg/ml lysozyme, 500 U DNase) at room temperature for 30 min. The lysate was centrifuged (1 h, 100,000×g, 4°C), and the supernatant was bound to glutathione (GSH) sepharose. The bound material was washed 4 times in 10 vol wash buffer (50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 10% glycerol, 1mM EDTA) for 15 min. The GST fusion protein was eluted with 6 ml elution buffer (breakage buffer plus 10 mM reduced glutathione).
The linear fusion between ubiquitin and sea urchin cyclin B was created by standard cloning techniques. Briefly, a PCR product encoding cyclin B was linked to the 3′ end of GST-ubiquitin or GST-K48R-ubiquitin. A 6xHIS tag was also introduced at the C-terminus of GST. Ubiquitin mutations in the fusion protein were created by site-directed mutagenesis. The fusion protein was purified as described for GST-K48R-ubiquitin, except that the GSH purification step was followed by metal affinity chromatography. Briefly, purified GST-fusions were dialyzed overnight into dialysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM MgCl2 and 10% glycerol) and incubated with IDA-sepharose beads charged with Co+2. Bound material was washed with dialysis buffer and eluted twice with 3 ml dialysis buffer containing 500 mM imidazole. Fractions were pooled and dialyzed.
All GST-tagged proteins also contain a TEV protease cleavage site at the C-terminus of GST, followed by a cAMP-dependent protein kinase A (PKA) phosphorylation site at the N-terminus of ubiquitin and its derivatives. To produce radiolabeled K48R-ubiquitin or ubiquitin-cyclin fusion protein, 50 μl of GST-K48R-ubiquitin or GST-ubiquitin-cyclin B was incubated with 10 μCi γ32P-ATP, 1.67 nmol ATP and 1 μl PKA (New England Biolabs) for 2 h at 30°C. After removal of ATP by gel filtration, the proteins were diluted to 250 μl (in 50 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 0.5 mM EDTA and 1 mM DTT) and incubated with 5 μl 6xHIS-TEV protease (5 mg/ml) for 1 h at 30°C. Cleaved material was incubated at 65°C for 15 min, followed by 5 min on ice. Precipitated material was removed by centrifugation, leaving the ubiquitin or fusion protein in the supernatant.
For detailed comparisons of ubiquitin with ubiquitin Y59 and K48 mutants, the desired point mutation was introduced into the GST-ubiquitin construct, and the resulting fusion protein was expressed and purified on GSH-sepharose as described above. Following cleavage with TEV protease (overnight at room temperature), ubiquitin was purified by two heat treatments (20 min at 65°C), each followed by centrifugation (25,000 rpm, 30 min, 4°C) to remove insoluble material. Ubiquitin was dialyzed into diubiquitin synthesis buffer (50 mM Citrate/Bis-Tris-Propane, pH 8.0, 100 mM NaCl, 10% Glycerol, 1 mM MgCl2) and concentrated to ∼3 mM with an Amicon ultra concentrator.
APC-dependent ubiquitination assays
Preparation of reaction components (E1, ATP, ubiquitin, APC, Cdh1, ubiquitin-E2 conjugates, and the N-terminal fragment [aa 13-110] of sea urchin cyclin B) was described previously (Carroll and Morgan, 2005; Rodrigo-Brenni and Morgan, 2007). 2.5 μg of Ubc1 or Ubc4 was used in each reaction (final concentration 5-8 μM) unless otherwise noted. APC (1.5 μl of 10 nM stock), Cdh1 (0.5 μl of 1.5 μM stock) and radiolabeled substrate (N-terminus of sea urchin cyclin B, N-terminus of yeast securin, or ubiquitin-cyclin B fusions) were mixed at room temperature. Reactions were initiated by combining the E1/E2 mix with the APC mix in a final volume of 20 μl. Reaction products were analyzed by SDS-PAGE and visualized with a PhosphorImager.
Diubiquitin synthesis assays
Ubc1 (0.05 μl of a 200 μM stock) was incubated with E1 (0.2 μl of a 9 μM stock), 32P-labeled K48R-ubiquitin (5 μl of 250 μl labeling reaction) and ATP (1 μl of a 20 mM stock) in 50 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM MgCl2 and 10% glycerol (total volume 7 μl) for 15 min at room temperature. The charging reaction was treated with 10 mM N-ethylmaleimide (NEM) and 50 mM EDTA for 15 min at room temperature. The treated reaction was incubated with either saturating amounts of ubiquitin (1 mM) or a range of ubiquitin concentrations in a final volume of 20 μl. Reactions were stopped by addition of nonreducing sample buffer containing 10 mM NEM, analyzed by SDS-PAGE and visualized by PhosphorImager. The appearance of diubiquitin and the remaining E2∼Ub conjugates were quantified with ImageQuant by drawing rectangles around the appropriate bands and converting counts detected to pmol of 32P-K48R ubiquitin.
Studies of pH dependency
Single discharge assays were carried out at a range of pH (6.86-10.26) as described above but with the following exceptions. Labeling of 32P-K48R-ubiquitin was carried out in 50 mM Citrate/Bis-Tris-Propane, pH 8.0, 100 mM NaCl, 1 mM MgCl2 and 10% glycerol. Charging reactions were stopped by treatment with NEM and EDTA. Treated reactions were added to reactions containing 1 mM ubiquitin at a range of pH values. The pH of the 50 mM Citrate/Bis-Tris-Propane was adjusted by adding different dilutions of HCl and NaOH (1/20 the volume of the final reaction). The final pH was measured at room temperature. Samples were removed at various times, denatured in nonreducing sample buffer containing 10 mM NEM, analyzed by SDS-PAGE and visualized with a PhosphorImager. The rate of diubiquitin formation at different pH values was calculated from linear curve fitting of the plots of pmol diubiquitin formed as a function of time. The rates were plotted against pH, and the p_Ka_ was estimated by nonlinear curve fitting using the sigmoidal function module of SigmaPlot. The shape of the curve suggests that only the basic form of a general base is involved in catalysis (Fersht, 1998).
Supplementary Material
Suppl Info
Acknowledgments
We thank M. Matyskiela, M. Lopez, J. Gross, D. Stanley, D. Mandell, T. Kortemme, and R. Deshaies for valuable discussions, and J. Gross and D. Stanley for comments on the manuscript. This work was supported by funding from the National Institute of General Medical Sciences (GM53270) and a fellowship from the National Science Foundation (to M.C.R.-B.).
Footnotes
Supplemental Information: The supplemental information includes seven figures, one table, and Supplemental References.
References
- Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- Capili AD, Lima CD. Taking it step by step: mechanistic insights from structural studies of ubiquitin/ubiquitin-like protein modification pathways. Curr Opin Struct Biol. 2007;17:726–735. doi: 10.1016/j.sbi.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Morgan DO. Enzymology of the Anaphase-Promoting Complex. Meth Enzymol. 2005;398:219–230. doi: 10.1016/S0076-6879(05)98018-X. [DOI] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Xu Y, Chau V. Tertiary structures of class I ubiquitin-conjugating enzymes are highly conserved: crystal structure of yeast Ubc4. Biochemistry. 1993;32:13809–13817. doi: 10.1021/bi00213a009. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA, USA: DeLano Scientific LLC; 2008. http://www.pymol.org. [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye BT, Schulman BA. Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu Rev Biophys Biomol Struct. 2007;36:131–150. doi: 10.1146/annurev.biophys.36.040306.132820. [DOI] [PubMed] [Google Scholar]
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat Struct Mol Biol. 2006;13:915–920. doi: 10.1038/nsmb1148. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and Mechanism in Protein Science. New York: W.H. Freeman and Co.; 1998. [Google Scholar]
- Hamilton KS, Ellison MJ, Barber KR, Williams RS, Huzil JT, McKenna S, Ptak C, Glover M, Shaw GS. Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure. 2001;9:897–904. doi: 10.1016/s0969-2126(01)00657-8. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Rodrigo-Brenni MC, Morgan DO. Mechanisms of ubiquitin transfer by the anaphase-promoting complex. J Biol. 2009;8:92. doi: 10.1186/jbiol184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkley N, Shaw GS. Solution structure of the flexible class II ubiquitin-conjugating enzyme Ubc1 provides insights for polyubiquitin chain assembly. J Biol Chem. 2004;279:47139–47147. doi: 10.1074/jbc.M409576200. [DOI] [PubMed] [Google Scholar]
- Ozkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci U S A. 2005;102:18890–18895. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Haldeman MT, Kasperek EM, Chen Z. Iodination of tyrosine 59 of ubiquitin selectively blocks ubiquitin's acceptor activity in diubiquitin synthesis catalyzed by E2(25K) J Biol Chem. 1992;267:14418–14423. [PubMed] [Google Scholar]
- Pierce NW, Kleiger G, Shan SO, Deshaies RJ. Detection of sequential polyubiquitylation on a millisecond timescale. Nature. 2009;462:615–619. doi: 10.1038/nature08595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708–714. doi: 10.1038/nsmb962. [DOI] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007;130:127–139. doi: 10.1016/j.cell.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. doi: 10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- Thornton BR, Toczyski DP. Precise destruction: an emerging picture of the APC. Genes Dev. 2006;20:3069–3078. doi: 10.1101/gad.1478306. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PY, Hanlon M, Eddins M, Tsui C, Rogers RS, Jensen JP, Matunis MJ, Weisman AM, Wolberger C, Pickart CM. A conserved catalytic residue in the ubiquitin-conjugating enzyme family. Embo J. 2003;22:5241–5250. doi: 10.1093/emboj/cdg501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunus AA, Lima CD. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat Struct Mol Biol. 2006;13:491–499. doi: 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl Info